

Extracellular Vesicle-Based Strategies for Tumor Immunotherapy


Abstract
1. Introduction
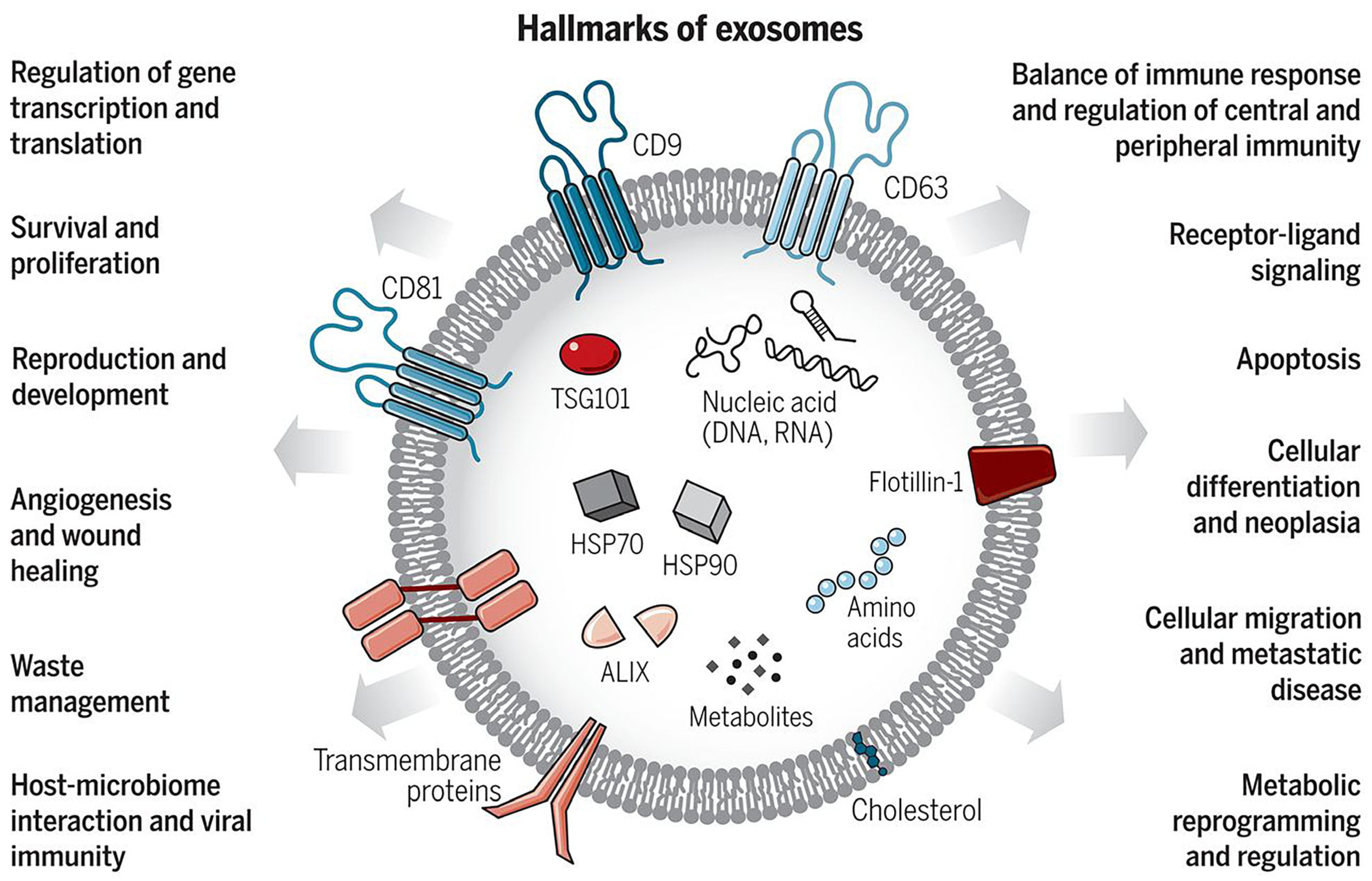
2. The Biogenesis and Characteristics of Extracellular Vesicles
2.1. Classification of Extracellular Vesicles
2.2. Isolation Methods for Extracellular Vesicles
2.3. Identification and Analysis of Extracellular Vesicles
3. Extracellular Vesicles for Diagnostic and Therapeutic Potentials
3.1. Extracellular Vesicles as Diagnostic Tool
3.2. Extracellular Vesicles for Tracking Cancer Progression
3.3. Extracellular Vesicles for Monitoring Treatment Response
4. Cancer Immunotherapy and Extracellular Vesicles
4.1. Immune Mechanisms
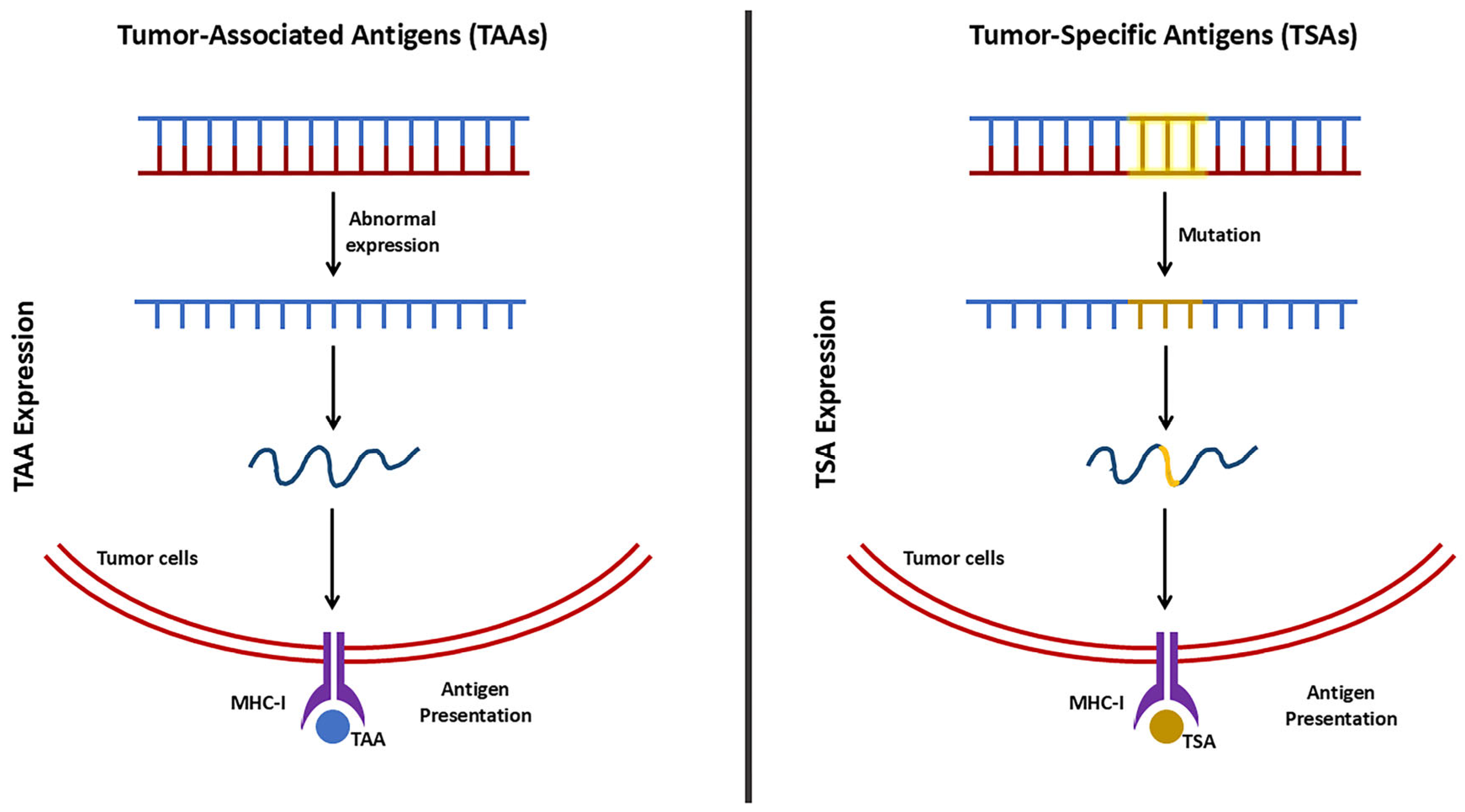
4.1.1. Anti-Tumor Immunity
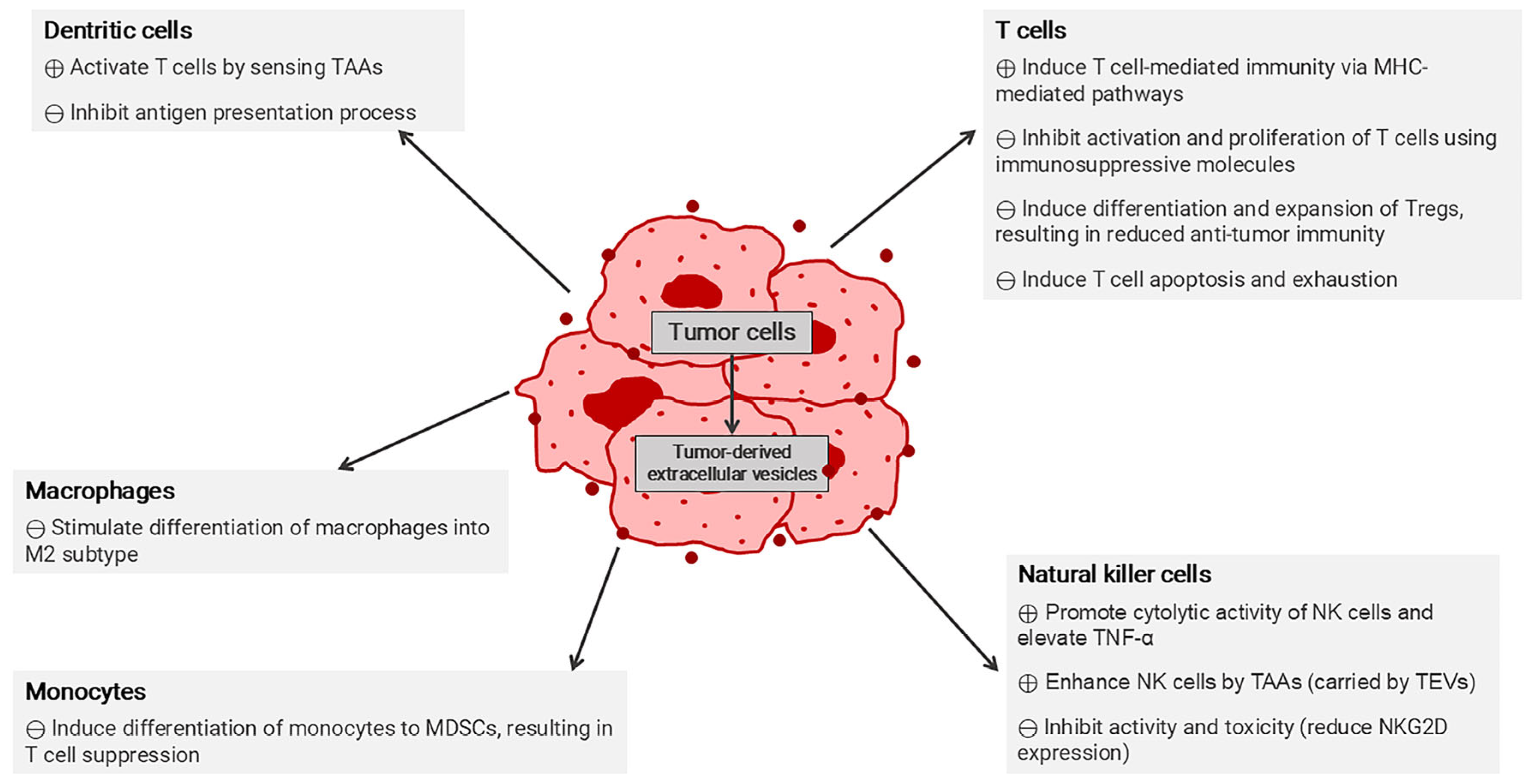
4.1.2. Immunosuppressive Mechanisms in Tumor Microenvironment
4.1.3. Immune Checkpoint Pathways in Tumor Immune Evasion
4.2. Extracellular Vesicles in Cancer Immunotherapy
4.2.1. Extracellular Vesicles for Targeted Delivery
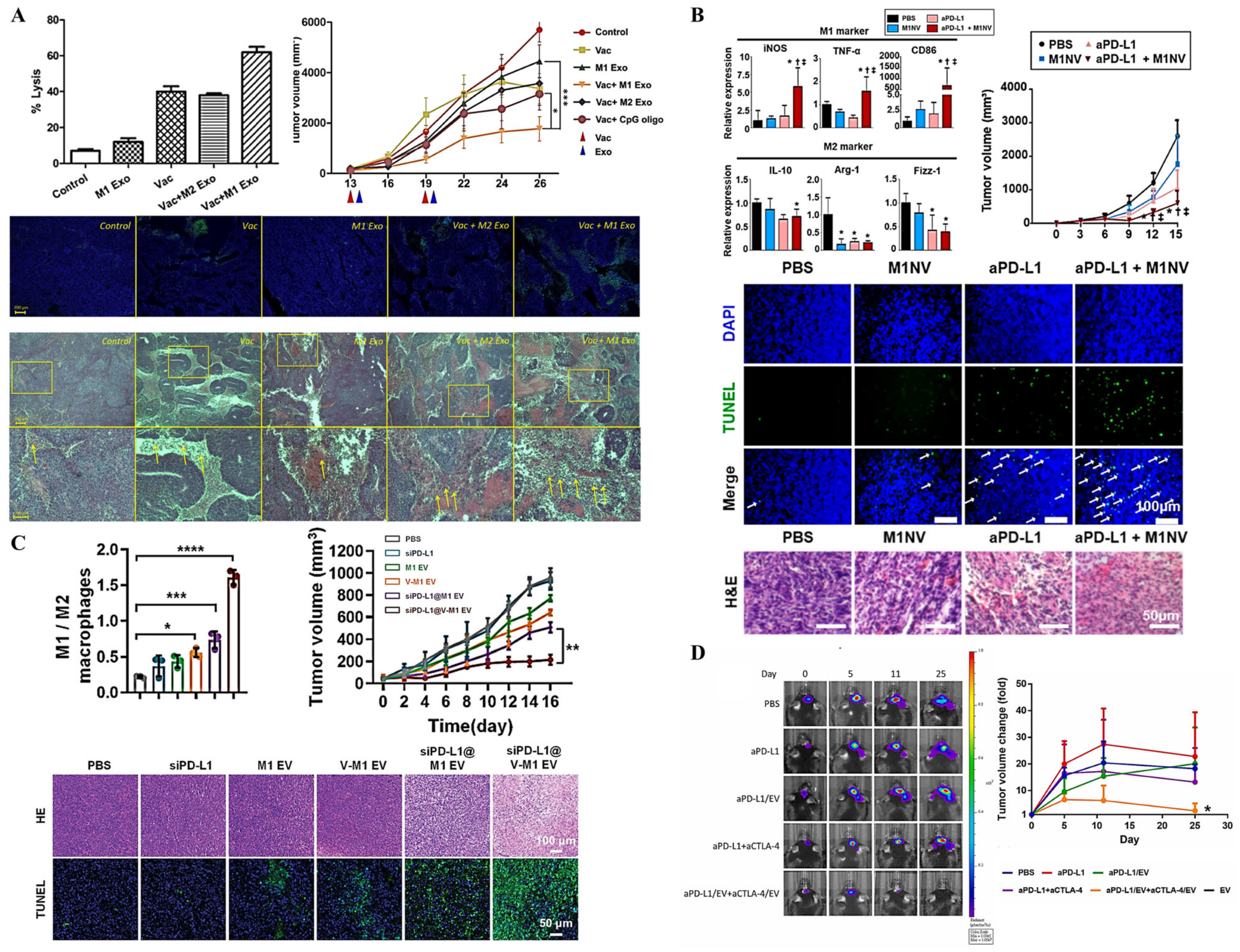
4.2.2. Extracellular Vesicles as Vaccines
4.2.3. Extracellular Vesicles for Overcoming Immunosuppressive Tumor Microenvironment
4.2.4. Combining Extracellular Vesicles with Immune Checkpoint Inhibitors
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAR | Chimeric Antigen Receptor |
| FDA | Food and Drug Administration |
| ICIs | Immune Checkpoint Inhibitors |
| EVs | Extracellular Vesicles |
| MISEV | Minimal Information for Studies of Extracellular Vesicles |
| DNA | Deoxyribonucleic Acid |
| RNA | Ribonucleic Acid |
| ILVs | Intraluminal Vesicles |
| MVEs | Multivesicular Endosomes |
| CD | Cluster of Differentiation |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| NTA | Nanoparticle Tracking Analysis |
| DLS | Dynamic Light Scattering |
| TEM | Transmission Electron Microscopy |
| SEM | Scanning Electron Microscopy |
| ARF6 | ADP-Ribosylation Factor 6 |
| SDS-PAGE | Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis |
| PBP | Polymer-based Precipitation |
| CA125 | Cancer Antigen 125 |
| TME | Tumor Microenvironment |
| TP53 | Tumor Protein p53 |
| CRC | Colorectal Cancer |
| HGS | Hepatocyte Growth Factor-Regulated Tyrosine Kinase Substrate |
| CSF | Cerebrospinal Fluid |
| PD | Parkinson’s Disease |
| EDIL3 | Epidermal Growth Factor-Like Repeats and Discoidin I-Like Domains 3 |
| EGFR | Epidermal Growth Factor Receptor |
| FAK | Focal Adhesion Kinase |
| CT | Computed Tomography |
| MRI | Magnetic Resonance Imaging |
| CA15-3 | Carcinoma Antigen 15-3 |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| TGF-β3 | Transforming Growth Factor-beta 3 |
| CRT | Chemoradiation Therapy |
| GWAS | Genome-Wide Association Studies |
| UBC | Urinary Bladder Cancer |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| TAAs | Tumor-Associated Antigens |
| TSAs | Tumor-Specific Antigens |
| MHC | Major Histocompatibility Complex |
| TCRs | T Cell Receptors |
| CD8+ T cells | Cytotoxic T Cells |
| Th cells | CD4+ Helper T Cells |
| APCs | Antigen Presenting Cells |
| DCs | Dendritic Cells |
| BCR | B Cell Receptor |
| MDSCs | Myeloid-Derived Suppressor Cells |
| TAMs | Tumor-Associated Macrophages |
| Tregs | Regulatory CD4+ T Cells |
| NK | Natural Killer |
| M-MDSCs | Monocytic MDSCs |
| PMN-MDSCs | Polymorphonuclear MDSCs |
| STAT | Signal Transducer and Activator of Transcription |
| ER | Endoplasmic Reticulum |
| NF-κB | Nuclear Factor Kappa B |
| cAMP | Cyclic Adenosine Monophosphate |
| IFN | Interferon |
| PTGES | Prostaglandin E Synthase |
| COX-2 | Cyclooxygenase-2 |
| C/EBPβ | CCAAT-Enhancer-Binding Protein-β |
| ARG1 | Arginase 1 |
| iNOS | Inducible Nitric Oxide Synthase |
| ROS | Reactive Oxygen Species |
| VEGF | Vascular Endothelial Growth Factor |
| IL | Interleukin |
| PGE2 | Prostaglandin E2 |
| PD-L1 | Programmed Cell Death-Ligand 1 |
| CCL18 | Chemokine (C-C motif) Ligand 18 |
| ECM | Extracellular Matrix |
| Teffs | Effector T Cells |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| PD-1 | Programmed Cell Death Protein 1 |
| IFN-γ | Interferon-Gamma |
| PI3K | Phosphatidylinositol 3-Kinase |
| RAS | Rat Sarcoma Virus |
| CTL | Cytotoxic T Lymphocyte |
| Akt | Serine/Threonine Protein Kinase |
| TNF-α | Tumor Necrosis Factor |
| NSCLC | Non-Small Cell Lung Cancer |
| miRNA | MicroRNA |
| LNP | Lipid Nanoparticles |
| Th1 | T-helper Cell Type 1 |
| TRP2 | Tyrosinase-Related Protein 2 |
| IMC | Immature Myeloid Cells |
| SIRPα | Signal Regulatory Protein Alpha |
| circUHRF1 | Circular Ubiquitin-Like with PHD and Ring Finger Domain 1 RNA |
| RGD | Arginine-Glycine-Aspartic Acid |
| MLKL | Mixed Lineage Kinase Domain-Like Protein |
| RIPK3 | Receptor-Interacting Protein Kinase 3 |
| GzmB | Granzyme B |
| FasL | Fas Ligand |
| TIM-3 | T Cell Immunoglobulin and Mucin Domain 3 |
| LAG-3 | Lymphocyte Activation Gene 3 |
References
- Luo, Q.; Zhang, L.; Luo, C.; Jiang, M. Emerging strategies in cancer therapy combining chemotherapy with immunotherapy. Cancer Lett. 2019, 454, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Kichloo, A.; Albosta, M.; Dahiya, D.; Guidi, J.C.; Aljadah, M.; Singh, J.; Shaka, H.; Wani, F.; Kumar, A.; Lekkala, M. Systemic adverse effects and toxicities associated with immunotherapy: A review. World J. Clin. Oncol. 2021, 12, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Thum, T. Exosomes: New players in cell–cell communication. Int. J. Biochem. Cell Biol. 2012, 44, 2060–2064. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Susa, F.; Limongi, T.; Dumontel, B.; Vighetto, V.; Cauda, V. Engineered Extracellular Vesicles as a Reliable Tool in Cancer Nanomedicine. Cancers 2019, 11, 1979. [Google Scholar] [CrossRef]
- Chargaff, E. Cell structure and the problem of blood coagulation. J. Biol. Chem. 1945, 160, 351–359. [Google Scholar] [CrossRef]
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [CrossRef]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Aaronson, S.; Behrens, U.; Orner, R.; Haines, T. Ultrastructure of intracellular and extracellular vesicles, membranes, and myelin figures produced by Ochromonas danica. J. Ultrastruct. Res. 1971, 35, 418–430. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. [Google Scholar] [CrossRef]
- Trams, E.G.; Lauter, C.J.; Salem, N., Jr.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Borges, F.T.; Reis, L.A.; Schor, N. Extracellular vesicles: Structure, function, and potential clinical uses in renal diseases. Braz. J. Med. Biol. Res. 2013, 46, 824–830. [Google Scholar] [CrossRef]
- Ronquist, G.; Nilsson, B.O.; Hjertën, S. Interaction Between Prostasomes and Spermatozoa From Human Semen. Arch. Androl. 1990, 24, 147–157. [Google Scholar] [CrossRef]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef]
- Gardiner, C.; Di Vizio, D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef]
- Grant, R.; Ansa-Addo, E.; Stratton, D.; Antwi-Baffour, S.; Jorfi, S.; Kholia, S.; Krige, L.; Lange, S.; Inal, J. A filtration-based protocol to isolate human Plasma Membrane-derived Vesicles and exosomes from blood plasma. J. Immunol. Methods 2011, 371, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Iii, N.E.G.; Chen, P.; Antes, T.J. Isolation of Extracellular Vesicles from Murine Bronchoalveolar Lavage Fluid Using an Ultrafiltration Centrifugation Technique. J. Vis. Exp. 2018, 141, e58310. [Google Scholar] [CrossRef]
- Zhang, M.; Jin, K.; Gao, L.; Zhang, Z.; Li, F.; Zhou, F.; Zhang, L. Methods and Technologies for Exosome Isolation and Characterization. Small Methods 2018, 2, 21. [Google Scholar] [CrossRef]
- Zeringer, E.; Barta, T.; Li, M.; Vlassov, A.V. Strategies for Isolation of Exosomes. Cold Spring Harb. Protoc. 2015, 2015, 319–323. [Google Scholar] [CrossRef]
- Palmieri, V.; Lucchetti, D.; Gatto, I.; Maiorana, A.; Marcantoni, M.; Maulucci, G.; Papi, M.; Pola, R.; De Spirito, M.; Sgambato, A. Dynamic light scattering for the characterization and counting of extracellular vesicles: A powerful noninvasive tool. J. Nanoparticle Res. 2014, 16, 2583. [Google Scholar] [CrossRef]
- Mathieu, M.; Névo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D.; et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021, 12, 4389. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Hoover, H.; Clancy, J.; Schweitzer, J.; Suckow, M.A.; Schroeder, V.; Castellino, F.J.; Schorey, J.S.; D’Souza-Schorey, C. ADP-Ribosylation Factor 6 Regulates Tumorigenic and Invasive Properties In Vivo. Cancer Res. 2009, 69, 2201–2209. [Google Scholar] [CrossRef]
- Sedgwick, A.E.; Clancy, J.W.; Balmert, M.O.; D’souza-Schorey, C. Extracellular microvesicles and invadopodia mediate non-overlapping modes of tumor cell invasion. Sci. Rep. 2015, 5, 14748. [Google Scholar] [CrossRef]
- Pellicciari, C.; Bottone, M.G.; Biggiogera, M. Detection of apoptotic cells by annexin V labeling at electron microscopy. Eur. J. Histochem. 1997, 41, 211–216. [Google Scholar]
- Ni, D.; Xu, P.; Gallagher, S. Immunoblotting and Immunodetection. Curr. Protoc. Mol. Biol. 2016, 114, 10.8.1–10.8.37. [Google Scholar] [CrossRef]
- Ko, J.; Carpenter, E.; Issadore, D. Detection and isolation of circulating exosomes and microvesicles for cancer monitoring and diagnostics using micro-/nano-based devices. Analyst 2016, 141, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Szatanek, R.; Baj-Krzyworzeka, M.; Zimoch, J.; Lekka, M.; Siedlar, M.; Baran, J. The Methods of Choice for Extracellular Vesicles (EVs) Characterization. Int. J. Mol. Sci. 2017, 18, 1153. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Pang, R.T.K.; Liu, W.; Li, Q.; Cheng, R.; Yeung, W.S.B. Polymer-based precipitation preserves biological activities of extracellular vesicles from an endometrial cell line. PLoS ONE 2017, 12, e0186534. [Google Scholar] [CrossRef] [PubMed]
- Huda, N.; Nafiujjaman; Deaguero, I.G.; Okonkwo, J.; Hill, M.L.; Kim, T. Nurunnabi Potential Use of Exosomes as Diagnostic Biomarkers and in Targeted Drug Delivery: Progress in Clinical and Preclinical Applications. ACS Biomater. Sci. Eng. 2021, 7, 2106–2149. [Google Scholar] [CrossRef]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef]
- Liang, B.; Peng, P.; Chen, S.; Li, L.; Zhang, M.; Cao, D.; Yang, J.; Li, H.; Gui, T.; Li, X.; et al. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J. Proteom. 2013, 80, 171–182. [Google Scholar] [CrossRef]
- Peng, P.; Zhang, W.; Cao, D.; Yang, J.; Shen, K. The proteomic comparison of peripheral circulation-derived exosomes from the epithelial ovarian carcinoma (EOC) patients and non-EOC subjects. Transl. Cancer Res. 2019, 8, 452–465. [Google Scholar] [CrossRef]
- Giusti, I.; D’ascenzo, S.; Dolo, V. Microvesicles as Potential Ovarian Cancer Biomarkers. BioMed Res. Int. 2013, 2013, 703048. [Google Scholar] [CrossRef]
- Zöller, M. Pancreatic cancer diagnosis by free and exosomal miRNA. World J. Gastrointest. Pathophysiol. 2013, 4, 74–90. [Google Scholar] [CrossRef]
- Choi, D.; Park, J.O.; Jang, S.C.; Yoon, Y.J.; Jung, J.W.; Choi, D.; Kim, J.; Kang, J.S.; Park, J.; Hwang, D.; et al. Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics 2011, 11, 2745–2751. [Google Scholar] [CrossRef]
- Baran, J.; Baj-Krzyworzeka, M.; Weglarczyk, K.; Szatanek, R.; Barbasz, J.; Czupryna, A.; Szczepanik, A.; Zembala, M. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol. Immunother. 2009, 59, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Pietrowska, M.; Zebrowska, A.; Gawin, M.; Marczak, L.; Sharma, P.; Mondal, S.; Mika, J.; Polańska, J.; Ferrone, S.; Kirkwood, J.M.; et al. Proteomic profile of melanoma cell-derived small extracellular vesicles in patients’ plasma: A potential correlate of melanoma progression. J. Extracell. Vesicles 2021, 10, e12063. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Tan, H.S.; Datta, A.; Lai, R.C.; Zhang, H.; Meng, W.; Lim, S.K.; Sze, S.K. Hypoxic Tumor Cell Modulates Its Microenvironment to Enhance Angiogenic and Metastatic Potential by Secretion of Proteins and Exosomes. Mol. Cell. Proteom. 2010, 9, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Vinik, Y.; Ortega, F.G.; Mills, G.B.; Lu, Y.; Jurkowicz, M.; Halperin, S.; Aharoni, M.; Gutman, M.; Lev, S. Proteomic analysis of circulating extracellular vesicles identifies potential markers of breast cancer progression, recurrence, and response. Sci. Adv. 2020, 6, eaba5714. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Hernandez, O.; Villegas-Comonfort, S.; Candanedo, F.; González-Vázquez, M.-C.; Chavez-Ocaña, S.; Jimenez-Villanueva, X.; Sierra-Martinez, M.; Salazar, E.P. Elevated Concentration of Microvesicles Isolated from Peripheral Blood in Breast Cancer Patients. Arch. Med. Res. 2013, 44, 208–214. [Google Scholar] [CrossRef]
- Shibata, C.; Otsuka, M.; Seimiya, T.; Ishigaki, K.; Miyakawa, Y.; Kishikawa, T.; Fujishiro, M. Smaller extracellular vesicles are released from pancreatic cancer cells by the alteration of the lipid composition under low glucose conditions. Biochem. Biophys. Res. Commun. 2022, 637, 314–321. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, W.; Guo, Z.; Ju, Q.; Zhu, L.; Gao, J.; Zhou, L.; Liu, F.; Xu, Y.; Zhan, Q.; et al. A novel TP53 pathway influences the HGS-mediated exosome formation in colorectal cancer. Sci. Rep. 2016, 6, 28083. [Google Scholar] [CrossRef]
- Royo, F.; Théry, C.; Falcón-Pérez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for Separation and Characterization of Extracellular Vesicles: Results of a Worldwide Survey Performed by the ISEV Rigor and Standardization Subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef]
- Tavoosidana, G.; Ronquist, G.; Darmanis, S.; Yan, J.; Carlsson, L.; Wu, D.; Conze, T.; Ek, P.; Semjonow, A.; Eltze, E.; et al. Multiple recognition assay reveals prostasomes as promising plasma biomarkers for prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 8809–8814. [Google Scholar] [CrossRef]
- Beckham, C.J.; Olsen, J.; Yin, P.-N.; Wu, C.-H.; Ting, H.-J.; Hagen, F.K.; Scosyrev, E.; Messing, E.M.; Lee, Y.-F. Bladder Cancer Exosomes Contain EDIL-3/Del1 and Facilitate Cancer Progression. J. Urol. 2014, 192, 583–592. [Google Scholar] [CrossRef]
- Nilsson, J.; Skog, J.; Nordstrand, A.; Baranov, V.; Minchevanilsson, L.; Breakefield, X.O.; Widmark, A. Prostate cancer-derived urine exosomes: A novel approach to biomarkers for prostate cancer. Br. J. Cancer 2009, 100, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Zhang, J.; Allison, R.; Gay, H.; Yang, W.; Bhowmick, N.A.; Frelix, G.; Shappell, S.; Chen, Y. Identification of extracellular δ-catenin accumulation for prostate cancer detection. Prostate 2008, 69, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Sun, I.O.; Lerman, L.O. Urinary Extracellular Vesicles as Biomarkers of Kidney Disease: From Diagnostics to Therapeutics. Diagnostics 2020, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kojima, K.; Mobley, J.A.; West, A.B. Proteomic analysis of urinary extracellular vesicles reveal biomarkers for neurologic disease. EBioMedicine 2019, 45, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, R.; Shetty, A.K. Extracellular Vesicles for the Diagnosis and Treatment of Parkinson’s Disease. Aging Dis. 2021, 12, 1438–1450. [Google Scholar] [CrossRef]
- Lee, J.-E.; Moon, P.-G.; Cho, Y.-E.; Kim, Y.-B.; Kim, I.-S.; Park, H.; Baek, M.-C. Identification of EDIL3 on extracellular vesicles involved in breast cancer cell invasion. J. Proteom. 2016, 131, 17–28. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, S.; Liu, C.; Han, Z.; Liu, Y.; Deng, J.; Li, Y.; Wu, X.; Cai, L.; Qin, L.; et al. Protein analysis of extracellular vesicles to monitor and predict therapeutic response in metastatic breast cancer. Nat. Commun. 2021, 12, 2536. [Google Scholar] [CrossRef]
- Fazel, R.; Krumholz, H.M.; Wang, Y.; Ross, J.S.; Chen, J.; Ting, H.H.; Shah, N.D.; Nasir, K.; Einstein, A.J.; Nallamothu, B.K. Exposure to Low-Dose Ionizing Radiation from Medical Imaging Procedures. N. Engl. J. Med. 2009, 361, 849–857. [Google Scholar] [CrossRef]
- Duffy, M.J.; Evoy, D.; McDermott, E.W. CA 15-3: Uses and limitation as a biomarker for breast cancer. Clin. Chim. Acta 2010, 411, 1869–1874. [Google Scholar] [CrossRef]
- Ebeling, F.G.; Stieber, P.; Untch, M.; Nagel, D.; Konecny, G.E.; Schmitt, U.M.; Fateh-Moghadam, A.; Seidel, D. Serum CEA and CA 15-3 as prognostic factors in primary breast cancer. Br. J. Cancer 2002, 86, 1217–1222. [Google Scholar] [CrossRef]
- Rodrigues-Junior, D.M.; Tan, S.S.; Lim, S.K.; Leong, H.S.; Melendez, M.E.; Ramos, C.R.N.; Viana, L.D.S.; Tan, D.S.W.; Carvalho, A.L.; Iyer, N.G.; et al. Circulating extracellular vesicle-associated TGFβ3 modulates response to cytotoxic therapy in head and neck squamous cell carcinoma. Carcinogenesis 2019, 40, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ni, J.; Beretov, J.; Wasinger, V.C.; Graham, P.; Li, Y. Recent advances of small extracellular vesicle biomarkers in breast cancer diagnosis and prognosis. Mol. Cancer 2023, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Grotenhuis, A.J.; Vermeulen, S.H.; Kiemeney, L.A. Germline Genetic Markers for Urinary Bladder Cancer Risk, Prognosis and Treatment Response. Future Oncol. 2010, 6, 1433–1460. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. Npj Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef]
- Zhang, E.; Ding, C.; Li, S.; Zhou, X.; Aikemu, B.; Fan, X.; Sun, J.; Zheng, M.; Yang, X. Roles and mechanisms of tumour-infiltrating B cells in human cancer: A new force in immunotherapy. Biomark. Res. 2023, 11, 28. [Google Scholar] [CrossRef]
- Deola, S.; Panelli, M.C.; Maric, D.; Selleri, S.; Dmitrieva, N.I.; Voss, C.Y.; Klein, H.; Stroncek, D.; Wang, E.; Marincola, F.M. Helper B Cells Promote Cytotoxic T Cell Survival and Proliferation Independently of Antigen Presentation through CD27/CD70 Interactions. J. Immunol. 2008, 180, 1362–1372. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions About Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; da Costa, M.P.; e Sousa, C.R. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef]
- Modak, M.; Mattes, A.-K.; Reiss, D.; Skronska-Wasek, W.; Langlois, R.; Sabarth, N.; Konopitzky, R.; Ramirez, F.; Lehr, K.; Mayr, T.; et al. CD206+ tumor-associated macrophages cross-present tumor antigen and drive antitumor immunity. J. Clin. Investig. 2022, 7, 155022. [Google Scholar] [CrossRef]
- Truffi, M.; Sorrentino, L.; Corsi, F. Fibroblasts in the tumor microenvironment. Tumor Microenviron. Non-Hematop. Cells 2020, 1234, 15–29. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Lu, Y.; Liu, C.; Yang, W. Tumor microenvironment-mediated immune tolerance in development and treatment of gastric cancer. Front. Immunol. 2022, 13, 1016817. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Cederbaum, S.D.; Yu, H.; Grody, W.W.; Kern, R.M.; Yoo, P.; Iyer, R.K. Arginases I and II: Do their functions overlap? Mol. Genet. Metab. 2004, 81, 38–44. [Google Scholar] [CrossRef]
- Mundy-Bosse, B.L.; Lesinski, G.B.; Jaimeramirez, A.C.; Benninger, K.; Khan, M.; Kuppusamy, P.; Guenterberg, K.; Kondadasula, S.V.; Chaudhury, A.R.; La Perle, K.M.; et al. Myeloid-Derived Suppressor Cell Inhibition of the IFN Response in Tumor-Bearing Mice. Cancer Res. 2011, 71, 5101–5110. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Eruslanov, E.; Kübler, H.; Tseng, T.; Sakai, Y.; Su, Z.; Kaliberov, S.; Heiser, A.; Rosser, C.; Dahm, P.; et al. Oxidative Stress Regulates Expression of VEGFR1 in Myeloid Cells: Link to Tumor-Induced Immune Suppression in Renal Cell Carcinoma. J. Immunol. 2008, 181, 346–353. [Google Scholar] [CrossRef]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- McRitchie, B.R.; Akkaya, B. Exhaust the exhausters: Targeting regulatory T cells in the tumor microenvironment. Front. Immunol. 2022, 13, 940052. [Google Scholar] [CrossRef]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell Activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef]
- Markowitz, J.; Wang, J.; VanGundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Bied, M.; Ho, W.W.; Ginhoux, F.; Blériot, C. Roles of macrophages in tumor development: A spatiotemporal perspective. Cell. Mol. Immunol. 2023, 20, 983–992. [Google Scholar] [CrossRef]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8+ T Cell-Derived Interferon-γ. Immunity 2019, 51, 381–397.e6. [Google Scholar] [CrossRef]
- Krneta, T.; Gillgrass, A.; Poznanski, S.; Chew, M.; Lee, A.J.; Kolb, M.; A Ashkar, A. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol. 2017, 101, 285–295. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, B.S.; Lee, S.-K. Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune Netw. 2020, 20, e4. [Google Scholar] [CrossRef]
- Tekguc, M.; Wing, J.B.; Osaki, M.; Long, J.; Sakaguchi, S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2023739118. [Google Scholar] [CrossRef]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; Sakaguchi, N.; Itoh, M.; Iwata, M.; Shimizu, J.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998, 10, 1969–1980. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Cui, J.W.; Li, Y.; Yang, Y.; Yang, H.K.; Dong, J.M.; Xiao, Z.H.; He, X.; Guo, J.H.; Wang, R.Q.; Dai, B.; et al. Tumor immunotherapy resistance: Revealing the mechanism of PD-1 / PD-L1-mediated tumor immune escape. Biomed. Pharmacother. 2024, 171, 116203. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Chikuma, S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. Curr. Top. Microbiol. Immunol. 2017, 410, 99–126. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Hong, M.M.Y.; Vareki, S.M. Addressing the Elephant in the Immunotherapy Room: Effector T-Cell Priming versus Depletion of Regulatory T-Cells by Anti-CTLA-4 Therapy. Cancers 2022, 14, 1580. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Salvi, S.; Fontana, V.; Boccardo, S.; Merlo, D.F.; Margallo, E.; Laurent, S.; Morabito, A.; Rijavec, E.; Bello, M.G.D.; Mora, M.; et al. Evaluation of CTLA-4 expression and relevance as a novel prognostic factor in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2012, 61, 1463–1472. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ling, Y.; Sun, Y.; Xiao, F.; Wang, L. Extracellular Vesicles Derived from Mesenchymal Stem Cells Promote Wound Healing and Skin Regeneration by Modulating Multiple Cellular Changes: A Brief Review. Genes 2023, 14, 1516. [Google Scholar] [CrossRef]
- Ruan, S.; Erwin, N.; He, M. Light-induced high-efficient cellular production of immune functional extracellular vesicles. J. Extracell. Vesicles 2022, 11, e12194. [Google Scholar] [CrossRef]
- Wan, Y.Y. Regulatory T cells: Immune suppression and beyond. Cell. Mol. Immunol. 2010, 7, 204–210. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef]
- Malek, T.R.; Castro, I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef]
- Jung, D.; Shin, S.; Kang, S.; Jung, I.; Ryu, S.; Noh, S.; Choi, S.; Jeong, J.; Lee, B.Y.; Kim, K.; et al. Reprogramming of T cell-derived small extracellular vesicles using IL2 surface engineering induces potent anti-cancer effects through miRNA delivery. J. Extracell. Vesicles 2022, 11, 12287. [Google Scholar] [CrossRef]
- Feng, C.; Tan, P.; Nie, G.; Zhu, M. Biomimetic and bioinspired nano-platforms for cancer vaccine development. Exploration 2023, 3, 20210263. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, J.; Lu, L.; Deng, D.; Huang, J.; Tang, Z.; Shi, X.; Lo, P.; Lovell, J.F.; Zheng, Y.; et al. Tumor cell membrane-based vaccines: A potential boost for cancer immunotherapy. Exploration 2024, 4, 20230171. [Google Scholar] [CrossRef]
- Zhang, B.; Sim, W.K.; Shen, T.-L.; Lim, S.K. Engineered EVs with pathogen proteins: Promising vaccine alternatives to LNP-mRNA vaccines. J. Biomed. Sci. 2024, 31, 9. [Google Scholar] [CrossRef]
- Jiang, J.; Mei, J.; Ma, Y.; Jiang, S.; Zhang, J.; Yi, S.; Feng, C.; Liu, Y.; Liu, Y. Tumor hijacks macrophages and microbiota through extracellular vesicles. Exploration 2022, 2, 20210144. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cai, S.; Li, M.; Salma, K.I.; Zhou, X.; Han, F.; Chen, J.; Huyan, T. Tumor-Derived Extracellular Vesicles: Their Role in Immune Cells and Immunotherapy. Int. J. Nanomed. 2021, 16, 5395–5409. [Google Scholar] [CrossRef]
- Horrevorts, S.K.; Stolk, D.A.; van de Ven, R.; Hulst, M.; Hof, B.v.H.; Duinkerken, S.; Heineke, M.H.; Ma, W.; Dusoswa, S.A.; Nieuwland, R.; et al. Glycan-Modified Apoptotic Melanoma-Derived Extracellular Vesicles as Antigen Source for Anti-Tumor Vaccination. Cancers 2019, 11, 1266. [Google Scholar] [CrossRef]
- Koyama, Y.; Ito, T.; Hasegawa, A.; Eriguchi, M.; Inaba, T.; Ushigusa, T.; Sugiura, K. Exosomes derived from tumor cells genetically modified to express Mycobacterium tuberculosis antigen: A novel vaccine for cancer therapy. Biotechnol. Lett. 2016, 38, 1857–1866. [Google Scholar] [CrossRef]
- Munich, S.; Sobo-Vujanovic, A.; Buchser, W.J.; Beer-Stolz, D.; Vujanovic, N.L. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. OncoImmunology 2012, 1, 1074–1083. [Google Scholar] [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. OncoImmunology 2016, 5, 1071008. [Google Scholar] [CrossRef]
- Halpert, M.M.; Konduri, V.; Liang, D.; Chen, Y.; Wing, J.B.; Paust, S.; Levitt, J.M.; Decker, W.K. Dendritic Cell-Secreted Cytotoxic T-Lymphocyte-Associated Protein-4 Regulates the T-cell Response by Downmodulating Bystander Surface B7. Stem Cells Dev. 2016, 25, 774–787. [Google Scholar] [CrossRef]
- Hoffmann, O.; Wormland, S.; Bittner, A.-K.; Collenburg, M.; Horn, P.A.; Kimmig, R.; Kasimir-Bauer, S.; Rebmann, V. Programmed death receptor ligand-2 (PD-L2) bearing extracellular vesicles as a new biomarker to identify early triple-negative breast cancer patients at high risk for relapse. J. Cancer Res. Clin. Oncol. 2023, 149, 1159–1174. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.-E.; Sung, K.J.; Sung, Y.H.; Pack, C.-G.; Jung, M.-K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.-W.; Chan, L.-C.; Wei, Y.; Hsu, J.-M.; Xia, W.; Cha, J.-H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-Polarized Macrophages Potentiate the Cancer Vaccine by Creating a Pro-inflammatory Microenvironment in the Lymph Node. Mol. Ther. 2017, 25, 1665–1675. [Google Scholar] [CrossRef]
- Choo, Y.W.; Kang, M.; Kim, H.Y.; Han, J.; Kang, S.; Lee, J.-R.; Jeong, G.-J.; Kwon, S.P.; Song, S.Y.; Go, S.; et al. M1 Macrophage-Derived Nanovesicles Potentiate the Anticancer Efficacy of Immune Checkpoint Inhibitors. ACS Nano 2018, 12, 8977–8993. [Google Scholar] [CrossRef]
- Liu, H.; Huang, L.; Mao, M.; Ding, J.; Wu, G.; Fan, W.; Yang, T.; Zhang, M.; Huang, Y.; Xie, H. Viral Protein-Pseudotyped and siRNA-Electroporated Extracellular Vesicles for Cancer Immunotherapy. Adv. Funct. Mater. 2020, 30, 2006515. [Google Scholar] [CrossRef]
- Lin, S.-W.; Yu, C.-P.; Tsai, J.-C.; Shyong, Y.-J. Delivery of extracellular vesicles loaded with immune checkpoint inhibitors for immunotherapeutic management of glioma. Mater. Today Bio 2024, 28, 101244. [Google Scholar] [CrossRef]
- Filipazzi, P.; Bürdek, M.; Villa, A.; Rivoltini, L.; Huber, V. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Semin. Cancer Biol. 2012, 22, 342–349. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.; Hu, D.; Liang, Y.; Liu, Z.; Zhong, T.; Wang, X. Tumor-derived extracellular vesicles regulate macrophage polarization: Role and therapeutic perspectives. Front. Immunol. 2024, 15, 1346587. [Google Scholar] [CrossRef]
- Xun, J.; Du, L.; Gao, R.; Shen, L.; Wang, D.; Kang, L.; Chen, C.; Zhang, Z.; Zhang, Y.; Yue, S.; et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-associated macrophages through inhibition of KDM6B. Theranostics 2021, 11, 6847–6859. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Li, Q.; Li, Y.; Zhang, L.; Zhida, L.; Yuan, F.; Zhang, R. Tumor-derived extracellular vesicle drug delivery system for chemo-photothermal-immune combination cancer treatment. iScience 2024, 27, 108833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-F.; Gao, C.; Huang, X.-Y.; Lu, J.-C.; Guo, X.-J.; Shi, G.-M.; Cai, J.-B.; Ke, A.-W. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol. Cancer 2020, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Fang, R.; Lyu, K.; Liao, J.; Long, Y.; Yang, J.; Wen, W.; Sun, W. Exosomal PD-L1 derived from head and neck squamous cell carcinoma promotes immune evasion by activating the positive feedback loop of activated regulatory T cell-M2 macrophage. Oral Oncol. 2023, 145, 106532. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Patel, T.H.; Brewer, J.R.; Fan, J.; Cheng, J.; Shen, Y.-L.; Xiang, Y.; Zhao, H.; Lemery, S.J.; Pazdur, R.; Kluetz, P.G.; et al. FDA Approval Summary: Tremelimumab in Combination with Durvalumab for the Treatment of Patients with Unresectable Hepatocellular Carcinoma. Clin. Cancer Res. 2023, 30, 269–273. [Google Scholar] [CrossRef]
- Agarwal, R.; Agarwal, P. Future Target Molecules in Antiglaucoma Therapy: TGF-β May Have a Role to Play. Ophthalmic Res. 2009, 43, 1–10. [Google Scholar] [CrossRef]
- Liang, R.; Lu, H.; Zhu, H.; Liang, G.; Zhang, J.; Gao, J.; Tian, T. Radiation-primed TGF-β trapping by engineered extracellular vesicles for targeted glioblastoma therapy. J. Control Release 2024, 370, 821–834. [Google Scholar] [CrossRef]
- Shamshiripour, P.; Rahnama, M.; Nikoobakht, M.; Rad, V.F.; Moradi, A.-R.; Ahmadvand, D. Extracellular vesicles derived from dendritic cells loaded with VEGF-A siRNA and doxorubicin reduce glioma angiogenesis in vitro. J. Control Release 2024, 369, 128–145. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non–Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Grignani, G.; Rutkowski, P.; Lebbe, C.; Prinzi, N.; Grob, J.-J.; Tanda, E.T.; Guida, M.; Burgess, M.; Pulini, J.; Shankar, S.; et al. 545 A phase 2 study of retifanlimab in patients with advanced or metastatic merkel cell carcinoma (MCC) (POD1UM-201). In Regular and Young Investigator Award Abstracts; BMJ Publishing Group Ltd.: London, UK, 2021; pp. A574–A575. [Google Scholar] [CrossRef]
- Wang, F.-H.; Wei, X.-L.; Feng, J.; Li, Q.; Xu, N.; Hu, X.-C.; Liao, W.; Jiang, Y.; Lin, X.-Y.; Zhang, Q.-Y.; et al. Efficacy, Safety, and Correlative Biomarkers of Toripalimab in Previously Treated Recurrent or Metastatic Nasopharyngeal Carcinoma: A Phase II Clinical Trial (POLARIS-02). J. Clin. Oncol. 2021, 39, 704–712. [Google Scholar] [CrossRef]
- Mai, H.-Q.; Chen, Q.-Y.; Chen, D.; Hu, C.; Yang, K.; Wen, J.; Li, J.; Shi, Y.; Jin, F.; Xu, R.; et al. Toripalimab Plus Chemotherapy for Recurrent or Metastatic Nasopharyngeal Carcinoma. JAMA 2023, 330, 1961–1970. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.-T.; Friedlander, T.W.; Hoimes, C.J.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef]
- Necchi, A.; Joseph, R.; Loriot, Y.; Hoffman-Censits, J.; Perez-Gracia, J.; Petrylak, D.; Derleth, C.; Tayama, D.; Zhu, Q.; Ding, B.; et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: Post-progression outcomes from the phase II IMvigor210 study. Ann. Oncol. 2017, 28, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’angelo, S.P.; Shih, K.C.; Lebbé, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Brown, J.; Mathews, C.; Press, J.; Sabatier, R.; O’malley, D.M.; Samouelian, V.; Boni, V.; et al. Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) or proficient/stable (MMRp/MSS) endometrial cancer: Interim results from GARNET—A phase I, single-arm study. J. Immunother. Cancer 2022, 10, e003777. [Google Scholar] [CrossRef]
- Duangthim, N.; Lomphithak, T.; Saito-Koyama, R.; Miki, Y.; Inoue, C.; Sato, I.; Miyauchi, E.; Abe, J.; Sasano, H.; Jitkaew, S. Prognostic significance and response to immune checkpoint inhibitors of RIPK3, MLKL and necroptosis in non-small cell lung cancer. Sci. Rep. 2024, 14, 21625. [Google Scholar] [CrossRef]
- Li, Y.; Du, Y.; Xue, C.; Wu, P.; Du, N.; Zhu, G.; Xu, H.; Zhu, Z. Efficacy and safety of anti-PD-1/PD-L1 therapy in the treatment of advanced colorectal cancer: A meta-analysis. BMC Gastroenterol. 2022, 22, 431. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Xu, F.; Jiang, D.; Xu, J.; Dai, H.; Fan, Q.; Fei, Z.; Wang, B.; Zhang, Y.; Ma, Q.; Yang, Q.; et al. Engineering of dendritic cell bispecific extracellular vesicles for tumor-targeting immunotherapy. Cell Rep. 2023, 42, 113138. [Google Scholar] [CrossRef]
- Phung, C.D.; Pham, T.T.; Nguyen, H.T.; Nguyen, T.T.; Ou, W.; Jeong, J.-H.; Choi, H.-G.; Ku, S.K.; Yong, C.S.; Kim, J.O. Anti-CTLA-4 antibody-functionalized dendritic cell-derived exosomes targeting tumor-draining lymph nodes for effective induction of antitumor T-cell responses. Acta Biomater. 2020, 115, 371–382. [Google Scholar] [CrossRef]
- Li, B.; Fang, T.; Li, Y.; Xue, T.; Zhang, Z.; Li, L.; Meng, F.; Wang, J.; Hou, L.; Liang, X.; et al. Engineered T cell extracellular vesicles displaying PD-1 boost anti-tumor immunity. Nano Today 2022, 46, 101606. [Google Scholar] [CrossRef]
- Shin, S.; Jung, I.; Jung, D.; Kim, C.S.; Kang, S.-M.; Ryu, S.; Choi, S.-J.; Noh, S.; Jeong, J.; Lee, B.Y.; et al. Novel antitumor therapeutic strategy using CD4+ T cell-derived extracellular vesicles. Biomaterials 2022, 289, 121765. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Yeung, V.; Clayton, A. Extracellular vesicles as modulators of the cancer microenvironment. Semin. Cell Dev. Biol. 2015, 40, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, Y.; Hu, Q. Recent advances in overcoming barriers to cell-based delivery systems for cancer immunotherapy. Exploration 2022, 2, 20210106. [Google Scholar] [CrossRef]
- Fonseka, P.; Marzan, A.L.; Mathivanan, S. Introduction to the Community of Extracellular Vesicles. New Front. Extracell. Vesicles 2021, 97, 3–18. [Google Scholar] [CrossRef]
- Dai, H.; Fan, Q.; Wang, C. Recent applications of immunomodulatory biomaterials for disease immunotherapy. Exploration 2022, 2, 20210157. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exosomes | Microvesicles | Apoptotic Bodies | |
|---|---|---|---|
| Size | 30–150 nm | 40–1000 nm | 1–5 µm |
| Original source | Multivesicular bodies and fusion with cell membrane | Outward cleavage of plasma membrane | Protrusion of cell membrane of apoptotic cells |
| Specific markers | CD9, CD81, CD63, TSG101, Alix, HSP70 | CD40, integrins, selectins | Phosphatidylserine |
| Target | Drug Generic Name (First Approval) | Targeted Cancer (Clinical Trial) | Key Clinical Trial | Reference |
|---|---|---|---|---|
| PD-1 | Nivolumab (approved in 2014) | Unresectable/metastatic melanoma | NCT01721746 (CheckMate 037) | [151] |
| Advanced/metastatic squamous NSCLC | NCT01642004 (CheckMate 017) | [152] | ||
| Pembrolizumab (approved in 2014) | Advanced melanoma | NCT01295827 (KEYNOTE-001) | [153] | |
| Advanced NSCLC | NCT01295827 (KEYNOTE-001), NCT01905657 (KEYNOTE-010) | [154,155] | ||
| Cemiplimab (approved in 2018) | Advanced CSCC | NCT02760498 | [156] | |
| Retifanlimab (approved 2023) | Metastatic/locally advanced MCC | NCT03599713 (POD1UM-201) | [157] | |
| Toripalimab (approved in 2023) | Recurrent/metastatic NPC | NCT02915432 (POLARIS-02), NCT03581786 (JUNIPER-02) | [158,159] | |
| PD-L1 | Durvalumab (approved in 2017) | Locally advanced NSCLC | NCT02125461 (PACIFIC) | [160] |
| Advanced/metastatic urothelial carcinoma | NCT01693562 | [161] | ||
| Atezolizumab (approved in 2016) | Urothelial carcinoma | IMvigor 210 (NCT02108652) | [162] | |
| NSCLC | IMpower110 (NCT02409342) | [163] | ||
| Avelumab (approved in 2017) | Metastatic MCC | JAVELIN Merkel 200 (NCT02155647) | [164] | |
| Advanced/metastatic urothelial carcinoma | JAVELIN Bladder 100 (NCT02603432) | [165] | ||
| Dostarlimab (approved in 2023) | Endometrial cancer | GARNET (NCT02715284) | [166] | |
| CTLA-4 | Ipilimumab (approved in 2011) | Malignant/metastatic melanoma | NCT00094653 | [145] |
| Tremelimumab (approved in 2022, in combination with Durvalumab) | Unresectable HCC | HIMALAYA (NCT03298451) | [146] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiramonai, L.; Liang, X.-J.; Zhu, M. Extracellular Vesicle-Based Strategies for Tumor Immunotherapy. Pharmaceutics 2025, 17, 257. https://doi.org/10.3390/pharmaceutics17020257
Jiramonai L, Liang X-J, Zhu M. Extracellular Vesicle-Based Strategies for Tumor Immunotherapy. Pharmaceutics. 2025; 17(2):257. https://doi.org/10.3390/pharmaceutics17020257
Chicago/Turabian StyleJiramonai, Luksika, Xing-Jie Liang, and Mengliang Zhu. 2025. "Extracellular Vesicle-Based Strategies for Tumor Immunotherapy" Pharmaceutics 17, no. 2: 257. https://doi.org/10.3390/pharmaceutics17020257
APA StyleJiramonai, L., Liang, X.-J., & Zhu, M. (2025). Extracellular Vesicle-Based Strategies for Tumor Immunotherapy. Pharmaceutics, 17(2), 257. https://doi.org/10.3390/pharmaceutics17020257