
Emerging Approaches in Glioblastoma Treatment: Modulating the Extracellular Matrix Through Nanotechnology


 , and
, and 
Abstract
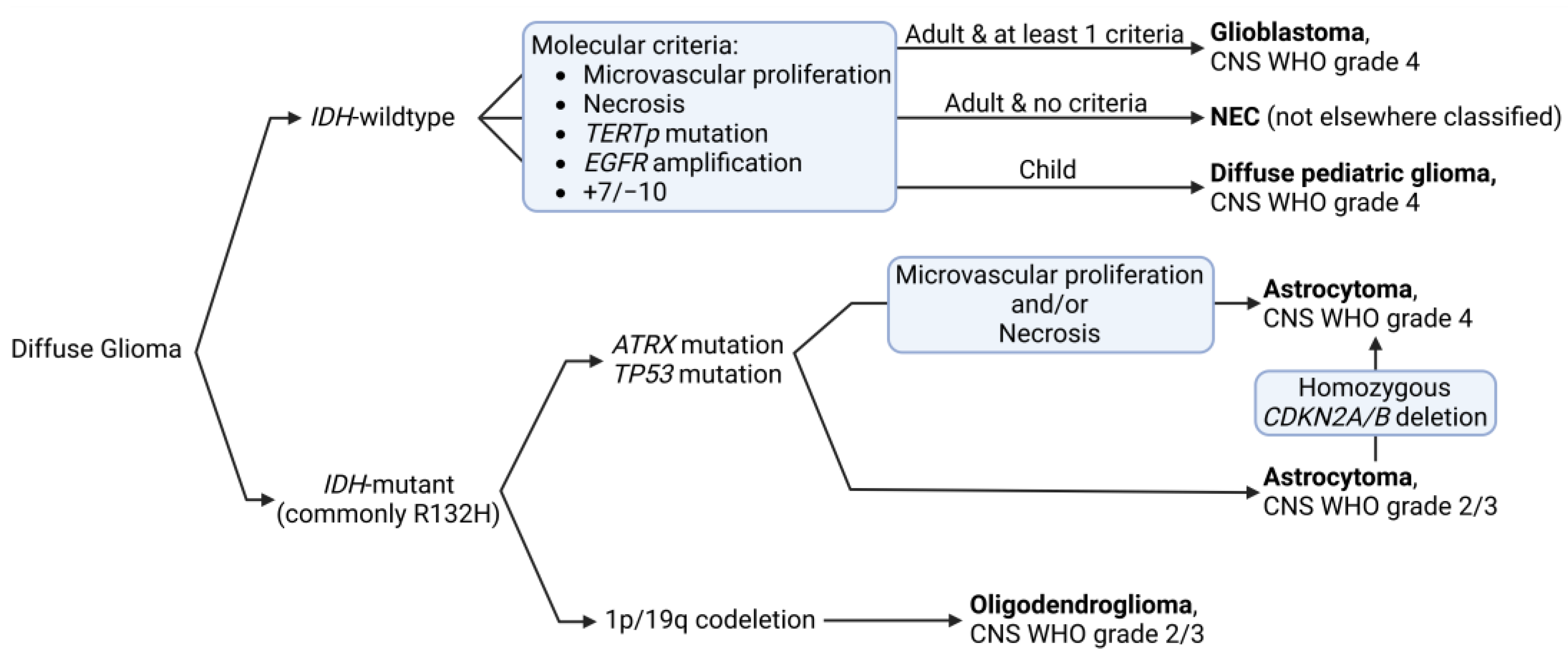
1. Glioblastoma: Epidemiology and Classification
2. Current Treatment Options
2.1. First-Line—Stupp Protocol
2.2. Second-Line Treatments
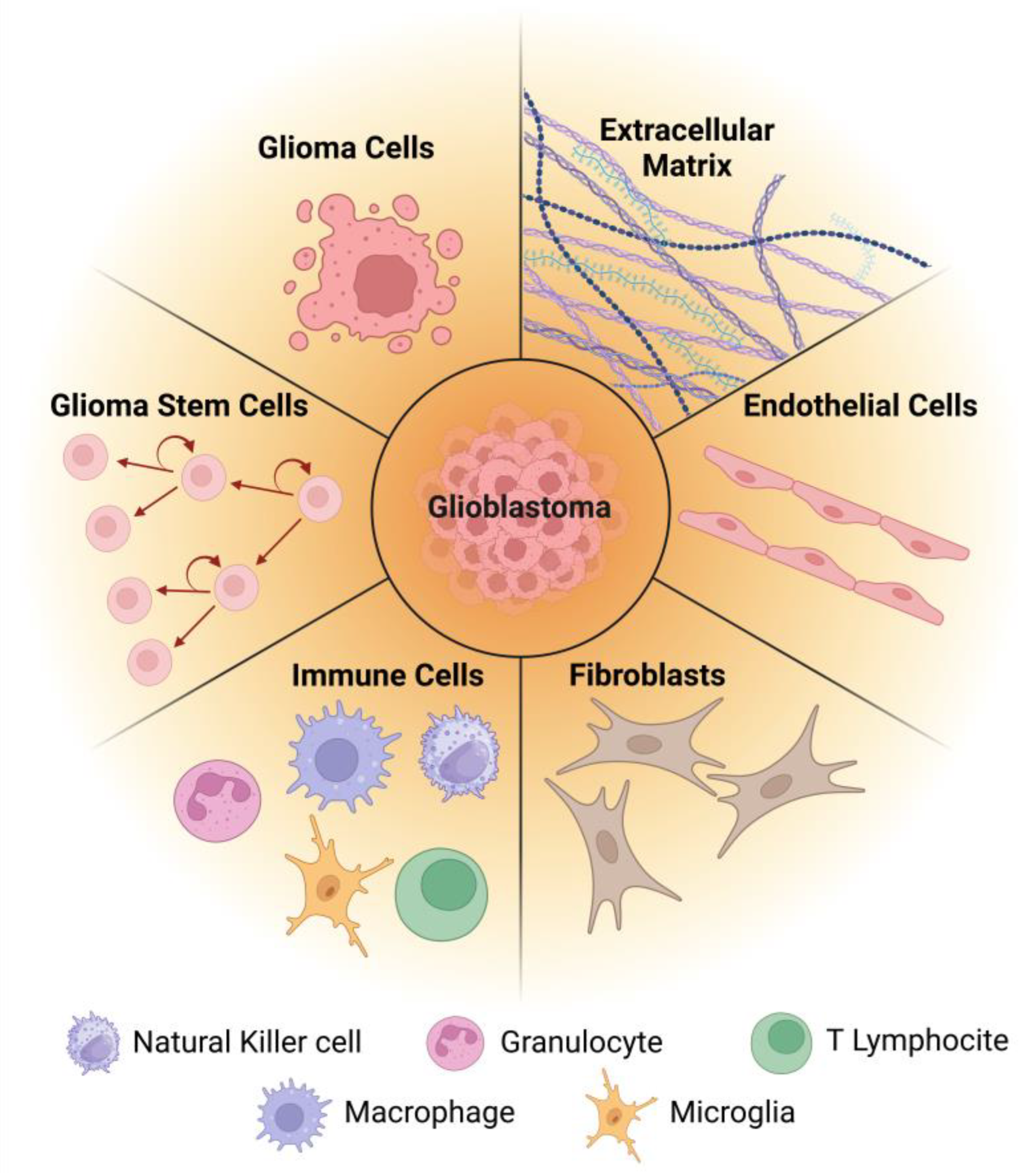
3. Glioblastoma and Its Microenvironment—Cellular and Molecular Features
3.1. Glioma Cells
3.2. Glioma Stem Cells
3.3. Immune Cells
3.4. Fibroblasts
3.5. Endothelial Cells
3.6. Extracellular Matrix
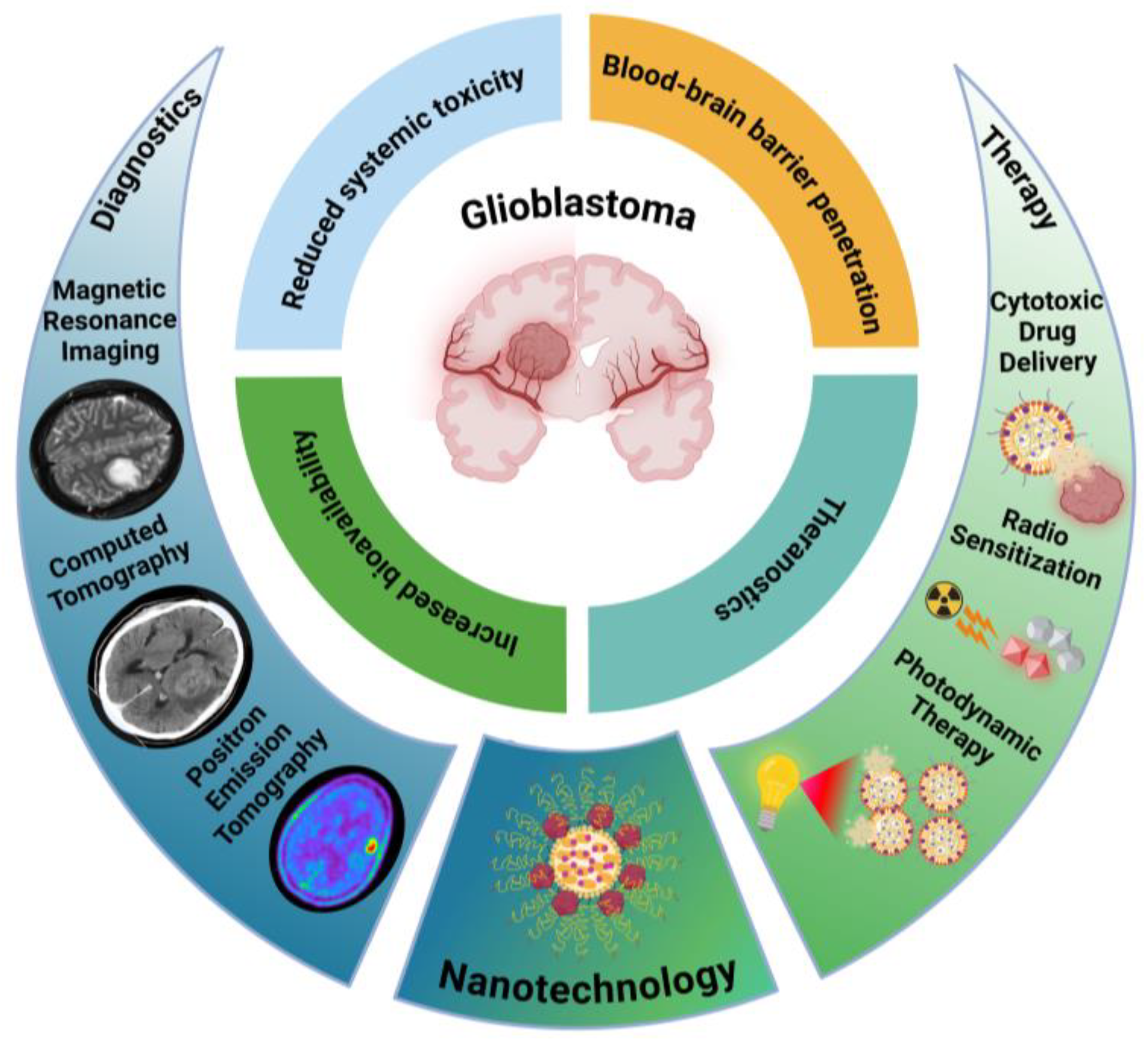
4. Nanotechnology in Glioblastoma
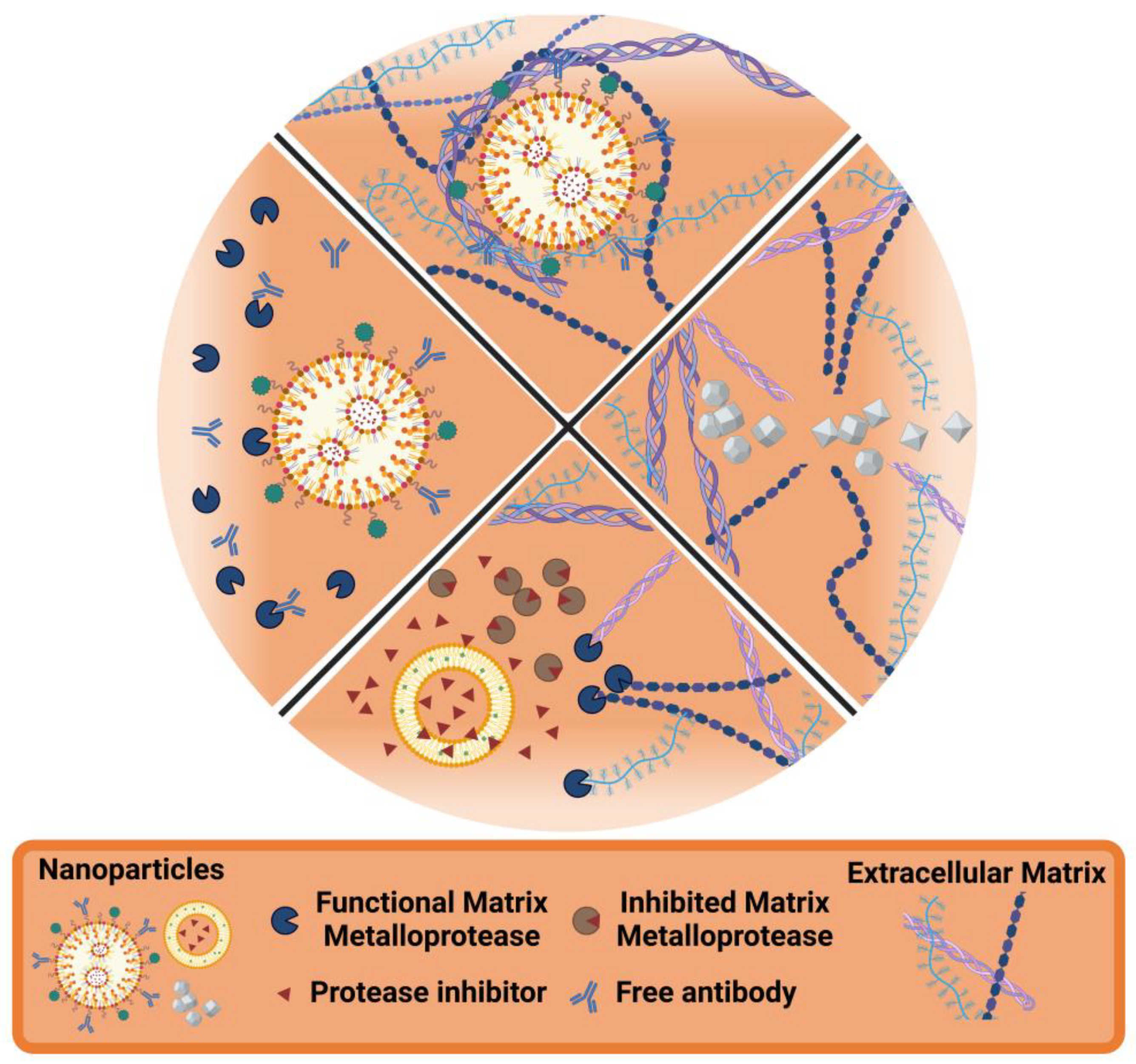
5. Nanotechnology and the ECM
5.1. ECM as the Target
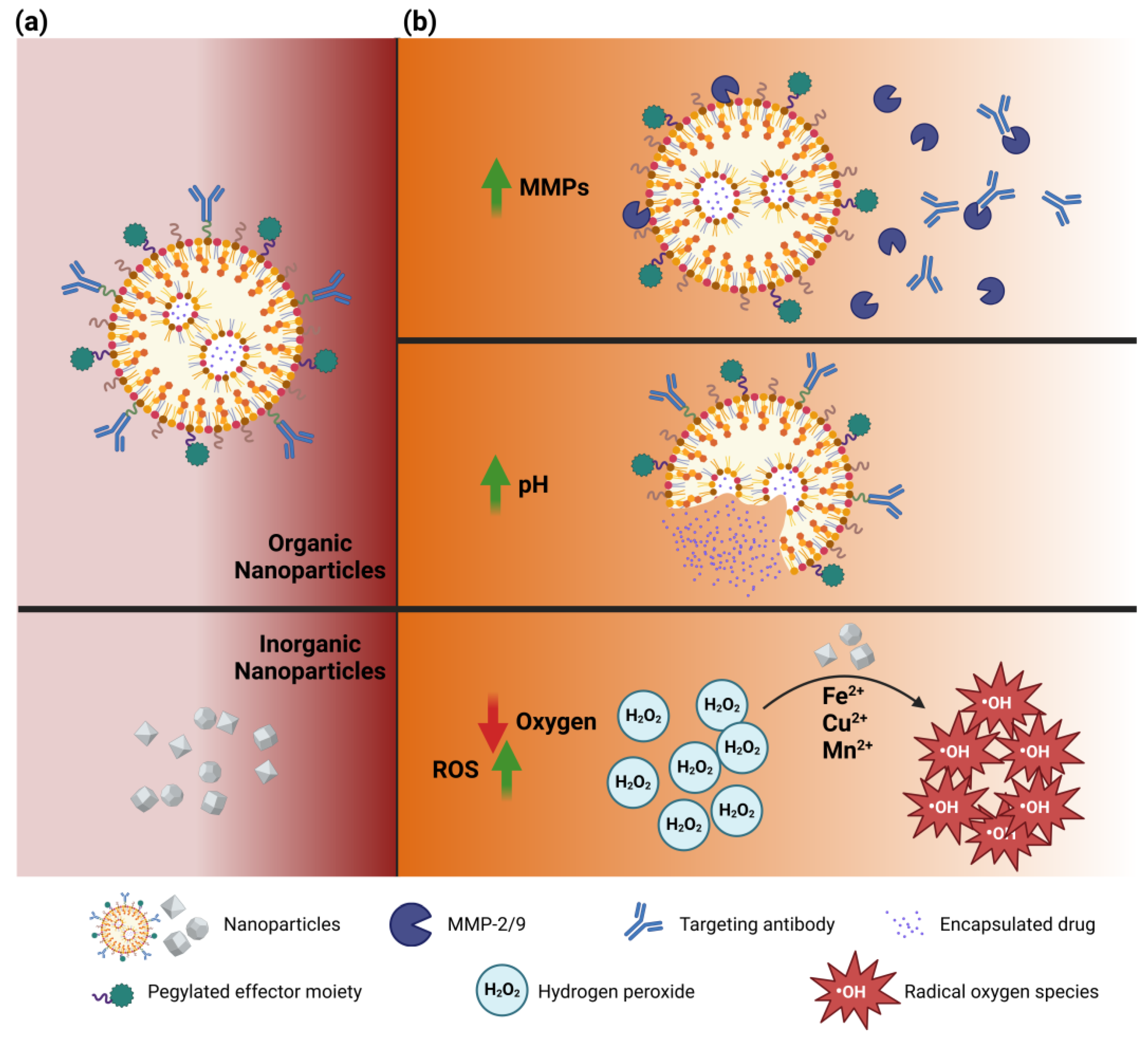
5.2. ECM-Responsive Nanoparticles
5.3. Enhancing ECM Degradation
5.4. Preventing ECM Degradation
6. Challenges and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GB | Glioblastoma |
| CNS | Central Nervous System |
| WHO | World Health Organization |
| IDH | Isocitrate Dehydrogenase |
| TERT | Telomerase Reverse Transcriptase |
| EGF | Epithelial Growth Factor |
| EGFR | Epithelial Growth Factor Receptor |
| NOS | Not Otherwise Specified |
| NEC | Not Elsewhere Classified |
| RT | Radiotherapy |
| TMZ | Temozolomide |
| MGMT | Methylguanine Methyltransferase |
| FDA | Food and Drug Administration |
| VEGF | Vascular Endothelial Growth Factor |
| BBB | Blood-Brain Barrier |
| TTFields | Tumor Treating Fields |
| TME | Tumor Microenvironment |
| TCGA | The Cancer Genome Atlas |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| TP53 | Tumor Protein P53 |
| SMO | Smoothened |
| GAS1 | Growth Arrest-Specific Protein 1 |
| GLI2 | GLI family zinc finger 2 |
| NOTCH3 | Neurogenic Locus Notch Homolog Protein 3 |
| JAG1 | Jagged 1 |
| LFNG | Beta-1,3-N-acetylglucosaminyltransferase Lunatic Fringe |
| NF1 | Neurofibromin |
| PTEN | Phosphatase and Tensin Homolog |
| PI3K | phosphatidylinositol 3-kinase |
| CHI3L1 | Chitinase-3-Like Protein 1 |
| CD | Cluster of Differentiation |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| PDGFRA | Platelet-Derived Growth Factor Receptor A |
| AC-like | Astrocyte-like |
| MES-like | Mesenchymal-like |
| OPC-like | Oligodendrocyte Progenitor-like |
| NPC-like | Neural Progenitor-like |
| CDK4 | Cyclin-Dependent Kinase 4 |
| GSCs | Glioma Stem Cells |
| SOX2 | Sex Determining Region Y Box 2 |
| DNA | Deoxyribonucleic Acid |
| ABC | ATP-Binding Cassette |
| OCT4 | Octamer-Binding Transcription Factor 4 |
| IL | Interleukin |
| TGF-β | Transforming Growth Factor Beta |
| TAM | Tumor-Associated Macrophage |
| CSF-1 | Colony Stimulating Factor 1 |
| GDNF | Glial Cell Line-Derived Neurotrophic Factor |
| CCL2 | Chemokine (CC Motif) Ligand 2 |
| SFD-1 | Stromal Cell-Derived Factor 1 |
| CXCL1 | Chemokine (CXC Motif) Ligand 1 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| IGF-1 | Insulin-like Growth Factor 1 |
| PDGF | Platelet-Derived Growth Factor |
| MDSC | Myeloid-Derived Suppressor Cells |
| NK | Natural Killer |
| ROS | Reactive Oxygen Species |
| CAF | Cancer-Associated Fibroblast |
| ERK | Extracellular Signal-Regulated Kinase |
| ECM | Extracellular Matrix |
| HA | Hyaluronic Acid |
| TN-C | Tenascin-C |
| MMP | Matrix Metalloproteinase |
| NP | Nanoparticle |
| DOX | Doxorubicin |
| EDB | Extra Domain B |
| CypA | Cyclophilin A |
| siRNA | Small Interfering Ribonucleic Acid |
| EPR | Enhanced Permeability and Retention |
| PTX | Paclitaxel |
| PLA | Polylactic Acid |
| NRP-1 | Neuropilin-1 |
| NW | Nanoworm |
| AgNP | Silver Nanoparticle |
| NIRF | Near-Infrared Fluorescence |
| PET | Positron Emission Tomography |
| MNP | Magnetic Nanoparticle |
| BSA | Bovine Serum Albumin |
| MRI | Magnetic Resonance Imaging |
| CLIO | Cross-Linked Iron Oxide |
| ICT | Azademethylcolchicine |
| PEG | Polyethylene Glycol |
| PCL | Polycaprolactone |
| dEGCG | Epigallocatechin-3-Gallate Dimer |
| ATP | Adenosine Triphosphate |
| HIF | Hypoxia-Inducible Factor |
| AS1411 | Anti-Nucleolin Aptamer |
| 5-FU | 5-Fluorouracil |
| TEM | Transmission Electron Microscopy |
| PLGA | Poly(Lactic-Co-Glycolic Acid) |
| LDLR | Low-Density Lipoprotein Receptor |
| LAT1 | L-type Amino Acid Transporter 1 |
| YAP | Yes-Associated Protein |
| VP | Verteporfin |
| h-MnO2 | Hollow Manganese Dioxide |
| H2O2 | Hydrogen Peroxide |
| Ce6 | Chlorine e6 |
| GSH | Glutathione |
| SS | Disulfide Bonds |
| MW | Molecular Weight |
| PD | Programmed Cell Death Protein |
| PD-L1 | Programmed Death-Ligand 1 |
| CG-HSANP | Collagenase-Modified Human Serum Albumin Nanoparticle |
| CTX | Chlorotoxin |
References
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Gittleman, H.; Boscia, A.; Ostrom, Q.T.; Truitt, G.; Fritz, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. Survivorship in adults with malignant brain and other central nervous system tumor from 2000–2014. Neuro-Oncology 2018, 20 (Suppl. 7), vii6–vii16. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20 (Suppl. 4), iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Reinders, A.N.; Koshy, M.; Korpics, M. The Patterns of Failure and Prognostic Impact of Tumor Location in Patients Undergoing Reirradiation for Glioblastoma. Cureus 2024, 16, e68820. [Google Scholar] [CrossRef]
- Carrano, A.; Juarez, J.J.; Incontri, D.; Ibarra, A.; Guerrero Cazares, H. Sex-Specific Differences in Glioblastoma. Cells 2021, 10, 1783. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, C.; Chen, J.; Lan, Y.; Zhang, W.; Kang, Z.; Zheng, Y.; Zhang, R.; Yu, J.; Li, W. Signaling pathways in brain tumors and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 8. [Google Scholar] [CrossRef]
- Gue, R.; Lakhani, D.A. The 2021 World Health Organization Central Nervous System Tumor Classification: The Spectrum of Diffuse Gliomas. Biomedicines 2024, 12, 1349. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, B.T.; Huse, J.T. Classification of adult-type diffuse gliomas: Impact of the World Health Organization 2021 update. Brain Pathol. 2022, 32, e13062. [Google Scholar] [CrossRef]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System tumours: A practical update on what neurosurgeons need to know—A minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G.S.; Lyutfi, E.; Georgieva, R.; Georgiev, R.; Dzhenkov, D.L.; Petkova, L.; Ivanov, B.D.; Kaprelyan, A.; Ghenev, P. Reclassification of Glioblastoma Multiforme According to the 2021 World Health Organization Classification of Central Nervous System Tumors: A Single Institution Report and Practical Significance. Cureus 2022, 14, e21822. [Google Scholar] [CrossRef]
- Osborn, A.G.; Louis, D.N.; Poussaint, T.Y.; Linscott, L.L.; Salzman, K.L. The 2021 World Health Organization Classification of Tumors of the Central Nervous System: What Neuroradiologists Need to Know. Am. J. Neuroradiol. 2022, 43, 928–937. [Google Scholar] [CrossRef]
- Colman, H.; Zhang, L.; Sulman, E.P.; McDonald, J.M.; Shooshtari, N.L.; Rivera, A.; Popoff, S.; Nutt, C.L.; Louis, D.N.; Cairncross, J.G.; et al. A multigene predictor of outcome in glioblastoma. Neuro-Oncology 2010, 12, 49–57. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Adel Fahmideh, M.; Cote, D.J.; Muskens, I.S.; Schraw, J.M.; Scheurer, M.E.; Bondy, M.L. Risk factors for childhood and adult primary brain tumors. Neuro-Oncology 2019, 21, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Perfetti, T.A.; Chokshi, C.; Venugopal, C.; Ashford, J.W.; Singh, S.K. Risk factors for glioblastoma are shared by other brain tumor types. Hum. Exp. Toxicol. 2024, 43, 09603271241241796. [Google Scholar] [CrossRef] [PubMed]
- Gunasegaran, B.; Ashley, C.L.; Marsh-Wakefield, F.; Guillemin, G.J.; Heng, B. Viruses in glioblastoma: An update on evidence and clinical trials. BJC Rep. 2024, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Tarev, I.; Cekov, A. Cerebellar Glioblastoma: A Literature Review and Case Analysis. Cureus 2024, 16, e55135. [Google Scholar] [CrossRef]
- Alharbi, B.; Alammar, H.; Alkhaibary, A.; Alharbi, A.; Khairy, S.; Alassiri, A.H.; AlSufiani, F.; Aloraidi, A.; Alkhani, A. Primary spinal cord glioblastoma: A rare cause of paraplegia. Surg. Neurol. Int. 2022, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fuenzalida, J.J.; Moyano-Valarezo, L.; Silva-Bravo, V.; Milos-Brandenberg, D.; Orellana-Donoso, M.; Nova-Baeza, P.; Suazo-Santibáñez, A.; Rodríguez-Luengo, M.; Oyanedel-Amaro, G.; Sanchis-Gimeno, J.; et al. Association between the Anatomical Location of Glioblastoma and Its Evaluation with Clinical Considerations: A Systematic Review and Meta-Analysis. J. Clin. Med. 2024, 13, 3460. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Virtuoso, A.; Papa, M.; Certo, F.; Barbagallo, G.M.V.; Altieri, R. Regional Development of Glioblastoma: The Anatomical Conundrum of Cancer Biology and Its Surgical Implication. Cells 2022, 11, 1349. [Google Scholar] [CrossRef] [PubMed]
- Fyllingen, E.H.; Bø, L.E.; Reinertsen, I.; Jakola, A.S.; Sagberg, L.M.; Berntsen, E.M.; Salvesen, Ø.; Solheim, O. Survival of glioblastoma in relation to tumor location: A statistical tumor atlas of a population-based cohort. Acta Neurochir. 2021, 163, 1895–1905. [Google Scholar] [CrossRef]
- Gilard, V.; Tebani, A.; Dabaj, I.; Laquerrière, A.; Fontanilles, M.; Derrey, S.; Marret, S.; Bekri, S. Diagnosis and Management of Glioblastoma: A Comprehensive Perspective. J. Pers. Med. 2021, 11, 258. [Google Scholar] [CrossRef]
- Rodgers, L.T.; Villano, J.L.; Hartz, A.M.S.; Bauer, B. Glioblastoma Standard of Care: Effects on Tumor Evolution and Reverse Translation in Preclinical Models. Cancers 2024, 16, 2638. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef] [PubMed]
- Jezierzański, M.; Nafalska, N.; Stopyra, M.; Furgoł, T.; Miciak, M.; Kabut, J.; Gisterek-Grocholska, I. Temozolomide (TMZ) in the Treatment of Glioblastoma Multiforme—A Literature Review and Clinical Outcomes. Curr. Oncol. 2024, 31, 3994–4002. [Google Scholar] [CrossRef] [PubMed]
- Furtak, J.; Kwiatkowski, A.; Śledzińska, P.; Bebyn, M.; Krajewski, S.; Szylberg, T.; Birski, M.; Druszcz, A.; Krystkiewicz, K.; Gasiński, P.; et al. Survival after reoperation for recurrent glioblastoma multiforme: A prospective study. Surg. Oncol. 2022, 42, 101771. [Google Scholar] [CrossRef] [PubMed]
- González, V.; Brell, M.; Fuster, J.; Moratinos, L.; Alegre, D.; López, S.; Ibáñez, J. Analyzing the role of reoperation in recurrent glioblastoma: A 15-year retrospective study in a single institution. World J. Surg. Oncol. 2022, 20, 384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-H.; Wang, Z.-F.; Pan, Z.-Y.; Péus, D.; Delgado-Fernandez, J.; Pallud, J.; Li, Z.-Q. A Meta-Analysis of Survival Outcomes Following Reoperation in Recurrent Glioblastoma: Time to Consider the Timing of Reoperation. Front. Neurol. 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Niyazi, M.; Alongi, F.; Navarria, P.; Belka, C. Current status and recent advances in reirradiation of glioblastoma. Radiat. Oncol. 2021, 16, 36. [Google Scholar] [CrossRef]
- Christ, S.M.; Youssef, G.; Tanguturi, S.K.; Cagney, D.; Shi, D.; McFaline-Figueroa, J.R.; Chukwueke, U.; Lee, E.Q.; Hertler, C.; Andratschke, N.; et al. Re-irradiation of recurrent IDH-wildtype glioblastoma in the bevacizumab and immunotherapy era: Target delineation, outcomes and patterns of recurrence. Clin. Transl. Radiat. Oncol. 2024, 44, 100697. [Google Scholar] [CrossRef] [PubMed]
- García-Cabezas, S.; Rivin Del Campo, E.; Solivera-Vela, J.; Palacios-Eito, A. Re-irradiation for high-grade gliomas: Has anything changed? World J. Clin. Oncol. 2021, 12, 767–786. [Google Scholar] [CrossRef] [PubMed]
- Bosio, A.; Cerretti, G.; Padovan, M.; Caccese, M.; Denaro, L.; Chioffi, F.; Della Puppa, A.; Aldegheri, V.; Guarneri, V.; Zagonel, V.; et al. Metronomic Temozolomide in Heavily Pretreated Patients with Recurrent Isocitrate Dehydrogenase Wild-type Glioblastoma: A Large Real-Life Mono-Institutional Study. Clin. Oncol. 2023, 35, e319–e327. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Tosoni, A.; Cavallo, G.; Bertorelle, R.; Gioia, V.; Franceschi, E.; Biscuola, M.; Blatt, V.; Crinò, L.; Ermani, M. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: Phase II study from gruppo italiano cooperativo di neuro-oncologia (GICNO). Br. J. Cancer 2006, 95, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Lamberti, G.; Visani, M.; Paccapelo, A.; Mura, A.; Tallini, G.; Pession, A.; De Biase, D.; Minichillo, S.; Tosoni, A.; et al. Temozolomide Rechallenge in Recurrent Glioblastoma: When is it Useful? Future Oncol. 2018, 14, 1063–1069. [Google Scholar] [CrossRef]
- Weller, M.; Tabatabai, G.; Kästner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Steinbach, J.P.; Küker, W.M.; Dichgans, J.; Bamberg, M.; Weller, M. One week on/one week off: A novel active regimen of temozolomide for recurrent glioblastoma. Neurology 2004, 62, 2113–2115. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Tosoni, A.; Amistà, P.; Nicolardi, L.; Grosso, D.; Berti, F.; Ermani, M. How effective is BCNU in recurrent glioblastoma in the modern era? A phase II trial. Neurology 2004, 63, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Tosoni, A.; Basso, U.; Reni, M.; Valduga, F.; Monfardini, S.; Amistà, P.; Nicolardi, L.; Sotti, G.; Ermani, M. Second-line chemotherapy with irinotecan plus carmustine in glioblastoma recurrent or progressive after first-line temozolomide chemotherapy: A phase II study of the Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). J. Clin. Oncol. 2004, 22, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Reithmeier, T.; Graf, E.; Piroth, T.; Trippel, M.; Pinsker, M.O.; Nikkhah, G. BCNU for recurrent glioblastoma multiforme: Efficacy, toxicity and prognostic factors. BMC Cancer 2010, 10, 30. [Google Scholar] [CrossRef]
- You, W.-C.; Lee, H.-D.; Pan, H.-C.; Chen, H.-C. Re-irradiation combined with bevacizumab for recurrent glioblastoma beyond bevacizumab failure: Survival outcomes and prognostic factors. Sci. Rep. 2023, 13, 9442. [Google Scholar] [CrossRef]
- Ameratunga, M.; Pavlakis, N.; Wheeler, H.; Grant, R.; Simes, J.; Khasraw, M. Anti-angiogenic therapy for high-grade glioma. Cochrane Database Syst. Rev. 2018, 11, CD008218. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Zhou, Z.; Huang, X.; Chen, Z.; Zhang, L.; Zhang, J.; Hua, W.; Mao, Y. Use of Bevacizumab in recurrent glioblastoma: A scoping review and evidence map. BMC Cancer 2023, 23, 544. [Google Scholar] [CrossRef]
- Ranchor, R.; Ramos, M.J.R.G.D.L.; Romao, R.M.; Mendes, A.S.; Pichel, R.C.; Coelho, J.Q.; Rosendo, E.M.; Magalhaes, M.J.; Araújo, A.M.F. 3P Bevacizumab plus irinotecan as second-line treatment of glioblastoma: Real-world evidence. ESMO Open 2023, 8, 101015. [Google Scholar] [CrossRef]
- Alves, B.; Peixoto, J.; Macedo, S.; Pinheiro, J.; Carvalho, B.; Soares, P.; Lima, J.; Lima, R.T. High VEGFA Expression Is Associated with Improved Progression-Free Survival after Bevacizumab Treatment in Recurrent Glioblastoma. Cancers 2023, 15, 2196. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, S.; Mangraviti, A.; Martini, M.; Cenci, T.; Mazzarella, C.; Gaudino, S.; Bracci, S.; Martino, A.; Della Pepa, G.M.; Offi, M.; et al. Clinical and NGS predictors of response to regorafenib in recurrent glioblastoma. Sci. Rep. 2022, 12, 16265. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Pallini, R.; D’Alessandris, Q.G.; Levi, A.; Falchetti, M.L. Regorafenib and glioblastoma: A literature review of preclinical studies, molecular mechanisms and clinical effectiveness. Expert Rev. Mol. Med. 2024, 26, e5. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Brar, H.K.; Jose, J.; Wu, Z.; Sharma, M. Tyrosine Kinase Inhibitors for Glioblastoma Multiforme: Challenges and Opportunities for Drug Delivery. Pharmaceutics 2022, 15, 59. [Google Scholar] [CrossRef]
- Rahban, M.; Joushi, S.; Bashiri, H.; Saso, L.; Sheibani, V. Characterization of prevalent tyrosine kinase inhibitors and their challenges in glioblastoma treatment. Front. Chem. 2024, 11, 1325214. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133, e163447. [Google Scholar] [CrossRef]
- Ser, M.H.; Webb, M.J.; Sener, U.; Campian, J.L. Immune Checkpoint Inhibitors and Glioblastoma: A Review on Current State and Future Directions. J. Immunother. Precis. Oncol. 2024, 7, 97–110. [Google Scholar] [CrossRef]
- Wang, H.; Yang, J.; Li, X.; Zhao, H. Current state of immune checkpoints therapy for glioblastoma. Heliyon 2024, 10, e24729. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, E.; Choi, J.; Tran, A.; Kim, L.H.; Lim, M. The landscape of immune checkpoint inhibitor clinical trials in glioblastoma: A systematic review. Neurooncol. Adv. 2024, 6, vdae174. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. 5), S2–S8. [Google Scholar] [CrossRef]
- Kirson, E.D.; Dbalý, V.; Tovarys, F.; Vymazal, J.; Soustiel, J.F.; Itzhaki, A.; Mordechovich, D.; Steinberg-Shapira, S.; Gurvich, Z.; Schneiderman, R.; et al. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 10152–10157. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taphoorn, M.; Dirven, L.; Taillibert, S.; Honnorat, J.; Chen, T.; Sroubek, J.; Paek, S.H.; Bruna Escuder, J.; Easaw, J.; et al. Tumor Treating Fields (TTFields)—A novel cancer treatment modality: Translating preclinical evidence and engineering into a survival benefit with delayed decline in quality of life. Ann. Oncol. 2017, 28, v112. [Google Scholar] [CrossRef]
- Wang, Y.; Pandey, M.; Ballo, M.T. Integration of Tumor-Treating Fields into the Multidisciplinary Management of Patients with Solid Malignancies. Oncologist 2019, 24, e1426–e1436. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Chang, C.; Chavarro, V.S.; Gerstl, J.V.E.; Blitz, S.E.; Spanehl, L.; Dubinski, D.; Valdes, P.A.; Tran, L.N.; Gupta, S.; Esposito, L.; et al. Recurrent Glioblastoma—Molecular Underpinnings and Evolving Treatment Paradigms. Int. J. Mol. Sci. 2024, 25, 6733. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Stupp, R.; Sonabend, A.M.; Dufour, H.; Chinot, O.; Mathon, B.; Ducray, F.; Guyotat, J.; Baize, N.; Menei, P.; et al. Repeated blood–brain barrier opening with a nine-emitter implantable ultrasound device in combination with carboplatin in recurrent glioblastoma: A phase I/II clinical trial. Nat. Commun. 2024, 15, 1650. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.L.; Solomon, I.H.; Landivar, A.M.; Nakashima, H.; Woods, J.K.; Santos, A.; Masud, N.; Fell, G.; Mo, X.; Yilmaz, A.S.; et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature 2023, 623, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Barthel, L.; Hadamitzky, M.; Dammann, P.; Schedlowski, M.; Sure, U.; Thakur, B.K.; Hetze, S. Glioma: Molecular signature and crossroads with tumor microenvironment. Cancer Metastasis Rev. 2022, 41, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef]
- Lin, N.; Yan, W.; Gao, K.; Wang, Y.; Zhang, J.; You, Y. Prevalence and Clinicopathologic Characteristics of the Molecular Subtypes in Malignant Glioma: A Multi-Institutional Analysis of 941 Cases. PLoS ONE 2014, 9, e94871. [Google Scholar] [CrossRef]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.M.B.; Kamel, A.; Ciubotaru, G.V.; Onose, G.; Sevastre, A.S.; Sfredel, V.; Danoiu, S.; Dricu, A.; Tataranu, L.G. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Seo, H.W.; Baek, J.-H.; Lim, S.H.; Hwang, S.-G.; Kim, E.H. Gene expression profiling of glioblastoma cell lines depending on TP53 status after tumor-treating fields (TTFields) treatment. Sci. Rep. 2020, 10, 12272. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Azam, Z.; To, S.-S.T.; Tannous, B.A. Mesenchymal Transformation: The Rosetta Stone of Glioblastoma Pathogenesis and Therapy Resistance. Adv. Sci. 2020, 7, 2002015. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.V.; Musina, G.R.; Pekov, S.I.; Kuzin, A.A.; Popov, I.A.; Belyaev, A.Y.; Kobyakov, G.L.; Usachev, D.Y.; Nikolaev, V.N.; Mikhailov, V.P. Cell-Population Dynamics in Diffuse Gliomas during Gliomagenesis and Its Impact on Patient Survival. Cancers 2022, 15, 145. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef]
- Almairac, F.; Turchi, L.; Sakakini, N.; Debruyne, D.N.; Elkeurti, S.; Gjernes, E.; Polo, B.; Bianchini, L.; Fontaine, D.; Paquis, P.; et al. ERK-Mediated Loss of miR-199a-3p and Induction of EGR1 Act as a “Toggle Switch” of GBM Cell Dedifferentiation into NANOG- and OCT4-Positive Cells. Cancer Res. 2020, 80, 3236–3250. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Bonaguidi, M.A.; Ming, G.L.; Song, H. Adult neural stem cells in the mammalian central nervous system. Cell Res 2009, 19, 672–682. [Google Scholar] [CrossRef]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Hersh, A.M.; Gaitsch, H.; Alomari, S.; Lubelski, D.; Tyler, B.M. Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy. Cancers 2022, 14, 3743. [Google Scholar] [CrossRef]
- Crivii, C.-B.; Boșca, A.B.; Melincovici, C.S.; Constantin, A.-M.; Mărginean, M.; Dronca, E.; Suflețel, R.; Gonciar, D.; Bungărdean, M.; Șovrea, A. Glioblastoma Microenvironment and Cellular Interactions. Cancers 2022, 14, 1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, C.; Wang, B.; Zhang, H.; Qin, G.; Li, C.; Cao, L.; Gao, Q.; Ping, Y.; Zhang, K.; et al. Regulatory T cells promote glioma cell stemness through TGF-β–NF-κB–IL6–STAT3 signaling. Cancer Immunol. Immunother. 2021, 70, 2601–2616. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, M.; Zhao, J.; Ren, T.; Yan, X.; Zhang, L.; Wang, X. Research Progress About Glioma Stem Cells in the Immune Microenvironment of Glioma. Front. Pharmacol. 2021, 12, 750857. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef] [PubMed]
- Persico, P.; Lorenzi, E.; Dipasquale, A.; Pessina, F.; Navarria, P.; Politi, L.S.; Santoro, A.; Simonelli, M. Checkpoint Inhibitors as High-Grade Gliomas Treatment: State of the Art and Future Perspectives. J. Clin. Med. 2021, 10, 1367. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Davis, Z.B.; Rechberger, J.S.; Toll, S.A.; Schwartz, J.D.; Daniels, D.J.; Miller, J.S.; Khatua, S. Advances in NK cell therapy for brain tumors. npj Precis. Oncol. 2023, 7, 17. [Google Scholar] [CrossRef]
- Galbo, P.M., Jr.; Madsen, A.T.; Liu, Y.; Peng, M.; Wei, Y.; Ciesielski, M.J.; Fenstermaker, R.A.; Graff, S.; Montagna, C.; Segall, J.E.; et al. Functional Contribution and Clinical Implication of Cancer-Associated Fibroblasts in Glioblastoma. Clin. Cancer Res. 2024, 30, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.; Zhang, S.; Chen, S.; Xiang, Y.; Yuan, Y.; Li, T.; Yang, W.; Wang, Z.; He, Y.; Li, W.; et al. Glioma-associated fibroblasts promote glioblastoma resistance to temozolomide through CCL2-CCR2 paracrine signaling. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lootens, T.; Roman, B.I.; Stevens, C.V.; De Wever, O.; Raedt, R. Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime? Int. J. Mol. Sci. 2024, 25, 2285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fei, Y.; Wang, H.; Hu, S.; Liu, C.; Hu, R.; Du, Q. CAFs orchestrates tumor immune microenvironment—A new target in cancer therapy? Front. Pharmacol. 2023, 14, 1113378. [Google Scholar] [CrossRef] [PubMed]
- Randles, A.; Wirsching, H.G.; Dean, J.A.; Cheng, Y.K.; Emerson, S.; Pattwell, S.S.; Holland, E.C.; Michor, F. Computational modelling of perivascular-niche dynamics for the optimization of treatment schedules for glioblastoma. Nat. Biomed. Eng. 2021, 5, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef]
- Hovis, G.; Chandra, N.; Kejriwal, N.; Hsieh, K.J.; Chu, A.; Yang, I.; Wadehra, M. Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments. Int. J. Mol. Sci. 2024, 25, 6118. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.; Martins, C.; Monteiro, J.; Baltazar, F.; Costa, B.M.; Sarmento, B. Glioblastoma Vasculature: From its Critical Role in Tumor Survival to Relevant in Vitro Modelling. Front. Drug Deliv. 2022, 2, 823412. [Google Scholar] [CrossRef]
- Maddison, K.; Bowden, N.A.; Graves, M.C.; Tooney, P.A. Characteristics of vasculogenic mimicry and tumour to endothelial transdifferentiation in human glioblastoma: A systematic review. BMC Cancer 2023, 23, 185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial Cells Create a Stem Cell Niche in Glioblastoma by Providing NOTCH Ligands That Nurture Self-Renewal of Cancer Stem-Like Cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.; Enright, H.A.; Cadena, J.; Peters, S.K.G.; Sales, A.P.; Osburn, J.J.; Soscia, D.A.; Kulp, K.S.; Wheeler, E.K.; Fischer, N.O. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Sci. Rep. 2019, 9, 4159. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Menna, G.; Di Bonaventura, R.; Lisi, L.; Mattogno, P.; Figà, F.; Bilgin, L.; D’Alessandris, Q.G.; Olivi, A.; Della Pepa, G.M. The Extracellular Matrix in Glioblastomas: A Glance at Its Structural Modifications in Shaping the Tumoral Microenvironment-A Systematic Review. Cancers 2023, 15, 1879. [Google Scholar] [CrossRef]
- Safarians, G.; Sohrabi, A.; Solomon, I.; Xiao, W.; Bastola, S.; Rajput, B.W.; Epperson, M.; Rosenzweig, I.; Tamura, K.; Singer, B.; et al. Glioblastoma Spheroid Invasion through Soft, Brain-Like Matrices Depends on Hyaluronic Acid–CD44 Interactions. Adv. Healthc. Mater. 2023, 12, 2203143. [Google Scholar] [CrossRef] [PubMed]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sinha, S.; Jiang, X.; Murphy, L.; Fitch, S.; Wilson, C.; Grant, G.; Yang, F. Matrix Stiffness Modulates Patient-Derived Glioblastoma Cell Fates in Three-Dimensional Hydrogels. Tissue Eng. Part A 2021, 27, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Erices, J.I.; Bizama, C.; Niechi, I.; Uribe, D.; Rosales, A.; Fabres, K.; Navarro-Martínez, G.; Torres, Á.; San Martín, R.; Roa, J.C.; et al. Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 7047. [Google Scholar] [CrossRef]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The Extracellular Matrix and Biocompatible Materials in Glioblastoma Treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef]
- Gupta, R.K.; Niklasson, M.; Bergström, T.; Segerman, A.; Betsholtz, C.; Westermark, B. Tumor-specific migration routes of xenotransplanted human glioblastoma cells in mouse brain. Sci. Rep. 2024, 14, 864. [Google Scholar] [CrossRef] [PubMed]
- Sood, D.; Tang-Schomer, M.; Pouli, D.; Mizzoni, C.; Raia, N.; Tai, A.; Arkun, K.; Wu, J.; Black, L.D., III; Scheffler, B.; et al. 3D extracellular matrix microenvironment in bioengineered tissue models of primary pediatric and adult brain tumors. Nat. Commun. 2019, 10, 4529. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.; Adhikary, K.; Acharjee, A.; Acharjee, P.; Trigun, S.K.; Mutlaq, A.S.; Ashique, S.; Yasmin, S.; Alshahrani, A.M.; Ansari, M.Y. Biological significance and pathophysiological role of Matrix Metalloproteinases in the Central Nervous System. Int. J. Biol. Macromol. 2024, 280, 135967. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The blood–brain barrier: Structure, regulation and drug delivery. Signal Transduct. Target. Ther. 2023, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Mărginean, L.; Ștefan, P.A.; Lebovici, A.; Opincariu, I.; Csutak, C.; Lupean, R.A.; Coroian, P.A.; Suciu, B.A. CT in the Differentiation of Gliomas from Brain Metastases: The Radiomics Analysis of the Peritumoral Zone. Brain Sci. 2022, 12, 109. [Google Scholar] [CrossRef]
- Manzarbeitia-Arroba, B.; Hodolic, M.; Pichler, R.; Osipova, O.; Soriano-Castrejón, Á.M.; García-Vicente, A.M. 18F-Fluoroethyl-L Tyrosine Positron Emission Tomography Radiomics in the Differentiation of Treatment-Related Changes from Disease Progression in Patients with Glioblastoma. Cancers 2024, 16, 195. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.; Almotairy, A.R.Z.; Henidi, H.; Alshehri, O.Y.; Aldughaim, M.S. Nanoparticles as Drug Delivery Systems: A Review of the Implication of Nanoparticles’ Physicochemical Properties on Responses in Biological Systems. Polymers 2023, 15, 1596. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Liu, R.; Zhou, Y.; Gao, H. Size-Tunable Strategies for a Tumor Targeted Drug Delivery System. ACS Cent. Sci. 2020, 6, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Molina, D.; Mao, X.; Alfonso-Triguero, P.; Lorenzo, J.; Bruna, J.; Yuste, V.J.; Candiota, A.P.; Novio, F. Advances in Preclinical/Clinical Glioblastoma Treatment: Can Nanoparticles Be of Help? Cancers 2022, 14, 4960. [Google Scholar] [CrossRef]
- Lai, G.; Wu, H.; Yang, K.; Hu, K.; Zhou, Y.; Chen, X.; Fu, F.; Li, J.; Xie, G.; Wang, H.-F.; et al. Progress of nanoparticle drug delivery system for the treatment of glioma. Front. Bioeng. Biotechnol. 2024, 12, 1403511. [Google Scholar] [CrossRef] [PubMed]
- Fabel, K.; Dietrich, J.; Hau, P.; Wismeth, C.; Winner, B.; Przywara, S.; Steinbrecher, A.; Ullrich, W.; Bogdahn, U. Long-term stabilization in patients with malignant glioma after treatment with liposomal doxorubicin. Cancer 2001, 92, 1936–1942. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.L.; Rosenthal, M.A.; Wong, S.S.; Ashley, D.M.; Woods, A.M.; Dowling, A.; Cher, L.M. Phase 2 study of temozolomide and Caelyx in patients with recurrent glioblastoma multiforme. Neuro-Oncology 2004, 6, 38–43. [Google Scholar] [CrossRef]
- Hau, P.; Fabel, K.; Baumgart, U.; Rümmele, P.; Grauer, O.; Bock, A.; Dietmaier, C.; Dietmaier, W.; Dietrich, J.; Dudel, C.; et al. Pegylated liposomal doxorubicin-efficacy in patients with recurrent high-grade glioma. Cancer 2004, 100, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Beier, C.P.; Schmid, C.; Gorlia, T.; Kleinletzenberger, C.; Beier, D.; Grauer, O.; Steinbrecher, A.; Hirschmann, B.; Brawanski, A.; Dietmaier, C.; et al. RNOP-09: Pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma—A phase II study. BMC Cancer 2009, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Ananda, S.; Nowak, A.K.; Cher, L.; Dowling, A.; Brown, C.; Simes, J.; Rosenthal, M.A. Phase 2 trial of temozolomide and pegylated liposomal doxorubicin in the treatment of patients with glioblastoma multiforme following concurrent radiotherapy and chemotherapy. J. Clin. Neurosci. 2011, 18, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.L.; Molinaro, A.M.; DeSilva, A.A.; Rabbitt, J.E.; Drummond, D.C.; Chang, S.M.; Butowski, N.A.; Prados, M. A phase I trial of intravenous liposomal irinotecan in patients with recurrent high-grade gliomas. J. Clin. Oncol. 2015, 33 (Suppl. 15), 2029. [Google Scholar] [CrossRef]
- Elinzano, H.; Toms, S.; Robison, J.; Mohler, A.; Carcieri, A.; Cielo, D.; Donnelly, J.; Disano, D.; Vatketich, J.; Baekey, J.; et al. Nanoliposomal Irinotecan and Metronomic Temozolomide for Patients with Recurrent Glioblastoma: BrUOG329, A Phase I Brown University Oncology Research Group Trial. Am. J. Clin. Oncol. 2021, 44, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Roque, D.; Cruz, N.; Ferreira, H.A.; Reis, C.P.; Matela, N.; Herculano-Carvalho, M.; Cascão, R.; Faria, C.C. Nanoparticle-Based Treatment in Glioblastoma. J. Pers. Med. 2023, 13, 1328. [Google Scholar] [CrossRef]
- Horta, M.; Soares, P.; Sarmento, B.; Leite Pereira, C.; Lima, R.T. Nanostructured lipid carriers for enhanced batimastat delivery across the blood–brain barrier: An in vitro study for glioblastoma treatment. Drug Deliv. Transl. Res. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Gu, G.; Xia, H.; Hu, Q.; Liu, Z.; Jiang, M.; Kang, T.; Miao, D.; Tu, Y.; Pang, Z.; Song, Q.; et al. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials 2013, 34, 196–208. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Qian, X.; Wang, Y.; Wang, J.; Li, J.; Li, Y.; Zhang, Z. Nanomedicine Strategies to Circumvent Intratumor Extracellular Matrix Barriers for Cancer Therapy. Adv. Healthc. Mater. 2022, 11, 2101428. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar]
- Mai, Z.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Modulating extracellular matrix stiffness: A strategic approach to boost cancer immunotherapy. Cell Death Dis. 2024, 15, 307. [Google Scholar] [CrossRef]
- Saw, P.E.; Zhang, A.; Nie, Y.; Zhang, L.; Xu, Y.; Xu, X. Tumor-Associated Fibronectin Targeted Liposomal Nanoplatform for Cyclophilin A siRNA Delivery and Targeted Malignant Glioblastoma Therapy. Front. Pharmacol. 2018, 9, 1194. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Cheng, Y.; Morshed, R.; Nord, K.; Han, Y.; Wegscheid, M.L.; Auffinger, B.; Wainwright, D.A.; Lesniak, M.S.; Tirrell, M.V. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials 2014, 35, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Zhu, Q.; Jiang, D.; Feng, X.; Feng, J.; Jiang, T.; Yao, J.; Jing, Y.; Song, Q.; Jiang, X.; et al. Synergistic targeting tenascin C and neuropilin-1 for specific penetration of nanoparticles for anti-glioblastoma treatment. Biomaterials 2016, 101, 60–75. [Google Scholar] [CrossRef]
- Lingasamy, P.; Tobi, A.; Kurm, K.; Kopanchuk, S.; Sudakov, A.; Salumäe, M.; Rätsep, T.; Asser, T.; Bjerkvig, R.; Teesalu, T. Tumor-penetrating peptide for systemic targeting of Tenascin-C. Sci. Rep. 2020, 10, 5809. [Google Scholar] [CrossRef] [PubMed]
- Lingasamy, P.; Põšnograjeva, K.; Kopanchuk, S.; Tobi, A.; Rinken, A.; General, I.J.; Asciutto, E.K.; Teesalu, T. PL1 Peptide Engages Acidic Surfaces on Tumor-Associated Fibronectin and Tenascin Isoforms to Trigger Cellular Uptake. Pharmaceutics 2021, 13, 1998. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Jiang, K.; Cole, D.; Jani, A.; Udayakumar, N.; Gillespie, G.Y.; Lu, G.; Dai, T.; Rosenthal, E.L.; Markert, J.M.; et al. Targeting MMP-14 for dual PET and fluorescence imaging of glioma in preclinical models. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Abakumov, M.A.; Nukolova, N.V.; Sokolsky-Papkov, M.; Shein, S.A.; Sandalova, T.O.; Vishwasrao, H.M.; Grinenko, N.F.; Gubsky, I.L.; Abakumov, A.M.; Kabanov, A.V.; et al. VEGF-targeted magnetic nanoparticles for MRI visualization of brain tumor. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 825–833. [Google Scholar] [CrossRef]
- Mohanty, S.; Chen, Z.; Li, K.; Morais, G.R.; Klockow, J.; Yerneni, K.; Pisani, L.; Chin, F.T.; Mitra, S.; Cheshier, S.; et al. A Novel Theranostic Strategy for MMP-14-Expressing Glioblastomas Impacts Survival. Mol. Cancer Ther. 2017, 16, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Chen, C.; Mu, M.; Chuan, D.; Liu, H.; Hou, H.; Huang, J.; Tong, A.; Guo, G.; Xu, J. Engineering MMP-2 Activated Nanoparticles Carrying B7-H3 Bispecific Antibodies for Ferroptosis-Enhanced Glioblastoma Immunotherapy. ACS Nano 2023, 17, 9126–9139. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, W.; Zhong, T.; Zhang, S.; Huang, D.; Guo, Y.; Yao, X.; Wang, C.; Zhang, W.-Q.; Zhang, X.; et al. Tumor-specific pH-responsive peptide-modified pH-sensitive liposomes containing doxorubicin for enhancing glioma targeting and anti-tumor activity. J. Control. Release 2016, 222, 56–66. [Google Scholar] [CrossRef]
- Sathiyaseelan, A.; Saravanakumar, K.; Mariadoss, A.V.A.; Wang, M.-H. pH-controlled nucleolin targeted release of dual drug from chitosan-gold based aptamer functionalized nano drug delivery system for improved glioblastoma treatment. Carbohydr. Polym. 2021, 262, 117907. [Google Scholar] [CrossRef]
- Martins, C.; Araujo, M.; Malfanti, A.; Pacheco, C.; Smith, S.J.; Ucakar, B.; Rahman, R.; Aylott, J.W.; Preat, V.; Sarmento, B. Stimuli-Responsive Multifunctional Nanomedicine for Enhanced Glioblastoma Chemotherapy Augments Multistage Blood-to-Brain Trafficking and Tumor Targeting. Small 2023, 19, e2300029. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhang, L.; Ren, Z.; Yuan, Y.; Yu, J.; Zhang, Y.; Gu, L.; Wang, X.; Wang, Y.; Xu, H.; et al. Stepwise-targeting and hypoxia-responsive liposome AMVY@NPs carrying siYAP and verteporfin for glioblastoma therapy. J. Nanobiotechnol. 2024, 22, 495. [Google Scholar] [CrossRef]
- Yang, G.; Xu, L.; Chao, Y.; Xu, J.; Sun, X.; Wu, Y.; Peng, R.; Liu, Z. Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat. Commun. 2017, 8, 902. [Google Scholar] [CrossRef]
- Tian, C.; Asghar, S.; Xu, Y.; Chen, Z.; Zhang, M.; Huang, L.; Ye, J.; Ping, Q.; Xiao, Y. The effect of the molecular weight of hyaluronic acid on the physicochemical characterization of hyaluronic acid-curcumin conjugates and in vitro evaluation in glioma cells. Colloids Surf. B Biointerfaces 2018, 165, 45–55. [Google Scholar] [CrossRef]
- Kiyokawa, J.; Kawamura, Y.; Ghouse, S.M.; Acar, S.; Barçın, E.; Martínez-Quintanilla, J.; Martuza, R.L.; Alemany, R.; Rabkin, S.D.; Shah, K.; et al. Modification of Extracellular Matrix Enhances Oncolytic Adenovirus Immunotherapy in Glioblastoma. Clin. Cancer Res. 2021, 27, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.K.; Behera, C.; Chakraborty, S.; Sandha, K.K.; Goswami, A.; Gupta, P.N. Tumor micro-environment targeted collagenase-modified albumin nanoparticles for improved drug delivery. J. Drug Deliv. Sci. Technol. 2022, 71, 103366. [Google Scholar] [CrossRef]
- Agarwal, S.; Mohamed, M.S.; Mizuki, T.; Maekawa, T.; Sakthi Kumar, D. Chlorotoxin modified morusin–PLGA nanoparticles for targeted glioblastoma therapy. J. Mater. Chem. B 2019, 7, 5896–5919. [Google Scholar] [CrossRef] [PubMed]
- Islam, Y.; Khalid, A.; Pluchino, S.; Sivakumaran, M.; Teixidò, M.; Leach, A.; Fatokun, A.A.; Downing, J.; Coxon, C.; Ehtezazi, T. Development of Brain Targeting Peptide Based MMP-9 Inhibiting Nanoparticles for the Treatment of Brain Diseases with Elevated MMP-9 Activity. J. Pharm. Sci. 2020, 109, 3134–3144. [Google Scholar] [CrossRef]
- Ventura, E.; Weller, M.; Macnair, W.; Eschbach, K.; Beisel, C.; Cordazzo, C.; Claassen, M.; Zardi, L.; Burghardt, I. TGF-β induces oncofetal fibronectin that, in turn, modulates TGF-β superfamily signaling in endothelial cells. J. Cell Sci. 2018, 131, jcs209619. [Google Scholar] [CrossRef]
- Lui, B.G.; Salomon, N.; Wüstehube-Lausch, J.; Daneschdar, M.; Schmoldt, H.-U.; Türeci, Ö.; Sahin, U. Targeting the tumor vasculature with engineered cystine-knot miniproteins. Nat. Commun. 2020, 11, 295. [Google Scholar] [CrossRef]
- Spaeth, N.; Wyss, M.T.; Pahnke, J.; Biollaz, G.; Trachsel, E.; Drandarov, K.; Treyer, V.; Weber, B.; Neri, D.; Buck, A. Radioimmunotherapy targeting the extra domain B of fibronectin in C6 rat gliomas: A preliminary study about the therapeutic efficacy of iodine-131-labeled SIP(L19). Nucl. Med. Biol. 2006, 33, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Dzikowski, L.; Mirzaei, R.; Sarkar, S.; Kumar, M.; Bose, P.; Bellail, A.; Hao, C.; Yong, V.W. Fibrinogen in the glioblastoma microenvironment contributes to the invasiveness of brain tumor-initiating cells. Brain Pathol. 2021, 31, e12947. [Google Scholar] [CrossRef]
- Zhang, N.; Ru, B.; Hu, J.; Xu, L.; Wan, Q.; Liu, W.; Cai, W.; Zhu, T.; Ji, Z.; Guo, R.; et al. Recent advances of CREKA peptide-based nanoplatforms in biomedical applications. J. Nanobiotechnol. 2023, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Brack, S.S.; Silacci, M.; Birchler, M.; Neri, D. Tumor-Targeting Properties of Novel Antibodies Specific to the Large Isoform of Tenascin-C. Clin. Cancer Res. 2006, 12, 3200–3208. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yu, X.; Sun, S.; Zhang, X.; Yang, W.; Zhang, J.; Zhang, X.; Jiang, Z. Increased expression of MMP-2 and MMP-9 indicates poor prognosis in glioma recurrence. Biomed. Pharmacother. 2019, 118, 109369. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, L.G.N.; Thode, H.; Eslambolchi, Y.; Chopra, S.; Young, D.; Gill, S.; Devel, L.; Dufour, A. Matrix Metalloproteinases: From Molecular Mechanisms to Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 2022, 74, 714–770. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.K.; Sorensen, M.D.; Aaberg-Jessen, C.; Hermansen, S.K.; Kristensen, B.W. Expression and prognostic impact of matrix metalloproteinase-2 (MMP-2) in astrocytomas. PLoS ONE 2017, 12, e0172234. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Cao, L.; Chen, X.Y.; Zhao, J.; Gao, L.; Li, S.Z.; Fei, Z. High expression of MMP9 in glioma affects cell proliferation and is associated with patient survival rates. Oncol. Lett. 2017, 13, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Conlon, G.A.; Murray, G.I. Recent advances in understanding the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2019, 247, 629–640. [Google Scholar] [CrossRef]
- Munaut, C.; Noel, A.; Hougrand, O.; Foidart, J.M.; Boniver, J.; Deprez, M. Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1-MMP, MMP-2 and MMP-9 in human glioblastomas. Int. J. Cancer 2003, 106, 848–855. [Google Scholar] [CrossRef]
- Ulasov, I.; Yi, R.; Guo, D.; Sarvaiya, P.; Cobbs, C. The emerging role of MMP14 in brain tumorigenesis and future therapeutics. Biochim. Biophys. Acta (BBA) Rev. Cancer 2014, 1846, 113–120. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef]
- Hatoum, A.; Mohammed, R.; Zakieh, O. The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag. Res. 2019, 11, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the Past, Present, and Future of Anti-Angiogenic Therapy in Glioblastoma. Cancers 2023, 15, 830. [Google Scholar] [CrossRef] [PubMed]
- Codrici, E.; Enciu, A.-M.; Popescu, I.-D.; Mihai, S.; Tanase, C. Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Targets. Stem Cells Int. 2016, 2016, 5728438. [Google Scholar] [CrossRef]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E., II; Jones, L.W.; Kirkpatrick, J.P.; et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J. Natl. Compr. Cancer Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef]
- Wu, Z.; Dai, L.; Tang, K.; Ma, Y.; Song, B.; Zhang, Y.; Li, J.; Lui, S.; Gong, Q.; Wu, M. Advances in magnetic resonance imaging contrast agents for glioblastoma-targeting theranostics. Regen. Biomater. 2021, 8, rbab062. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.J.; Perez, M.R.; Mikos, A.G. Extracellular matrix component-derived nanoparticles for drug delivery and tissue engineering. J. Control. Release 2023, 360, 888–912. [Google Scholar] [CrossRef] [PubMed]
- Karimi, N.; Kheiri, H.; Zarrinpour, V.; Forghanifard, M.M. Bioinformatic analysis of MMP family members in GBM. Inform. Med. Unlocked 2023, 39, 101240. [Google Scholar] [CrossRef]
- Talib, W.H.; Awajan, D.; Alqudah, A.; Alsawwaf, R.; Althunibat, R.; Abu AlRoos, M.; Al Safadi, A.a.; Abu Asab, S.; Hadi, R.W.; Al Kury, L.T. Targeting Cancer Hallmarks with Epigallocatechin Gallate (EGCG): Mechanistic Basis and Therapeutic Targets. Molecules 2024, 29, 1373. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, A.; Bogdanov, A.; Chubenko, V.; Volkov, N.; Moiseenko, F.; Moiseyenko, V. Tumor acidity: From hallmark of cancer to target of treatment. Front. Oncol. 2022, 12, 979154. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Ponneri Babuharisankar, A.; Lin, Y.-C.; Lien, H.-W.; Lo, Y.K.; Chou, H.-Y.; Tangeda, V.; Cheng, L.-C.; Cheng, A.N.; Lee, A.Y.-L. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J. Biomed. Sci. 2022, 29, 74. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2020, 7, 620677. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, X.; Zhang, D.; Mu, X. Research Progress of Disulfide Bond Based Tumor Microenvironment Targeted Drug Delivery System. Int. J. Nanomed. 2024, 19, 7547–7566. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huo, M.; Wang, J.; Zhou, J.; Mohammad, J.M.; Zhang, Y.; Zhu, Q.; Waddad, A.Y.; Zhang, Q. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials 2012, 33, 2310–2320. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, X.; Cao, J.; Gao, H. Overcoming the biological barriers in the tumor microenvironment for improving drug delivery and efficacy. J. Mater. Chem. B 2020, 8, 6765–6781. [Google Scholar] [CrossRef]
- Parsons, S.L.; Watson, S.A.; Steele, R.J. Phase I/II trial of batimastat, a matrix metalloproteinase inhibitor, in patients with malignant ascites. Eur. J. Surg. Oncol. 1997, 23, 526–531. [Google Scholar] [CrossRef]
- Groves, M.D.; Puduvalli, V.K.; Hess, K.R.; Jaeckle, K.A.; Peterson, P.; Yung, W.K.; Levin, V.A. Phase II trial of temozolomide plus the matrix metalloproteinase inhibitor, marimastat, in recurrent and progressive glioblastoma multiforme. J. Clin. Oncol. 2002, 20, 1383–1388. [Google Scholar] [CrossRef]
- Boguszewska-Czubara, A.; Budzynska, B.; Skalicka-Wozniak, K.; Kurzepa, J. Perspectives and New Aspects of Metalloproteinases’ Inhibitors in the Therapy of CNS Disorders: From Chemistry to Medicine. Curr. Med. Chem. 2019, 26, 3208–3224. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Sehedic, D.; Chourpa, I.; Tetaud, C.; Griveau, A.; Loussouarn, C.; Avril, S.; Legendre, C.; Lepareur, N.; Wion, D.; Hindre, F.; et al. Locoregional Confinement and Major Clinical Benefit of 188Re-Loaded CXCR4-Targeted Nanocarriers in an Orthotopic Human to Mouse Model of Glioblastoma. Theranostics 2017, 7, 4517–4536. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Farkas, S.; Cioca, D.; Murányi, J.; Hornyák, P.; Brunyánszki, A.; Szekér, P.; Boros, E.; Horváth, P.; Hujber, Z.; Rácz, G.Z.; et al. Chlorotoxin binds to both matrix metalloproteinase 2 and neuropilin 1. J. Biol. Chem. 2023, 299, 104998. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Approach | Key Features | Outcome | Ref. |
|---|---|---|---|---|
| ECM as the target | Fibronectin-targeted liposomal nanoplatform | Functionalized with EDB-specific aptide to deliver CypA siRNA | Improved cellular uptake and CypA silencing in GB cells; reduced tumor growth and increased survival rates in vivo | [143] |
| CREKA-micelle nanoparticles | Targets fibrin deposits in GB | Enhanced tumor homing; potential for targeted drug delivery | [144] | |
| Ft-PLA-PTX dual-targeting peptide system | Targets TN-C and NRP-1 | Enhanced internalization in glioma cells and penetration in 3D spheroids; Increased median survival compared to saline | [145] | |
| PL3-functionalized iron oxide nanoworms and silver nanoparticles | Targets TN-C and NRP-1 | Increased GB targeting; increased survival rates in glioma-bearing mice | [146] | |
| PL1-functionalized silver nanoparticles | Targets fibronectin EDB and TN-C | Strong binding to targets and facilitated cellular uptake | [147] | |
| MMP-14 targeting dual-modality imaging agent | Combines NIRF dye and PET radionuclide | Enhanced tumor specificity and imaging contrast for GB detection; potential for preoperative planning and real-time surgical guidance | [148] | |
| VEGF-targeting iron oxide magnetic nanoparticles | Coated with cross-linked BSA and functionalized with anti-VEGF antibodies | Improved tumor visualization using MRI; sustained contrast for 24 h post-injection | [149] | |
| ECM-responsive nanoparticles | CLIO-ICT nanoparticles | MMP-14-activated prodrug release system | Selective targeting of GB cells and GSCs; disrupted tumor blood vessels; induced apoptosis and reduced GSCs populations; real-time tumor response monitoring via MRI | [150] |
| Activatable cell-penetrating peptide-modified nanoparticles | MMP-2 and MMP-9 responsive design | Enhanced internalization and targeted PTX delivery within GB microenvironment; improved penetration in 3D GB spheroids; increased survival rates compared to conventional PTX formulations | [139] | |
| MMP-2-activated nanoparticles with bispecific antibodies | Combines immunotherapy with ferroptosis induction | Targeted B7-H3 and CD3; crossed BBB; accumulated in GB tissue; released cargo upon MMP-2 cleavage; activated T-cells and induced cytokine release | [151] | |
| pH-responsive DOX-loaded liposomes | Incorporates pH-sensitive peptide trigger | pH-triggered drug release; specific targeting of GB cells under acidic conditions; anti-tumor activity and anti-angiogenic effects | [152] | |
| Chitosan-based pH-responsive gold nanoparticles | Functionalized with anti-nucleolin aptamer (AS1411) | Enhanced drug release in acidic environments; dual-drug delivery (5-FU and DOX); induced greater cell death than single-drug formulations | [153] | |
| Acid-cleavable angiopep-2 functionalized nanoparticles | Dual-surface tailored PLGA and PEG based NPs | Brain-targeting in circulation; transformed into GB-accumulating after BBB crossing; enhanced cell uptake and cytotoxicity; improved BBB permeability | [154] | |
| Hypoxia-responsive liposomal system (AMVY@NPs) | Stepwise targeting and drug release | Crossed BBB; targeted GB cells; released cargo in hypoxic conditions; synergistic inhibition of GB cell growth and pluripotency | [155] | |
| Hollow manganese dioxide (h-MnO2) nanosystem | pH-responsive design | Alleviated tumor hypoxia; enabled targeted drug delivery, imaging, and TME modulation; enhanced photodynamic therapy efficacy | [156] | |
| Redox-sensitive HA-curcumin conjugate nanocarriers | GSH-responsive disulfide bonds | Rapid drug release in high GSH environments; enhanced curcumin solubility and stability; superior cytotoxicity and cellular uptake in GB cells | [157] | |
| Enhancing ECM degradation | Hyaluronidase-expressing oncolytic adenovirus (ICOVIR17) | Targets HA-rich ECM; increases tumor-infiltrating CD8+ T cells and macrophages | Prolonged survival in GB models; potential combination with anti-PD-1 therapies for enhanced efficacy | [158] |
| Collagenase-modified human serum albumin nanoparticles (CG-HSANPs) | Loaded with gemcitabine; improved tumor spheroid penetration | Overcame limitations of gemcitabine’s short half-life; enhanced drug delivery efficacy and induced higher levels of nuclear fragmentation and ROS generation | [159] | |
| Preventing ECM degradation | PLGA nanoparticles functionalized with chlorotoxin (CTX) | Targets MMP-2 and chloride channels overexpressed in GB cells; delivers morusin | Effectively crossed BBB; induced apoptosis and ROS generation in GB cells with low toxicity to normal cells | [160] |
| Amphiphilic peptide nanoparticles | Conjugated with MMP-9 inhibiting peptide; brain-targeting ligand included | Efficiently inhibited MMP-9 activity; demonstrated low toxicity while crossing the BBB | [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horta, M.; Soares, P.; Leite Pereira, C.; Lima, R.T. Emerging Approaches in Glioblastoma Treatment: Modulating the Extracellular Matrix Through Nanotechnology. Pharmaceutics 2025, 17, 142. https://doi.org/10.3390/pharmaceutics17020142
Horta M, Soares P, Leite Pereira C, Lima RT. Emerging Approaches in Glioblastoma Treatment: Modulating the Extracellular Matrix Through Nanotechnology. Pharmaceutics. 2025; 17(2):142. https://doi.org/10.3390/pharmaceutics17020142
Chicago/Turabian StyleHorta, Miguel, Paula Soares, Catarina Leite Pereira, and Raquel T. Lima. 2025. "Emerging Approaches in Glioblastoma Treatment: Modulating the Extracellular Matrix Through Nanotechnology" Pharmaceutics 17, no. 2: 142. https://doi.org/10.3390/pharmaceutics17020142
APA StyleHorta, M., Soares, P., Leite Pereira, C., & Lima, R. T. (2025). Emerging Approaches in Glioblastoma Treatment: Modulating the Extracellular Matrix Through Nanotechnology. Pharmaceutics, 17(2), 142. https://doi.org/10.3390/pharmaceutics17020142