
Effect of Staggered vs. Simultaneous Co-Administration of Bempedoic Acid on Pharmacokinetics of Pravastatin: Randomized, Cross-Over Clinical Trial in Healthy Volunteers

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Participants and Methods
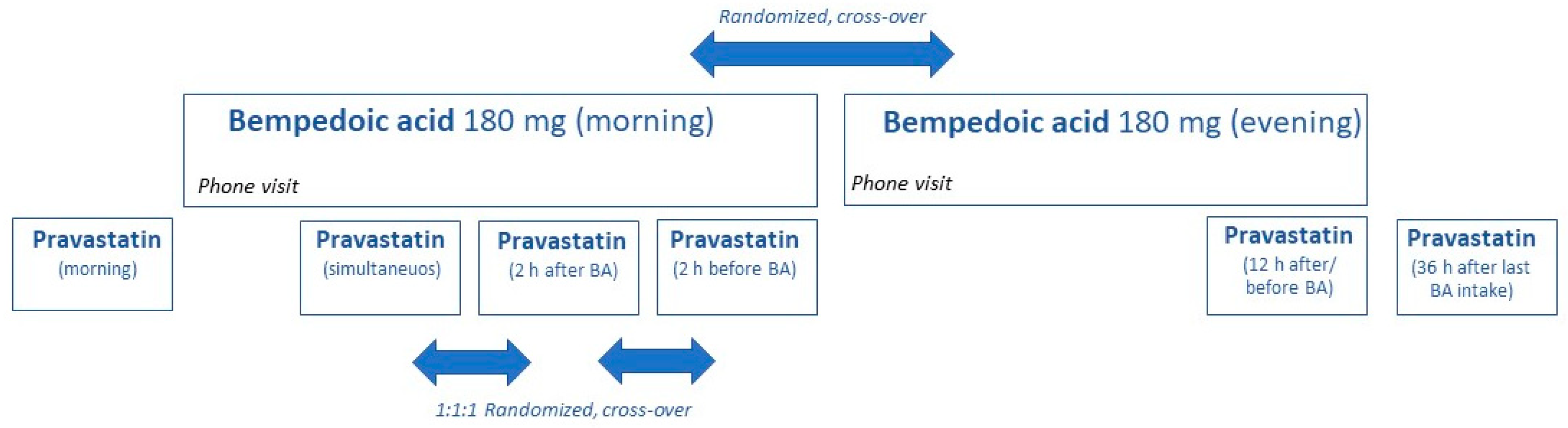
2.1. Trial Design and Participants
2.2. Quantification of Pravastatin, Pravastatin Lactone, and 3α-Hydroxy-Pravastatin
2.3. Quantification of BA
2.4. Genotyping of SLCO1B1
2.5. Pharmacokinetic and Statistical Analysis
3. Results
3.1. Study Population
3.2. Pravastatin Exposure
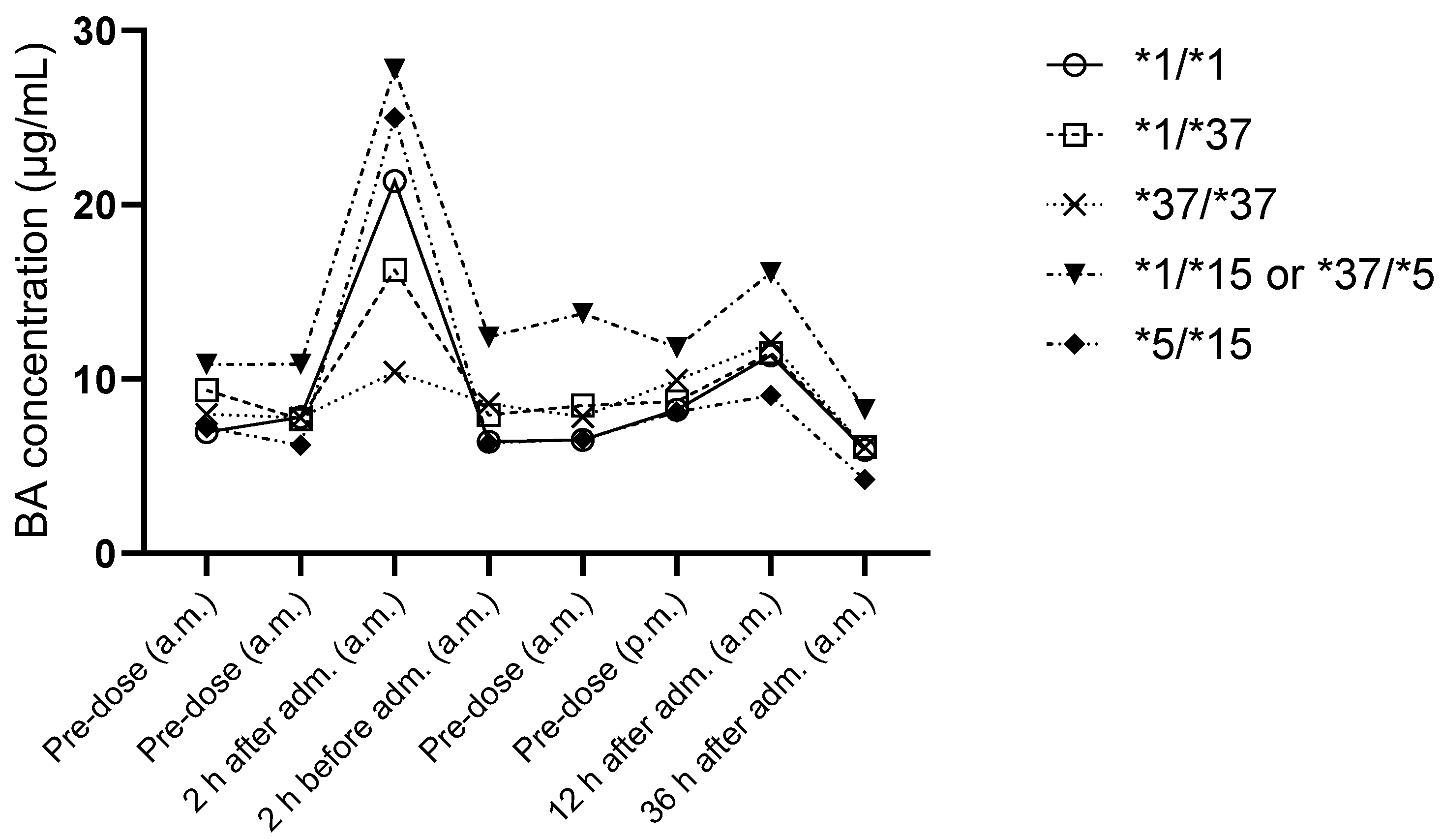
3.3. Bempedoic Acid
3.4. LDL-C-Lowering Effect
3.5. Evaluation of the Impact of SLCO1B1 Haplotypes
3.6. Adverse Events
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Medicines Agency (EMA). Nilemdo Summary of Product Characteristics; Daiichi Sankyo Europe GmbH: Munich, Germany, 2024. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/nilemdo#product-info (accessed on 26 November 2024).
- Nissen, S.E.; Lincoff, A.M.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N. Engl. J. Med. 2023, 388, 1353–1364. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Hernandez, A.V.; Banach, M.; Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group; the International Lipid Expert Panel (ILEP). Efficacy and safety of bempedoic acid for the treatment of hypercholesterolemia: A systematic review and meta-analysis. PLoS Med. 2020, 17, e1003121. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Ballantyne, C.M.; Banach, M.; Bays, H.; Catapano, A.L.; Duell, P.B.; Mancini, G.J. Efficacy and safety of bempedoic acid in patients not receiving statins in phase 3 clinical trials. J. Clin. Lipidol. 2022, 16, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.B.; Crass, R.L.; Chapel, S.; Kerschnitzki, M.; Sasiela, W.J.; Emery, M.G.; Amore, B.M.; Barrett, P.H.R.; Watts, G.F.; Catapano, A.L. Pharmacodynamic effect of bempedoic acid and statin combinations: Predictions from a dose-response model. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Amore, B.M.; Cramer, C.; MacDougall, D.; Emery, M.G. The Disposition and Metabolism of Bempedoic Acid, a Potent Inhibitor of ATP Citrate Lyase, in Healthy Human Subjects. Drug Metab. Dispos. 2023, 51, 599–609. [Google Scholar] [CrossRef]
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Lin, J.; Bi, Y.A.; Rotter, C.J.; Fahmi, O.A.; Lam, J.L.; El-Kattan, A.F.; Goosen, T.C.; Lai, Y. Quantitative prediction of repaglinide-rifampicin complex drug interactions using dynamic and static mechanistic models: Delineating differential CYP3A4 induction and OATP1B1 inhibition potential of rifampicin. Drug Metab. Dispos. 2013, 41, 966–974. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA’s Examples of Drugs That Interact with CYP Enzymes and Transporter Systems. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/healthcare-professionals-fdas-examples-drugs-interact-cyp-enzymes-and-transporter-systems (accessed on 27 November 2024).
- Climent, E.; Benaiges, D.; Pedro-Botet, J. Hydrophilic or Lipophilic Statins? Front. Cardiovasc. Med. 2021, 8, 687585. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.B.; Abdel-Rahman, S.; Gaedigk, R.; Gaedigk, A.; Raghuveer, G.; Staggs, V.S.; Kauffman, R.; Van Haandel, L.; Leeder, J.S. Impact of Genetic Variation on Pravastatin Systemic Exposure in Pediatric Hypercholesterolemia. Clin. Pharmacol. Ther. 2019, 105, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Everett, D.W.; Chando, T.J.; Didonato, G.C.; Singhvi, S.M.; Pan, H.Y.; Weinstein, S.H. Biotransformation of pravastatin sodium in humans. Drug Metab. Dispos. 1991, 19, 740–748. [Google Scholar]
- van Haandel, L.; Gibson, K.T.; Leeder, J.S.; Wagner, J.B. Quantification of pravastatin acid, lactone and isomers in human plasma by UHPLC-MS/MS and its application to a pediatric pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1012–1013, 169–177. [Google Scholar] [CrossRef]
- Pravastatin. SmPC; HEXAL®: Holzkirchen, Germany, 2023. [Google Scholar]
- Shepherd, J.; Cobbe, S.M.; Ford, I.; Isles, C.G.; Lorimer, A.R.; MacFarlane, P.W.; McKillop, J.H.; Packard, C.J.; West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med. 1995, 333, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Adams, S.P.; Alaeiilkhchi, N.; Tasnim, S.; Wright, J.M. Pravastatin for lowering lipids. Cochrane Database Syst. Rev. 2023, 9, CD013673. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonization of technical requirements for pharmaceutics for human use (ICH). Bioanalytical Method Validation and Study Sample Analysis M10; ICH Harmonised Guideline: Geneva, Switzerland, 2022; Available online: https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf (accessed on 26 November 2024).
- Aklillu, E.; Mugusi, S.; Ngaimisi, E.; Hoffmann, M.M.; Konig, S.; Ziesenitz, V.; Mikus, G.; Haefeli, W.E.; Weiss, J. Frequency of the SLCO1B1 388A>G and the 521T>C polymorphism in Tanzania genotyped by a new LightCycler(R)-based method. Eur. J. Clin. Pharmacol. 2011, 67, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on the Investigation of Bioequivalence. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (accessed on 21 August 2024).
- Ramsey, L.B.; Gong, L.; Lee, S.B.; Wagner, J.B.; Zhou, X.; Sangkuhl, K.; Adams, S.M.; Straka, R.J.; Empey, P.E.; Boone, E.C.; et al. PharmVar GeneFocus: SLCO1B1. Clin. Pharmacol. Ther. 2023, 113, 782–793. [Google Scholar] [CrossRef]
- Kivisto, K.T.; Niemi, M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharm. Res. 2007, 24, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Ieiri, I.; Yasuda, K.; Fujino, A.; Fujiwara, H.; Otsubo, K.; Hirano, M.; Watanabe, T.; Kitamura, Y.; Kusuhara, H.; et al. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin. Pharmacol. Ther. 2006, 79, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Group, S.C.; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Turongkaravee, S.; Jittikoon, J.; Lukkunaprasit, T.; Sangroongruangsri, S.; Chaikledkaew, U.; Thakkinstian, A. A systematic review and meta-analysis of genotype-based and individualized data analysis of SLCO1B1 gene and statin-induced myopathy. Pharmacogenom. J. 2021, 21, 296–307. [Google Scholar] [CrossRef]
- Niemi, M.; Schaeffeler, E.; Lang, T.; Fromm, M.F.; Neuvonen, M.; Kyrklund, C.; Backman, J.T.; Kerb, R.; Schwab, M.; Neuvonen, P.J.; et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics 2004, 14, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.B.; Ruggiero, M.; Leeder, J.S.; Hagenbuch, B. Functional Consequences of Pravastatin Isomerization on OATP1B1-Mediated Transport. Drug Metab. Dispos. 2020, 48, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Igel, M.; Arnold, K.A.; Niemi, M.; Hofmann, U.; Schwab, M.; Lutjohann, D.; von Bergmann, K.; Eichelbaum, M.; Kivisto, K.T. Impact of the SLCO1B1 polymorphism on the pharmacokinetics and lipid-lowering efficacy of multiple-dose pravastatin. Clin. Pharmacol. Ther. 2006, 79, 419–426. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M.; Trial, C.H. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Laufs, U.; Ray, K.K.; Leiter, L.A.; Bays, H.E.; Goldberg, A.C.; Stroes, E.S.; MacDougall, D.; Zhao, X.; Catapano, A.L. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur. J. Prev. Cardiol. 2020, 27, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.C.; Leiter, L.A.; Stroes, E.S.G.; Baum, S.J.; Hanselman, J.C.; Bloedon, L.T.; Lalwani, N.D.; Patel, P.M.; Zhao, X.; Duell, P.B. Effect of Bempedoic Acid vs Placebo Added to Maximally Tolerated Statins on Low-Density Lipoprotein Cholesterol in Patients at High Risk for Cardiovascular Disease: The CLEAR Wisdom Randomized Clinical Trial. JAMA 2019, 322, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Winsemius, A.; Ansquer, J.C.; Olbrich, M.; van Amsterdam, P.; Aubonnet, P.; Beckmann, K.; Driessen, S.; van Assche, H.; Piskol, G.; Lehnick, D.; et al. Pharmacokinetic interaction between simvastatin and fenofibrate with staggered and simultaneous dosing: Does it matter? J. Clin. Pharmacol. 2014, 54, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Jia, Y.Y.; Li, F.; Liu, W.X.; Lu, C.T.; Zhu, Y.R.; Yang, J.; Ding, L.K.; Yang, L.; Wen, A.D. The effect of staggered administration of zinc sulfate on the pharmacokinetics of oral cephalexin. Br. J. Clin. Pharmacol. 2012, 73, 422–427. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rs2306283 | rs4149056 | Haplotypes | SLCO1B1 Phenotype (1) | No. of Participants (Per-Protocol Population) |
|---|---|---|---|---|
| AA | TT | *1/*1 | Normal function | 5 (4) |
| AG | TT | *1/*37 | Normal function (2) | 7 (6) |
| GG | TT | *37/*37 | Normal function (2) | 1 |
| AG | TC | *1/*15 or *37/*5 | Intermediate function | 2 |
| AG | CC | *5/*15 | Low function | 1 |
| GG | CC | *15/*15 | Low function | 1 (0) |
| PRA Mono | PRA + BA sim. | BA 2 h Before PRA | BA 2 h After PRA | BA 12 h Before PRA | BA 36 h Before PRA | ||
|---|---|---|---|---|---|---|---|
| PRA | AUC∞ (min*ng/mL) | 7303 (4302–12,398) | 13,123 (7512–22,926) | 9496 (4620–19,519) | 9252 (3545–24,149) | 9241 (3773–22,638) | 10,854 (5293–22,257) |
| GMR (90% CI) | 1.80 (1.31–2.46) | 1.38 (0.82–2.33) | 1.42 (0.66–3.06) | 1.42 (0.70–2.87) | 1.21 (0.69–2.10) | ||
| Cmax (ng/mL) | 55.7 (31.4–98.6) | 108 (59.5–198) | 61.7 (26.9–141) | 66.5 (24.7–179) | 72.2 (27.7–188) | 85.4 (41.3–176) | |
| GMR (90% CI) | 1.95 1.40–2.72 | 1.76 (0.92–3.37) | 1.63 (0.72–3.69) | 1.50 (0.65–3.49) | 1.27 (0.75–2.16) | ||
| Tmax (min) | 67.9 (58.5–77.4) | 67.7 (46.5–88.9) | 85.9 (62.7–109) | 69.7 (59.8–79.6) | 81.5 (57.5–106) | 66.5 (50.0–83.0) | |
| PRA 3-iso | AUC∞ (min*ng/mL) | 1746 (732–4162) | 2394 (1322–4335) | 3720 (1706–8110) | 3762 (1972–7178) | 3863 (2387–6251) | 2822 (1547–5146) |
| GMR (90% CI) | 1.37 (0.35–5.33) | 0.64 (0.20–2.54) | 0.64 (0.30–1.35) | 0.62 (0.29–1.32) | 0.85 (0.34–2.09) | ||
| Cmax (ng/mL) | 15.3 (5.96–39.1) | 23.4 (12.7–43.1) | 31.0 (12.7–76.1) | 27.6 (12.4–61.6) | 32.5 (19.2–55.1) | 24.6 (13.5–45.1) | |
| GMR (90% CI) | 1.53 (0.36–6.51) | 0.75 (0.21–3.43) | 0.85 (0.30–2.41) | 0.72 (0.32–1.63) | 0.95 (0.41–2.18) | ||
| Tmax (min) | 66.6 (56.1–77.1) | 69.8 (51.0–88.6) | 85.8 (59.4–112) | 77.1 (58.3–96.0) | 85.9 (55.8–116) | 65.4 (49.3–81.6) | |
| PRA lactone | AUC∞ (min*ng/mL) | 120 (68.8–209) | 154 (81–291) | 164 (116–233) | 175 (107–288) | 181 (108–303) | 149 (65.8–339) |
| GMR (90% CI) | 1.28 (0.98–1.68) | 0.94 (0.78–1.67) | 0.88 (0.71–1.61) | 0.85 (0.72–1.47) | 1.03 (0.54–1.97) | ||
| Cmax (ng/mL) | (0.39–1.51) | 1.03 (0.50–2.13) | 1.05 (0.72–1.53) | 1.12 (0.67–1.86) | 1.18 (0.70–2.0) | 0.95 (0.43–2.09) | |
| GMR (90% CI) | 1.34 (0.91–1.97) | 0.98 (0.80–1.91) | 0.92 (0.77–1.73) | 0.87 (0.75–1.60) | 1.09 (0.54–2.19) | ||
| Tmax (min) | 69.1 (57.5–80.7) | 67.6 (50.7–84.6) | 81.4 (57.5–105) | 60.1 (45.3–74.9) | 75.1 (44.9–105) | 62.3 (48.8–75.8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoll, F.; Amato, S.; Sauter, M.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Blank, A. Effect of Staggered vs. Simultaneous Co-Administration of Bempedoic Acid on Pharmacokinetics of Pravastatin: Randomized, Cross-Over Clinical Trial in Healthy Volunteers. Pharmaceutics 2025, 17, 60. https://doi.org/10.3390/pharmaceutics17010060
Stoll F, Amato S, Sauter M, Burhenne J, Weiss J, Haefeli WE, Blank A. Effect of Staggered vs. Simultaneous Co-Administration of Bempedoic Acid on Pharmacokinetics of Pravastatin: Randomized, Cross-Over Clinical Trial in Healthy Volunteers. Pharmaceutics. 2025; 17(1):60. https://doi.org/10.3390/pharmaceutics17010060
Chicago/Turabian StyleStoll, Felicitas, Salvatore Amato, Max Sauter, Jürgen Burhenne, Johanna Weiss, Walter E. Haefeli, and Antje Blank. 2025. "Effect of Staggered vs. Simultaneous Co-Administration of Bempedoic Acid on Pharmacokinetics of Pravastatin: Randomized, Cross-Over Clinical Trial in Healthy Volunteers" Pharmaceutics 17, no. 1: 60. https://doi.org/10.3390/pharmaceutics17010060
APA StyleStoll, F., Amato, S., Sauter, M., Burhenne, J., Weiss, J., Haefeli, W. E., & Blank, A. (2025). Effect of Staggered vs. Simultaneous Co-Administration of Bempedoic Acid on Pharmacokinetics of Pravastatin: Randomized, Cross-Over Clinical Trial in Healthy Volunteers. Pharmaceutics, 17(1), 60. https://doi.org/10.3390/pharmaceutics17010060