
Inhibition of Aβ Aggregation by Cholesterol-End-Modified PEG Vesicles and Micelles


Abstract

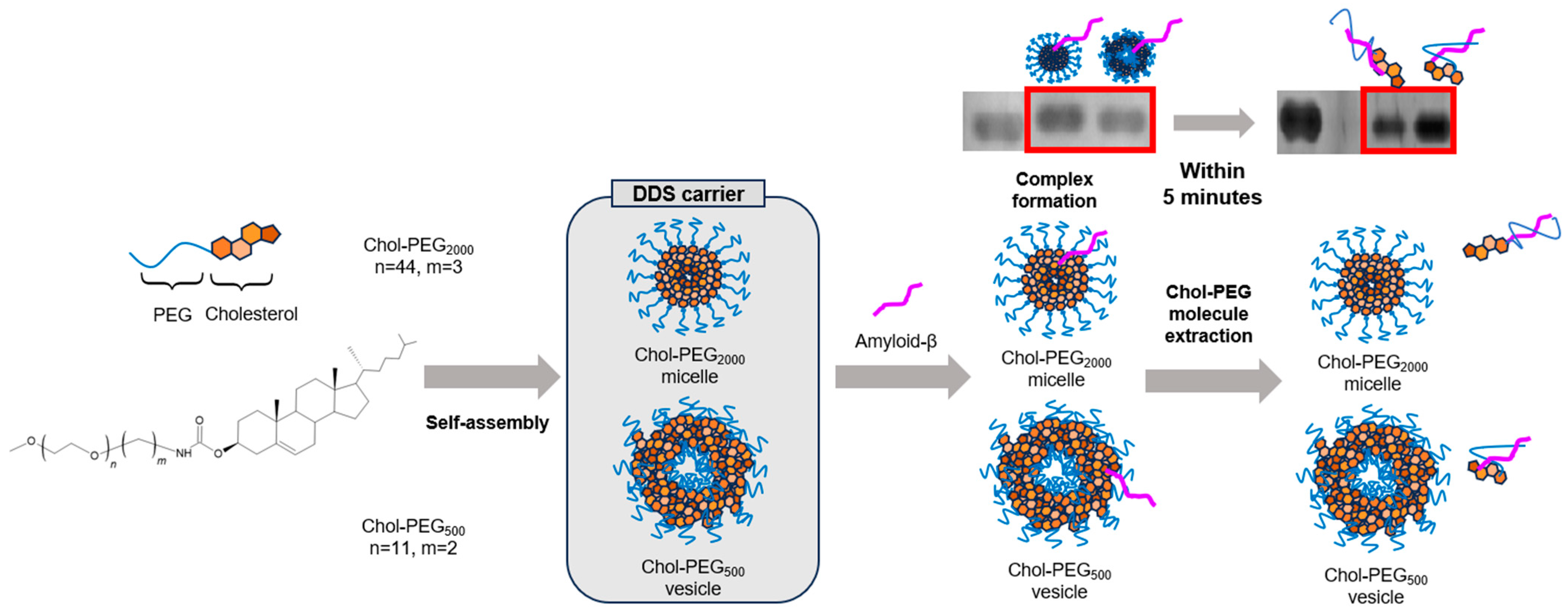
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Chol-PEG Assemblies
2.3. Particle Size and Zeta Potential Measurement
2.4. Transmission Electron Microscopy (TEM) Observation
2.5. Thioflavin T (ThT) Assay
2.6. Circular Dichroism (CD) Measurement
2.7. Naive Polyacrylamide Gel Electrophoresis (Native-PAGE)
3. Results
3.1. Physical Properties of Chol-PEG Assemblies
3.2. ThT Assay of Aβ40/Chol-PEG Assemblies
3.3. Evaluation of β-Sheet Formation with CD Measurement
3.4. Aggregation Evaluation Using Native-PAGE
3.5. Disaggregation Effect of Chol-PEG Assembly on Aggregated Aβ
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beshir, S.A.; Hussain, N.; Menon, V.B.; Al Haddad, A.H.I.; Al Zeer, R.A.K.; Elnour, A.A. Advancements and Challenges in Antiamyloid Therapy for Alzheimer’s Disease: A Comprehensive Review. Int. J. Alzheimer’s Dis. 2024, 2024, 2052142. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.-Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Grøntvedt, G.R.; Schröder, T.N.; Sando, S.B.; White, L.; Bråthen, G.; Doeller, C.F. Alzheimer’s disease. Curr. Biol. 2018, 28, R645–R649. [Google Scholar] [CrossRef]
- Aleksis, R.; Oleskovs, F.; Jaudzems, K.; Pahnke, J.; Biverstål, H. Structural studies of amyloid-β peptides: Unlocking the mechanism of aggregation and the associated toxicity. Biochimie 2017, 140, 176–192. [Google Scholar] [CrossRef]
- Leong, Y.Q.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab. Brain Dis. 2019, 35, 11–30. [Google Scholar] [CrossRef]
- Choi, H.; Kim, C.; Song, H.; Cha, M.; Cho, H.J.; Son, S.M.; Kim, H.J.; Mook-Jung, I. Amyloid β-induced elevation of O-GlcNAcylated c-Fos promotes neuronal cell death. Aging Cell 2018, 18, e12872. [Google Scholar] [CrossRef]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Yamin, G.; Ono, K.; Inayathullah, M.; Teplow, D.B. Amyloid β -Protein Assembly as a Therapeutic Target of Alzheimers Disease. Curr. Pharm. Des. 2008, 14, 3231–3246. [Google Scholar] [CrossRef]
- Kapadia, A.; Sharma, K.K.; Maurya, I.K.; Singh, V.; Khullar, M.; Jain, R. Structural and mechanistic insights into the inhibition of amyloid-β aggregation by Aβ39-42 fragment derived synthetic peptides. Eur. J. Med. Chem. 2020, 212, 113126. [Google Scholar] [CrossRef]
- Loeffler, D.A. Antibody-Mediated Clearance of Brain Amyloid-β: Mechanisms of Action, Effects of Natural and Monoclonal Anti-Aβ Antibodies, and Downstream Effects. J. Alzheimer’s Dis. Rep. 2023, 7, 873–899. [Google Scholar] [CrossRef]
- Amano, A.; Sanjo, N.; Araki, W.; Anraku, Y.; Nakakido, M.; Matsubara, E.; Tomiyama, T.; Nagata, T.; Tsumoto, K.; Kataoka, K.; et al. Peripheral administration of nanomicelle-encapsulated anti-Aβ oligomer fragment antibody reduces various toxic Aβ species in the brain. J. Nanobiotechnol. 2023, 21, 36. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K.; Mariam, Z.; Magoola, M. Engineered Antibodies to Improve Efficacy against Neurodegenerative Disorders. Int. J. Mol. Sci. 2024, 25, 6683. [Google Scholar] [CrossRef]
- Xie, J.; Shen, Z.; Anraku, Y.; Kataoka, K.; Chen, X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials 2019, 224, 119491. [Google Scholar] [CrossRef]
- Lee, D.; Minko, T. Nanotherapeutics for Nose-to-Brain Drug Delivery: An Approach to Bypass the Blood Brain Barrier. Pharmaceutics 2021, 13, 2049. [Google Scholar] [CrossRef]
- Wang, Z.; Xiong, G.; Tsang, W.C.; Schätzlein, A.G.; Uchegbu, I.F.; Tsang, A. Nose-to-Brain Delivery. J. Pharmacol. Exp. Ther. 2019, 370, 593–601. [Google Scholar] [CrossRef]
- Adscheid, S.A.; Türeli, A.E.; Günday-Türeli, N.; Schneider, M. Nanotechnological approaches for efficient N2B delivery: From small-molecule drugs to biopharmaceuticals. Beilstein J. Nanotechnol. 2024, 15, 1400–1414. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.-F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef]
- Lazar, A.N.; Bich, C.; Panchal, M.; Desbenoit, N.; Petit, V.W.; Touboul, D.; Dauphinot, L.; Marquer, C.; Laprévote, O.; Brunelle, A.; et al. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) imaging reveals cholesterol overload in the cerebral cortex of Alzheimer disease patients. Acta Neuropathol. 2013, 125, 133–144. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Abad-Rodriguez, J.; Galvan, C.; Biondi, E.; Navarro, P.; Delacourte, A.; Dingwall, C.; Dotti, C.G. Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Rep. 2003, 4, 1190–1196. [Google Scholar] [CrossRef]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef]
- Rushworth, J.V.; Hooper, N.M. Lipid Rafts: Linking Alzheimer′s Amyloid-β Production, Aggregation, and Toxicity at Neuronal Membranes. Int. J. Alzheimer’s Dis. 2010, 2011, 603052. [Google Scholar] [CrossRef] [PubMed]
- Avdulov, N.A.; Chochina, S.V.; Igbavboa, U.; Warden, C.S.; Vassiliev, A.V.; Wood, W.G. Lipid Binding to Amyloid β-Peptide Aggregates: Preferential Binding of Cholesterol as Compared with Phosphatidylcholine and Fatty Acids. J. Neurochem. 1997, 69, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Panchal, M.; Loeper, J.; Cossec, J.-C.; Perruchini, C.; Lazar, A.; Pompon, D.; Duyckaerts, C. Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 2010, 51, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-Amyloid Peptides with Cholesterol: Mechanistic Insights into Amyloid Pore Formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.R. Cholesterol binding to amyloid-β fibrils: A TEM study. Micron 2008, 39, 1192–1196. [Google Scholar] [CrossRef]
- Martínez-Senac, M.d.M.; Villalaín, J.; Gómez-Fernández, J.C. Structure of the Alzheimer β-amyloid peptide (25–35) and its interaction with negatively charged phospholipid vesicles. Eur. J. Biochem. 1999, 265, 744–753. [Google Scholar] [CrossRef]
- Ahyayauch, H.; Masserini, M.E.; Alonso, A.; Goñi, F.M. Understanding Aβ Peptide Binding to Lipid Membranes: A Biophysical Perspective. Int. J. Mol. Sci. 2024, 25, 6401. [Google Scholar] [CrossRef]
- Asayama, S.; Nagashima, K.; Negishi, Y.; Kawakami, H. Byproduct-Free Intact Modification of Insulin by Cholesterol End-Modified Poly(ethylene glycol) for in Vivo Protein Delivery. Bioconjugate Chem. 2018, 29, 67–73. [Google Scholar] [CrossRef]
- Watanabe, S.; Asayama, S. Cholesterol-end-modified–PEG vesicle: DDS carrier with long-term stability to encapsulate a drug model by facile preparation. Chem. Lett. 2024, 53, upae166. [Google Scholar] [CrossRef]
- Kurano, T.; Kanazawa, T.; Ooba, A.; Masuyama, Y.; Maruhana, N.; Yamada, M.; Iioka, S.; Ibaraki, H.; Kosuge, Y.; Kondo, H.; et al. Nose-to-brain/spinal cord delivery kinetics of liposomes with different surface properties. J. Control. Release 2022, 344, 225–234. [Google Scholar] [CrossRef]
- Zhang, Q.-Z.; Zha, L.-S.; Zhang, Y.; Jiang, W.-M.; Lu, W.; Shi, Z.-Q.; Jiang, X.-G.; Fu, S.-K. The brain targeting efficiency following nasally applied MPEG-PLA nanoparticles in rats. J. Drug Target. 2006, 14, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, X.; Yu, S.; Gong, G.; Shu, H. Research progress in brain-targeted nasal drug delivery. Front. Aging Neurosci. 2024, 15, 1341295. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The carboxy terminus of the .beta. amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Cukalevski, R.; Yang, X.; Meisl, G.; Weininger, U.; Bernfur, K.; Frohm, B.; Knowles, T.P.J.; Linse, S. The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 2015, 6, 4215–4233. [Google Scholar] [CrossRef]
- Wang, L.; Eom, K.; Kwon, T. Different Aggregation Pathways and Structures for Aβ40 and Aβ42 Peptides. Biomolecules 2021, 11, 198. [Google Scholar] [CrossRef]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils—Current status. J. Chem. Biol. 2009, 3, 1–18. [Google Scholar] [CrossRef]
- LeVine, H., 3rd. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods En Mol. 1999, 309, 274. [Google Scholar] [CrossRef]
- Batzli, K.M.; Love, B.J. Agitation of amyloid proteins to speed aggregation measured by ThT fluorescence: A call for standardization. Mater. Sci. Eng. C 2015, 48, 359–364. [Google Scholar] [CrossRef]
- Tikader, B.; Maji, S.K.; Kar, S. A generic approach to decipher the mechanistic pathway of heterogeneous protein aggregation kinetics. Chem. Sci. 2021, 12, 13530. [Google Scholar] [CrossRef]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef]
- Hashemi, M.; Banerjee, S.; Lyubchenko, Y.L. Free Cholesterol Accelerates Aβ Self-Assembly on Membranes at Physiological Concentration. Int. J. Mol. Sci. 2022, 23, 2803. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K. Cholesterol and Aß Aggregation. Pharmacopsychiatry 2003, 36, 127–129. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef]
- Tsuchie, Y.; Kusuda, S.; Kawabe, H.; Mori, W.; Lindgren, M.; Watanabe, Y.; Zako, T. Polyallylamine Binds to Aβ Amyloid and Inhibits Antibody Recognition. Int. J. Mol. Sci. 2024, 25, 3112. [Google Scholar] [CrossRef]
- Kawahara, M.; Muramoto, K.; Kobayashi, K.; Mori, H.; Kuroda, Y. Aluminum Promotes the Aggregation of Alzheimer′s Amyloid β-Protein in Vitro. Biochem. Biophys. Res. Commun. 1994, 198, 531. [Google Scholar] [CrossRef]
- Nakano, H.; Hamaguchi, T.; Ikeda, T.; Watanabe-Nakayama, T.; Ono, K.; Yamada, M. Inactivation of seeding activity of amyloid β-protein aggregates in vitro. J. Neurochem. 2022, 160, 499–516. [Google Scholar] [CrossRef]
- Pachahara, S.K.; Adicherla, H.; Nagaraj, R. Self-Assembly of Aβ40, Aβ42 and Aβ43 Peptides in Aqueous Mixtures of Fluorinated Alcohols. PLoS ONE 2015, 10, e0136567. [Google Scholar] [CrossRef]
- Cheung, D.L. Surface Hydrophobicity Strongly Influences Adsorption and Conformation of Amyloid Beta Derived Peptides. Molecules 2024, 29, 3634. [Google Scholar] [CrossRef]
- Qu, L.; Fudo, S.; Matsuzaki, K.; Hoshino, T. Computational Study on the Assembly of Amyloid β-Peptides in the Hydrophobic Environment. Chem. Pharm. Bull. 2019, 67, 959–965. [Google Scholar] [CrossRef]
- Mueller, C.; Capelle, M.A.; Seyrek, E.; Martel, S.; Carrupt, P.-A.; Arvinte, T.; Borchard, G. Noncovalent PEGylation: Different Effects of Dansyl-, l-Tryptophan–, Phenylbutylamino-, Benzyl- and Cholesteryl-PEGs on the Aggregation of Salmon Calcitonin and Lysozyme. J. Pharm. Sci. 2012, 101, 1995–2008. [Google Scholar] [CrossRef]
- Ahyayauch, H.; García-Arribas, A.B.; Masserini, M.E.; Pantano, S.; Goñi, F.M.; Alonso, A. β-Amyloid (1–42) peptide adsorbs but does not insert into ganglioside-containing phospholipid membranes in the liquid-disordered state: Modelling and experimental studies. Int. J. Biol. Macromol. 2020, 164, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, C.; Klimov, D.K. The Alzheimer’s disease A β peptide binds to the anionic DMPS lipid bilayer. Biochim. Biophys. Acta (BBA)—Biomembr. 2016, 1858, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Dey, C.; Roy, M.; Ghosh, R.; Pal, P.; Roy, D.; Dey, S.G. Active Site Environment and Reactivity of Copper-Aβ in Membrane Mimetic SDS Micellar Environment. Chem.—Eur. J. 2024, 30, e202401531. [Google Scholar] [CrossRef]
- Salahuddin, P.; Fatima, M.T.; Abdelhameed, A.S.; Nusrat, S.; Khan, R.H. Structure of amyloid oligomers and their mechanisms of toxicities: Targeting amyloid oligomers using novel therapeutic approaches. Eur. J. Med. Chem. 2016, 114, 41. [Google Scholar] [CrossRef]
- Pozzi, D.; Colapicchioni, V.; Caracciolo, G.; Piovesana, S.; Capriotti, A.L.; Palchetti, S.; De Grossi, S.; Riccioli, A.; Amenitsch, H.; Laganà, A. Effect of polyethyleneglycol (PEG) chain length on the bio–nano-interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, J. Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef]
- van Gils, J.H.M.; van Dijk, E.; Peduzzo, A.; Hofmann, A.; Vettore, N.; Schützmann, M.P.; Groth, G.; Mouhib, H.; Otzen, D.E.; Buell, A.K.; et al. The hydrophobic effect characterises the thermodynamic signature of amyloid fibril growth. PLOS Comput. Biol. 2020, 16, e1007767. [Google Scholar] [CrossRef]
- Banerjee, S.; Hashemi, M.; Zagorski, K.; Lyubchenko, Y.L. Cholesterol in Membranes Facilitates Aggregation of Amyloid β Protein at Physiologically Relevant Concentrations. ACS Chem. Neurosci. 2021, 12, 506–516. [Google Scholar] [CrossRef]
- Elbassal, E.A.; Liu, H.; Morris, C.; Wojcikiewicz, E.P.; Du, D. Effects of Charged Cholesterol Derivatives on Aβ40 Amyloid Formation. J. Phys. Chem. B 2016, 120, 59–68. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, S.; Ueda, M.; Asayama, S. Inhibition of Aβ Aggregation by Cholesterol-End-Modified PEG Vesicles and Micelles. Pharmaceutics 2025, 17, 1. https://doi.org/10.3390/pharmaceutics17010001
Watanabe S, Ueda M, Asayama S. Inhibition of Aβ Aggregation by Cholesterol-End-Modified PEG Vesicles and Micelles. Pharmaceutics. 2025; 17(1):1. https://doi.org/10.3390/pharmaceutics17010001
Chicago/Turabian StyleWatanabe, Shota, Motoki Ueda, and Shoichiro Asayama. 2025. "Inhibition of Aβ Aggregation by Cholesterol-End-Modified PEG Vesicles and Micelles" Pharmaceutics 17, no. 1: 1. https://doi.org/10.3390/pharmaceutics17010001
APA StyleWatanabe, S., Ueda, M., & Asayama, S. (2025). Inhibition of Aβ Aggregation by Cholesterol-End-Modified PEG Vesicles and Micelles. Pharmaceutics, 17(1), 1. https://doi.org/10.3390/pharmaceutics17010001