

Polymeric Nanoparticles Enable mRNA Transfection and Its Translation in Intervertebral Disc and Human Joint Cells, Except for M1 Macrophages

 , , , and
, , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Cell Culture
2.1.1. Human Nucleus Pulposus and Annulus Fibrosus Cells
2.1.2. Human OA Chondrocytes
2.1.3. Human Fibroblast-like Synoviocytes
2.1.4. Human Macrophages
2.2. Nanoparticle Formulation and Characterization
2.3. Viability, Cell Number and Transfection Efficiency
2.4. Nanoparticle Internalization
2.5. Statistics
3. Results
3.1. Nanoparticle Characterization
Uncoated and PGA-PEG-Coated Nanoparticles Show Different Physical Characteristics
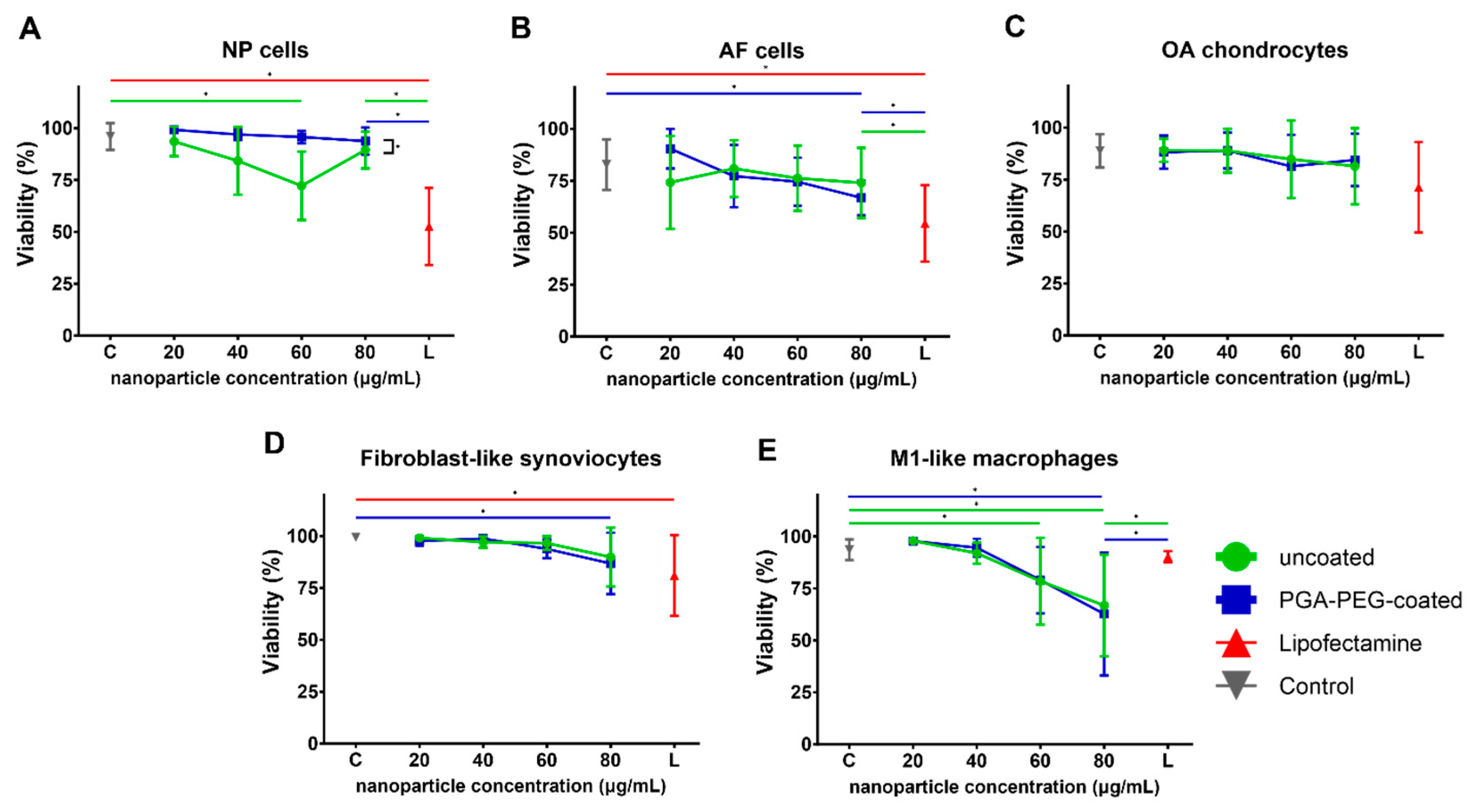
3.2. Viability
3.2.1. Toxicity of Nanoparticles Is Minor in Chondrogenic Cells and FLS
3.2.2. Toxicity in Macrophages Is Dose-Dependent
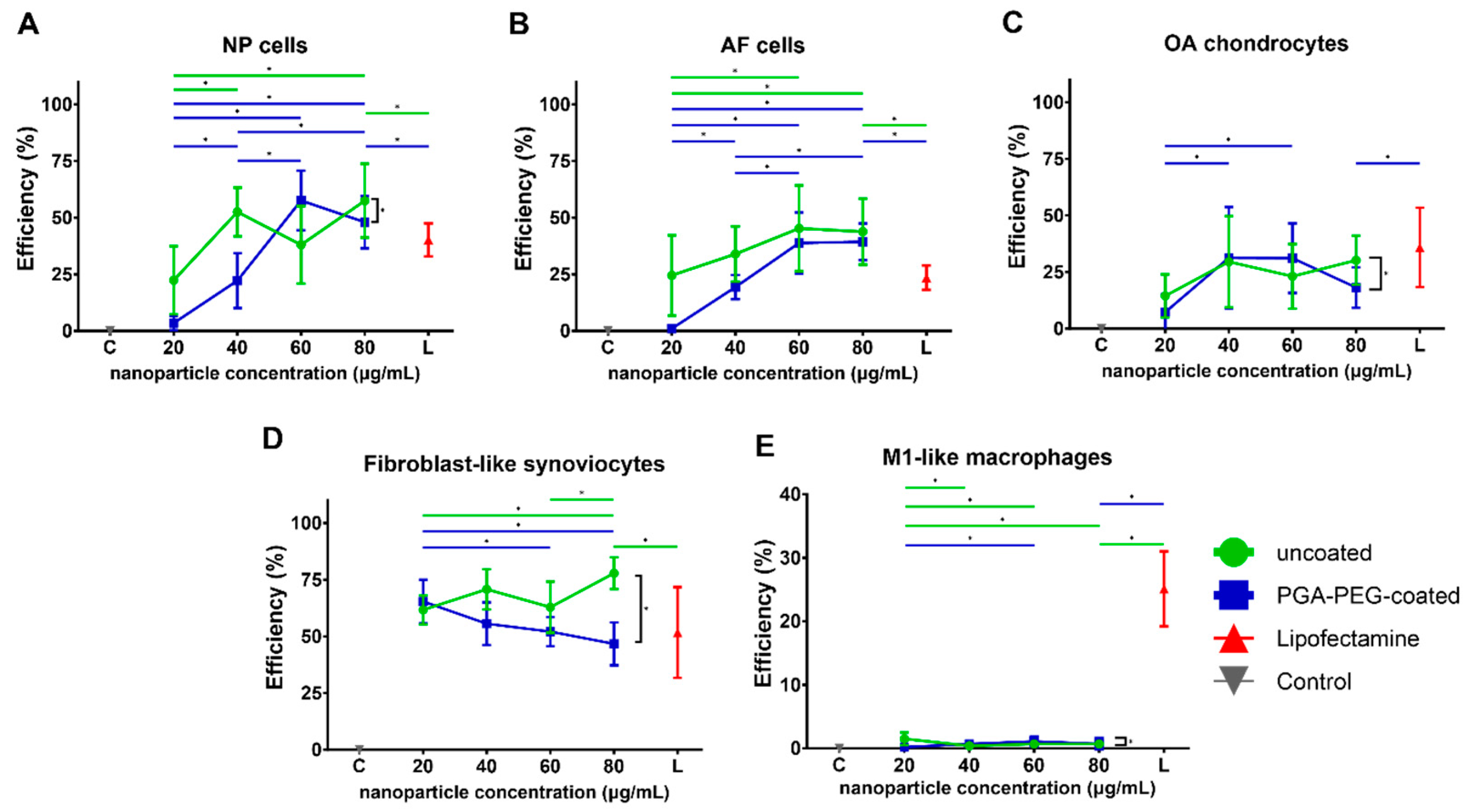
3.3. Transfection Efficiency
3.3.1. Transfection Shows EGFP Expression in Chondrogenic Cells and FLS
3.3.2. Transfection Efficiency in Macrophages Is Negligible
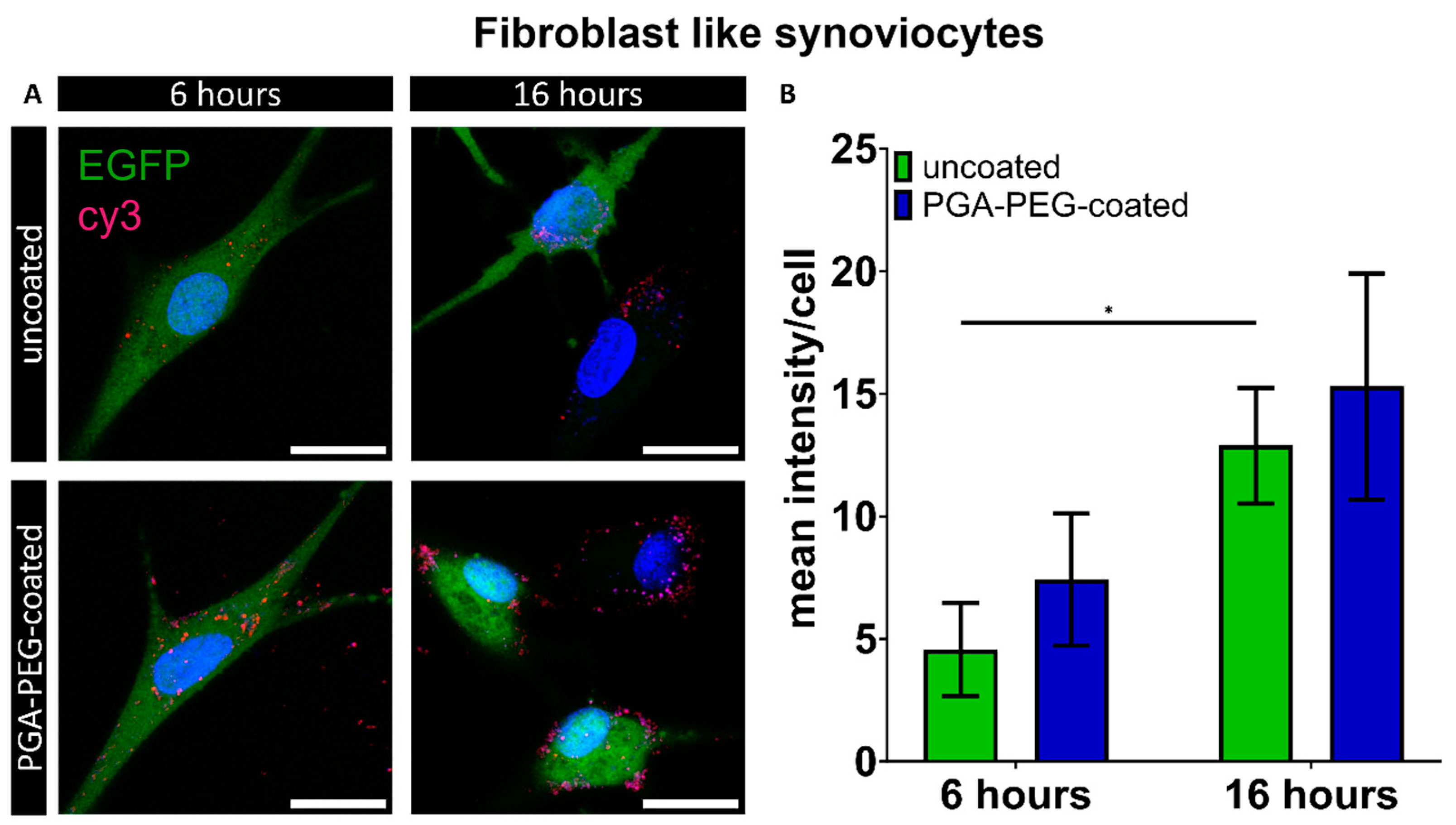
3.4. Internalization of Nanoparticles
Macrophages Internalize Nanoparticles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rustenburg, C.M.E.; Emanuel, K.S.; Peeters, M.; Lems, W.F.; Vergroesen, P.A.; Smit, T.H. Osteoarthritis and intervertebral disc degeneration: Quite different, quite similar. JOR Spine 2018, 1, e1033. [Google Scholar] [CrossRef] [PubMed]
- Meucci, R.D.; Fassa, A.G.; Faria, N.M.X. Prevalence of chronic low back pain: Systematic review. Rev. Saude Publica 2015, 49, 1. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; Lin, J.; Guo, A. Prevalence Trends of Site-Specific Osteoarthritis From 1990 to 2019: Findings From the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Batt, M. An update on the pathophysiology of osteoarthritis. Ann. Phys. Rehabilitat. Med. 2016, 59, 333–339. [Google Scholar] [CrossRef]
- Richard, M.J.; Driban, J.B.; McAlindon, T.E. Pharmaceutical treatment of osteoarthritis. Osteoarthr. Cartil. 2023, 31, 458–466. [Google Scholar] [CrossRef]
- Hsu, E.; Murphy, S.; Chang, D.; Cohen, S.P. Expert opinion on emerging drugs: Chronic low back pain. Expert Opin. Emerg. Drugs 2014, 20, 103–127. [Google Scholar] [CrossRef]
- Rand, J.A.; Trousdale, R.T.; Ilstrup, D.M.; Harmsen, W.S. Factors Affecting the Durability of Primary Total Knee Prostheses. JBJS 2003, 85, 259–265. [Google Scholar] [CrossRef]
- Zhao, L.; Manchikanti, L.; Kaye, A.D.; Abd-Elsayed, A. Treatment of Discogenic Low Back Pain: Current Treatment Strategies and Future Options-a Literature Review. Curr. Pain Headache Rep. 2019, 23, 86. [Google Scholar] [CrossRef]
- Sarzi-Puttini, P.; Cimmino, M.A.; Scarpa, R.; Caporali, R.; Parazzini, F.; Zaninelli, A.; Atzeni, F.; Canesi, B. Osteoarthritis: An overview of the disease and its treatment strategies. In Seminars in Arthritis and Rheumatism; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1–10. [Google Scholar]
- Knezevic, N.N.; Mandalia, S.; Raasch, J.; Knezevic, I.; Candido, K.D. Treatment of chronic low back pain–new approaches on the horizon. J. Pain Res. 2017, 10, 1111. [Google Scholar] [CrossRef]
- Park, P.; Garton, H.J.; Gala, V.C.; Hoff, J.T.; McGillicuddy, J.E. Adjacent Segment Disease after Lumbar or Lumbosacral Fusion: Review of the Literature. Spine (Phila Pa 1976) 2004, 29, 1938–1944. [Google Scholar] [CrossRef]
- Steinert, A.F.; Nöth, U.; Tuan, R.S. Concepts in gene therapy for cartilage repair. Injury 2008, 39, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulas, T.; Lambe, T.; Raphael, J.H.; Kitas, G.D.; Duarte, R.V. Biologic drugs as analgesics for the management of low back pain and sciatica. Pain Med. 2019, 20, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, Y.; Jiang, Y.; Yao, Q.; Chen, R.; Zhao, Y.-Z.; Kou, L. Recombinant protein drugs-based intra articular drug delivery systems for osteoarthritis therapy. Eur. J. Pharm. Biopharm. 2023, 183, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Jiskoot, W. Immunogenicity of therapeutic proteins. In Pharmaceutical Biotechnology; Crommelin, D., Sindelar, R., Meibohm, B., Eds.; Springer Science+ Business Media: New York, NY, USA, 2013. [Google Scholar]
- Schillberg, S.; Raven, N.; Spiegel, H.; Rasche, S.; Buntru, M. Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front. Plant Sci. 2019, 10, 720. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Lux, C.T.; Scharenberg, A.M. Therapeutic Gene Editing Safety and Specificity. Hematol./Oncol. Clin. N. Am. 2017, 31, 787–795. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Namvar, A.; Bolhassani, A.; Khairkhah, N.; Motevalli, F. Physicochemical properties of polymers: An important system to overcome the cell barriers in gene transfection. Biopolymers 2015, 103, 363–375. [Google Scholar] [CrossRef]
- Lin, C.; Engbersen, J.F.J. PEGylated bioreducible poly(amido amine)s for non-viral gene delivery. Mater. Sci. Eng. C 2011, 31, 1330–1337. [Google Scholar] [CrossRef]
- Lin, C.; Zhong, Z.; Lok, M.C.; Jiang, X.; Hennink, W.E.; Feijen, J.; Engbersen, J.F.J. Novel bioreducible poly(amido amine)s for highly efficient gene delivery. Bioconjug. Chem. 2007, 18, 138–145. [Google Scholar] [CrossRef]
- Sturm, L.; Schwemberger, B.; Menzel, U.; Häckel, S.; Albers, C.E.; Plank, C.; Rip, J.; Alini, M.; Traweger, A.; Grad, S.; et al. In vitro evaluation of a nanoparticle-based mrna delivery system for cells in the joint. Biomedicines 2021, 9, 794. [Google Scholar] [CrossRef]
- García, K.P.; Zarschler, K.; Barbaro, L.; Barreto, J.A.; O’Malley, W.; Spiccia, L.; Stephan, H.; Graham, B. Zwitterionic-coated “stealth” nanoparticles for biomedical applications: Recent advances in countering biomolecular corona formation and uptake by the mononuclear phagocyte system. Small 2014, 10, 2516–2529. [Google Scholar] [CrossRef]
- Bajpayee, A.G.; Grodzinsky, A.J. Cartilage-targeting drug delivery: Can electrostatic interactions help? Nat. Rev. Rheumatol. 2017, 13, 183–193. [Google Scholar] [CrossRef]
- Pontes, A.P.; van der Wal, S.; Roelofs, K.; Grobbink, A.; Creemers, L.B.; Engbersen, J.F.; Rip, J. A poly(amidoamine)-based polymeric nanoparticle platform for efficient in vivo delivery of mRNA. Biomater. Adv. 2024, 156, 213713. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, W.; Yong, H.; He, M.; Yang, Y.; Deng, Z.; Li, Y. Macrophages in osteoarthritis: Pathophysiology and therapeutics. Am. J. Transl. Res. 2020, 12, 261–268. [Google Scholar] [PubMed]
- Grotenhuis, N.; Bayon, Y.; Lange, J.F.; Van Osch, G.J.V.M.; Bastiaansen-Jenniskens, Y.M. A culture model to analyze the acute biomaterial-dependent reaction of human primary macrophages. Biochem. Biophys. Res. Commun. 2013, 433, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zhong, Z.; Lok, M.C.; Jiang, X.; Hennink, W.E.; Feijen, J.; Engbersen, J.F. Linear poly (amido amine) s with secondary and tertiary amino groups and variable amounts of disulfide linkages: Synthesis and in vitro gene transfer properties. J. Control. Release 2006, 116, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, J.K.; Alumasa, J.N.; Yearick, K.; Ekoue-Kovi, K.A.; Casabianca, L.B.; De Dios, A.C.; Wolf, C.; Roepe, P.D. 4-N-, 4-S-, and 4-O-chloroquine analogues: Influence of side chain length and quinolyl nitrogen p K a on activity vs chloroquine resistant malaria. J. Med. Chem. 2008, 51, 3466–3479. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sharmin, R.; Sigaeva, A.; Klijn, C.W.M.; Mzyk, A.; Schirhagl, R. Not all cells are created equal–endosomal escape in fluorescent nanodiamonds in different cells. Nanoscale 2021, 13, 13294–13300. [Google Scholar] [CrossRef] [PubMed]
- Floyd, T.G.; Song, J.I.; Hapeshi, A.; Laroque, S.; Hartlieb, M.; Perrier, S. Bottlebrush copolymers for gene delivery: Influence of architecture, charge density, and backbone length on transfection efficiency. J. Mater. Chem. B 2022, 10, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Pontes, A.P.; van der Wal, S.; Ranamalla, S.R.; Roelofs, K.; Tomuta, I.; Creemers, L.B.; Rip, J. Cell uptake and intracellular trafficking of bioreducible poly(amidoamine) nanoparticles for efficient mRNA translation in chondrocytes. Front. Bioeng. Biotechnol. 2023, 11, 1290871. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99 Pt A, 28–51. [Google Scholar] [CrossRef]
- Sanchez, L.; Yi, Y.; Yu, Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale 2017, 9, 288–297. [Google Scholar] [CrossRef]
- De Haes, W.; Van Mol, G.; Merlin, C.; De Smedt, S.C.; Vanham, G.; Rejman, J. Internalization of mRNA lipoplexes by dendritic cells. Mol. Pharm. 2012, 9, 2942–2949. [Google Scholar] [CrossRef]
- Lu, J.J.; Langer, R.; Chen, J. A Novel Mechanism Is Involved in Cationic Lipid-Mediated Functional siRNA Delivery. Mol. Pharm. 2009, 6, 763–771. [Google Scholar] [CrossRef]
- Vercauteren, D.; Piest, M.; van der Aa, L.J.; Al Soraj, M.; Jones, A.T.; Engbersen, J.F.; De Smedt, S.C.; Braeckmans, K. Flotillin-dependent endocytosis and a phagocytosis-like mechanism for cellular internalization of disulfide-based poly(amido amine)/DNA polyplexes. Biomaterials 2011, 32, 3072–3084. [Google Scholar] [CrossRef]
- Schmidt, F.; Thywißen, A.; Goldmann, M.; Cunha, C.; Cseresnyés, Z.; Schmidt, H.; Rafiq, M.; Galiani, S.; Gräler, M.H.; Chamilos, G.; et al. Flotillin-Dependent Membrane Microdomains Are Required for Functional Phagolysosomes against Fungal Infections. Cell Rep. 2020, 32, 108017. [Google Scholar] [CrossRef]
- Moradian, H.; Roch, T.; Lendlein, A.; Gossen, M. mRNA transfection-induced activation of primary human monocytes and macrophages: Dependence on carrier system and nucleotide modification. Sci. Rep. 2020, 10, 4181. [Google Scholar] [CrossRef] [PubMed]
- Wissing, T.B.; Van Haaften, E.E.; Koch, S.E.; Ippel, B.D.; Kurniawan, N.A.; Bouten, C.V.C.; Smits, A.I.P.M. Hemodynamic loads distinctively impact the secretory profile of biomaterial-activated macrophages-implications for in situ vascular tissue engineering. Biomater. Sci. 2020, 8, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Kraus, V.B.; Setton, L.A. Progress in intra-articular therapy. Nat. Rev. Rheumatol. 2014, 10, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Deloney, M.; Smart, K.; Christiansen, B.A.; Panitch, A. Thermoresponsive, hollow, degradable core-shell nanoparticles for intra-articular delivery of anti-inflammatory peptide. J. Control. Release 2020, 323, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Bedingfield, S.K.; Colazo, J.M.; Yu, F.; Liu, D.D.; Jackson, M.A.; Himmel, L.E.; Cho, H.; Crofford, L.J.; Hasty, K.A.; Duvall, C.L. Amelioration of post-traumatic osteoarthritis via nanoparticle depots delivering small interfering RNA to damaged cartilage. Nat. Biomed. Eng. 2021, 5, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Pistiner, J.; Adjei, I.M.; Sharma, B. Nanoparticle Properties for Delivery to Cartilage: The Implications of Disease State, Synovial Fluid, and Off-Target Uptake. Mol. Pharm. 2019, 16, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, G.; Callipo, L.; De Sanctis, S.C.; Cavaliere, C.; Pozzi, D.; Laganà, A. Surface adsorption of protein corona controls the cell internalization mechanism of DC-Chol-DOPE/DNA lipoplexes in serum. Biochim. Biophys. Acta Biomembr. 2010, 1798, 536–543. [Google Scholar] [CrossRef]
- Berrecoso, G.; Crecente-Campo, J.; Alonso, M.J. Unveiling the pitfalls of the protein corona of polymeric drug nanocarriers. Drug Deliv. Transl. Res. 2020, 10, 730–750. [Google Scholar] [CrossRef]
- Digiacomo, L.; Cardarelli, F.; Pozzi, D.; Palchetti, S.; Digman, M.A.; Gratton, E.; Capriotti, A.L.; Mahmoudi, M.; Caracciolo, G. An apolipoprotein-enriched biomolecular corona switches the cellular uptake mechanism and trafficking pathway of lipid nanoparticles. Nanoscale 2017, 9, 17254–17262. [Google Scholar] [CrossRef]
- Garcia, J.P.; Stein, J.; Cai, Y.; Riemers, F.; Wexselblatt, E.; Wengel, J.; Tryfonidou, M.; Yayon, A.; Howard, K.A.; Creemers, L.B. Fibrin-hyaluronic acid hydrogel-based delivery of antisense oligonucleotides for ADAMTS5 inhibition in co-delivered and resident joint cells in osteoarthritis. J. Control. Release 2019, 294, 247–258. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP Type | ps-PAAQ:mRNA Ratio (w/w) | Peak 1 Mean by Intensity (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) |
|---|---|---|---|---|
| Uncoated | 25 | 112.9 | 0.341 ± 0.079 | +20.4 ± 2.4 |
| Coated | 25 | 58.8 | 0.105 ± 0.026 | −0.1 ± 1.2 |
| Uncoated + Cy3-siRNA | 25 | 108.7 | 0.317 ± 0.056 | +24.8 ± 5.3 |
| Coated + Cy3-siRNA | 25 | 67.4 | 0.205 ± 0.066 | −4.4 ± 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muenzebrock, K.A.; Ho, F.Y.W.; Pontes, A.P.; Jorquera-Cordero, C.; Utomo, L.; Garcia, J.P.; Willems, P.C.; Welting, T.J.M.; Rip, J.; Creemers, L.B. Polymeric Nanoparticles Enable mRNA Transfection and Its Translation in Intervertebral Disc and Human Joint Cells, Except for M1 Macrophages. Pharmaceutics 2024, 16, 438. https://doi.org/10.3390/pharmaceutics16040438
Muenzebrock KA, Ho FYW, Pontes AP, Jorquera-Cordero C, Utomo L, Garcia JP, Willems PC, Welting TJM, Rip J, Creemers LB. Polymeric Nanoparticles Enable mRNA Transfection and Its Translation in Intervertebral Disc and Human Joint Cells, Except for M1 Macrophages. Pharmaceutics. 2024; 16(4):438. https://doi.org/10.3390/pharmaceutics16040438
Chicago/Turabian StyleMuenzebrock, Katrin Agnes, Fiona Y. W. Ho, Adriano P. Pontes, Carla Jorquera-Cordero, Lizette Utomo, Joao Pedro Garcia, Paul C. Willems, Tim J. M. Welting, Jaap Rip, and Laura B. Creemers. 2024. "Polymeric Nanoparticles Enable mRNA Transfection and Its Translation in Intervertebral Disc and Human Joint Cells, Except for M1 Macrophages" Pharmaceutics 16, no. 4: 438. https://doi.org/10.3390/pharmaceutics16040438
APA StyleMuenzebrock, K. A., Ho, F. Y. W., Pontes, A. P., Jorquera-Cordero, C., Utomo, L., Garcia, J. P., Willems, P. C., Welting, T. J. M., Rip, J., & Creemers, L. B. (2024). Polymeric Nanoparticles Enable mRNA Transfection and Its Translation in Intervertebral Disc and Human Joint Cells, Except for M1 Macrophages. Pharmaceutics, 16(4), 438. https://doi.org/10.3390/pharmaceutics16040438