
Design of Experiments to Tailor the Potential of BSA-Coated Peptide Nanocomplexes for Temozolomide/p53 Gene Co-Delivery

 , ,
, ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
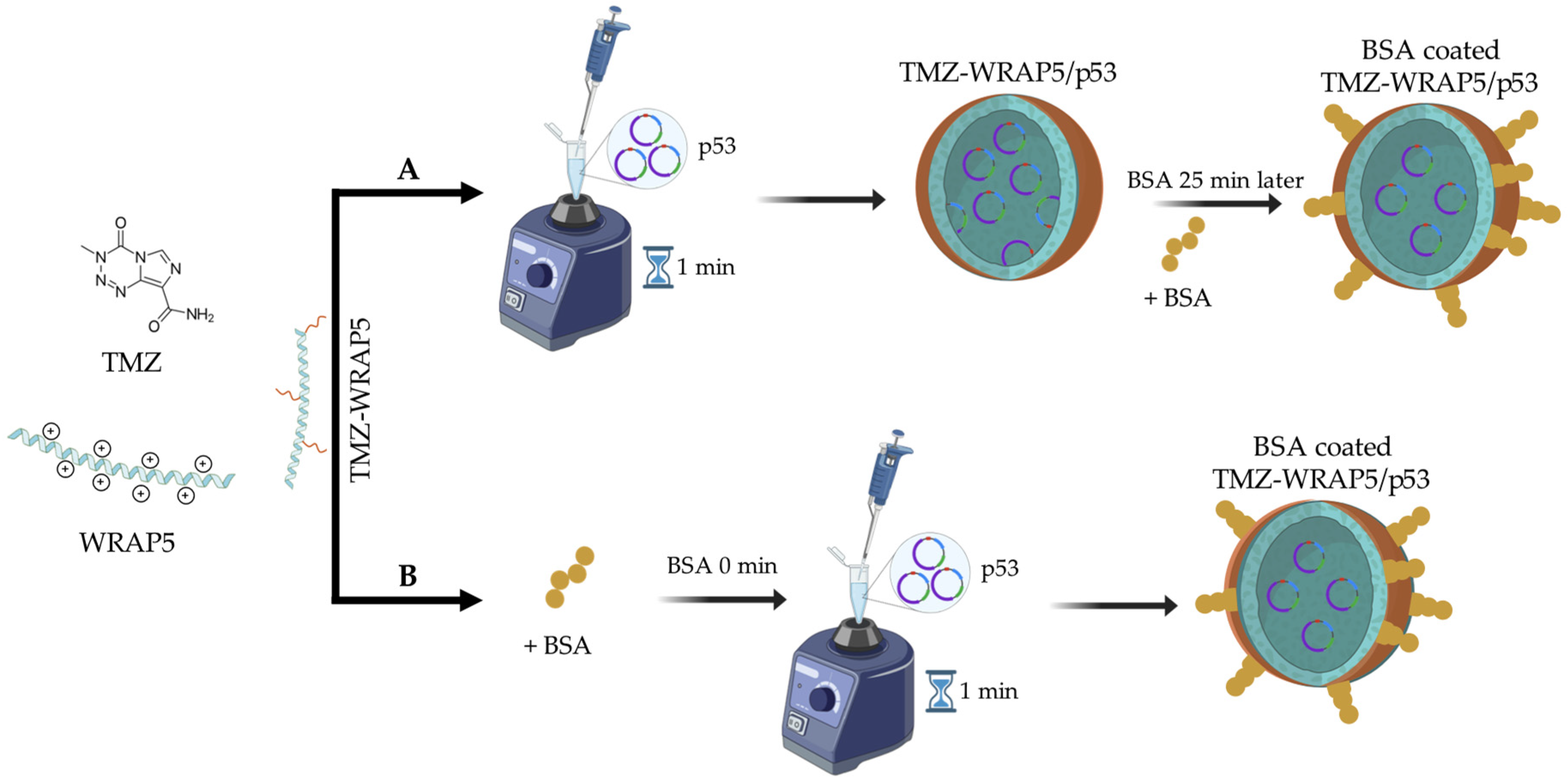
2.2.1. Preparation of a Combined Aqueous Solution of TMZ and Peptide
2.2.2. Formation of TMZ-WRAP5/p53 Complexes
2.2.3. BSA Coating
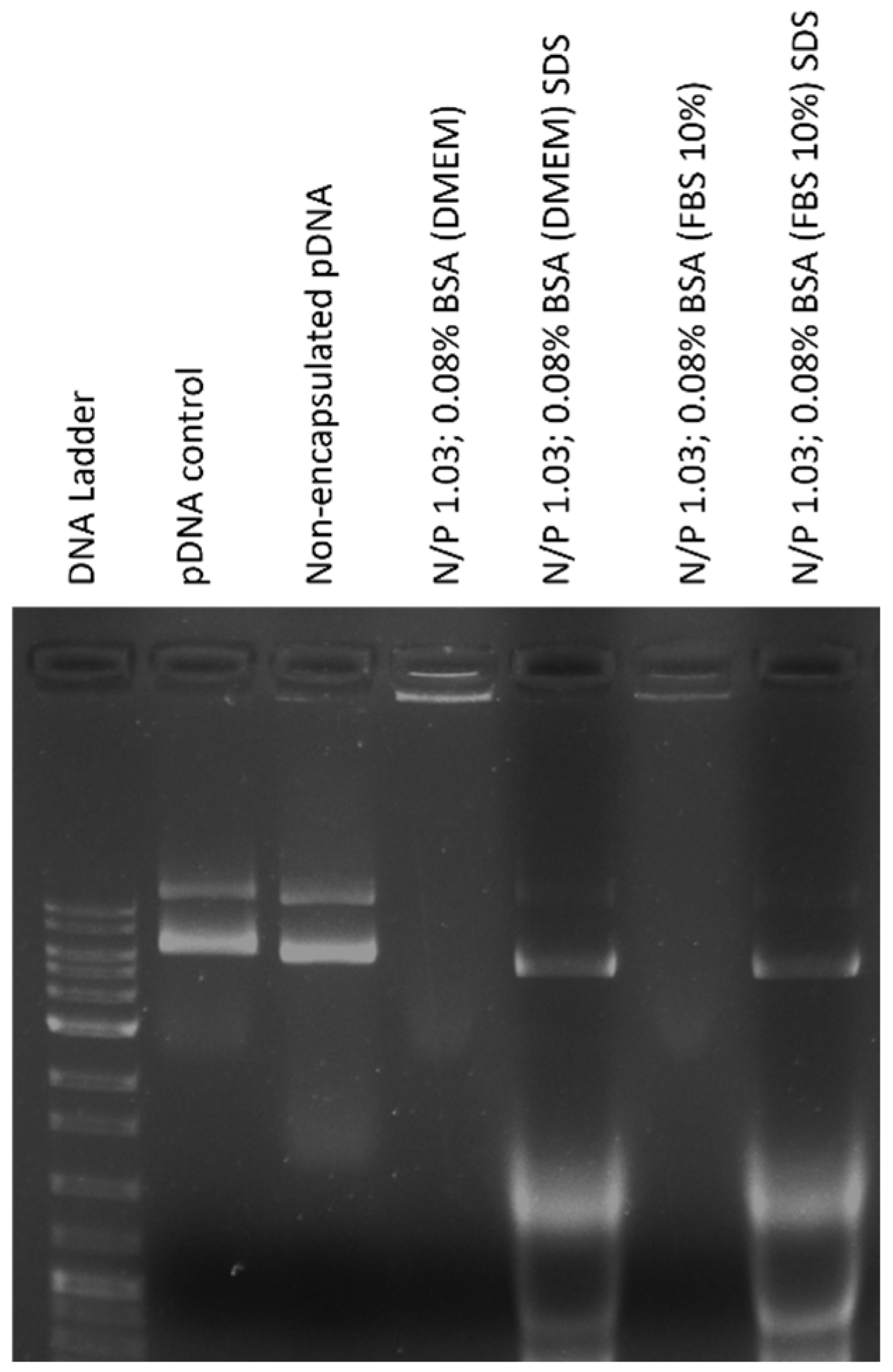
2.2.4. Agarose Gel Electrophoresis
2.2.5. Design of Experiments (DoE)
2.2.6. Determination of Particle Size and Charge
2.2.7. pDNA Complexation Capacity
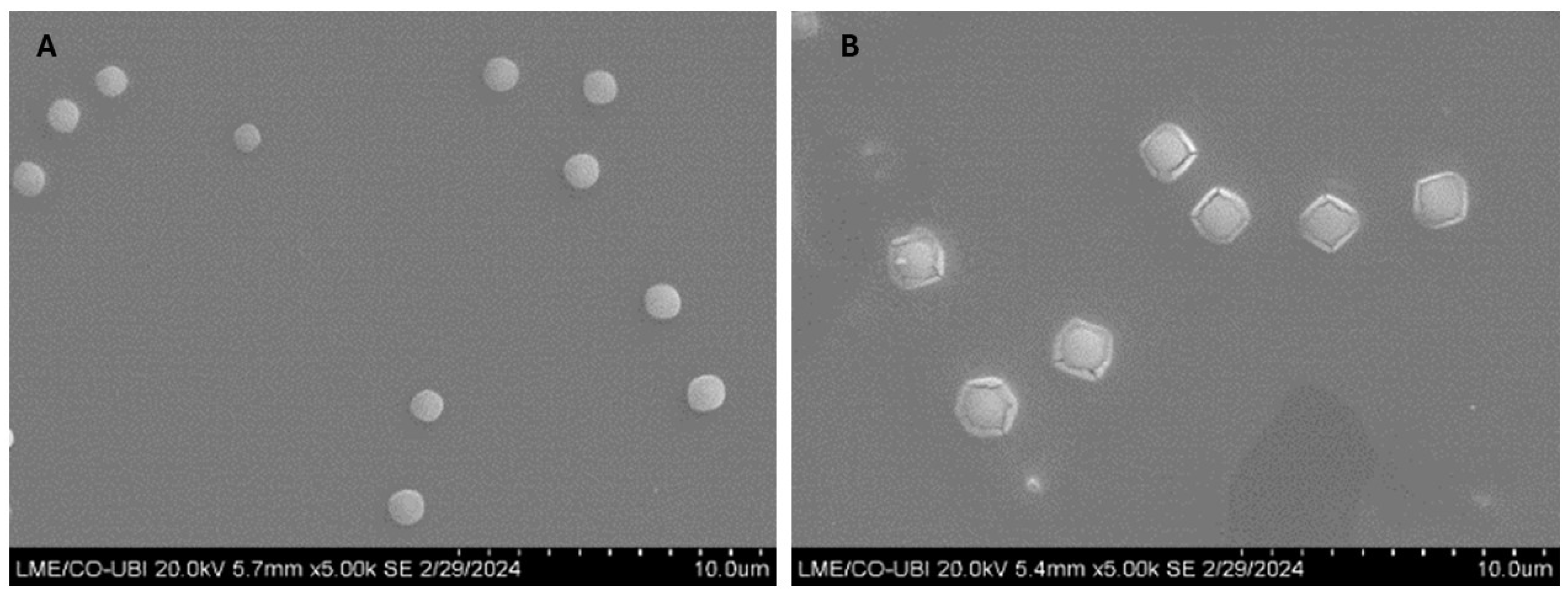
2.2.8. Scanning Electron Microscopy
2.2.9. Cell Culture
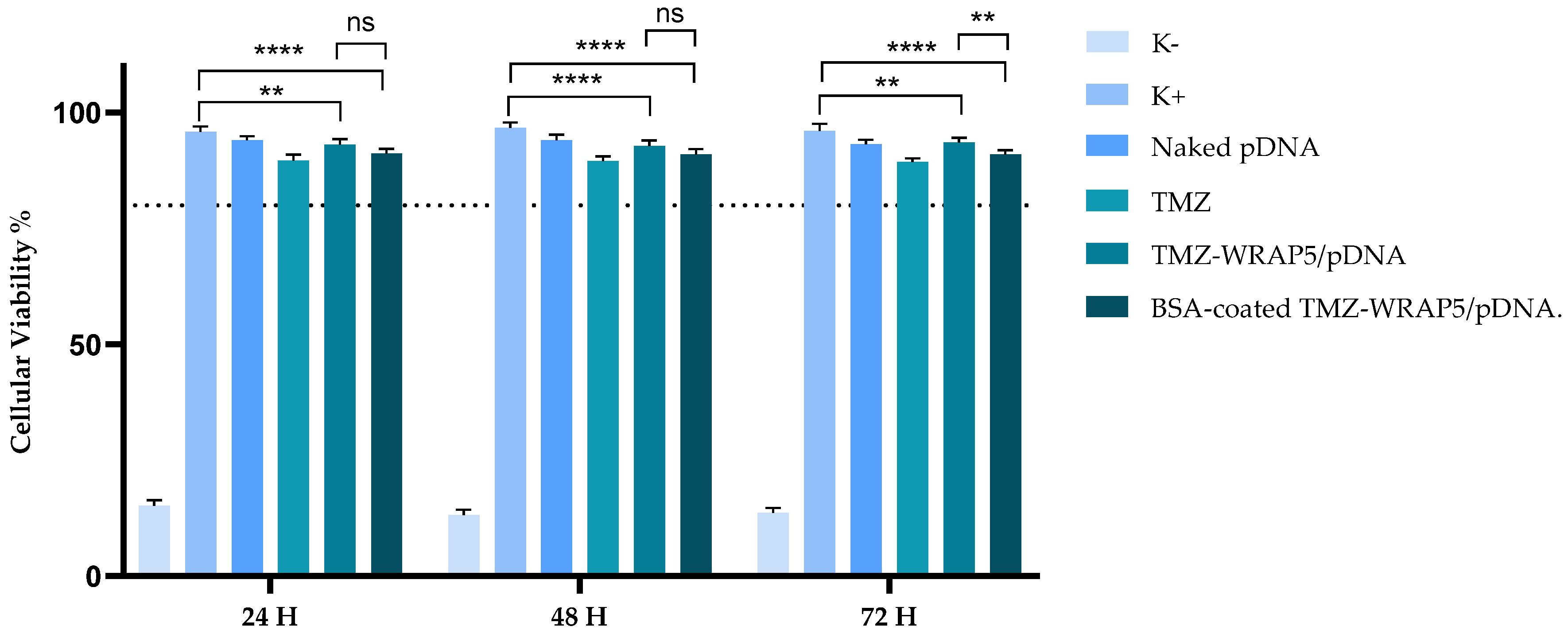
2.2.10. Evaluation of Cellular Viability
2.2.11. Stability in Media
2.2.12. Hemolysis Test
2.2.13. Gene Transcription in U-87 Cells
2.2.14. Statistical Analysis
3. Results and Discussion
3.1. Rationality of BSA-Coated TMZ-WRAP5/p53 Complexes
3.2. DoE: Identification of Tuning Parameters
3.3. DoE Application: Finding the Optimal BSA-Coated TMZ-WRAP5/p53 Formulation
3.4. Study of Morphology
3.5. The Optimal BSA-Coated Complexes Display a Non-Cytotoxic Profile
3.6. The Optimal BSA-Coated Complexes Are Stable and Protect pDNA
3.7. The Optimal BSA-Coated Complexes Are Hemocompatible
3.8. The Optimal BSA-Coated Complexes Inhibit Glioblastoma Cell Viability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pineda, E.; Domenech, M.; Hernández, A.; Comas, S.; Balaña, C. Recurrent Glioblastoma: Ongoing Clinical Challenges and Future Prospects. OncoTargets Ther. 2023, 16, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Salgado, M.A.; Villamayor, M.; Albarrán, V.; Alía, V.; Sotoca, P.; Chamorro, J.; Rosero, D.; Barrill, A.M.; Martín, M.; Fernandez, E.; et al. Recurrent Glioblastoma: A Review of the Treatment Options. Cancers 2023, 15, 4279. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yang, Q.; Xu, P.; Deng, M.; Jiang, T.; Cai, L.; Li, J.; Sai, K.; Xi, S.; Ouyang, H.; et al. Adjuvant Temozolomide Chemotherapy With or Without Interferon Alfa Among Patients with Newly Diagnosed High-grade Gliomas: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2253285. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, D.; Savage, J.; Patel, A.; Collinson, F.; Mant, R.; Boele, F.; Brazil, L.; Meade, S.; Buckle, P.; Lax, S.; et al. A randomised phase II trial of temozolomide with or without cannabinoids in patients with recurrent glioblastoma (ARISTOCRAT): Protocol for a multi-centre, double-blind, placebo-controlled trial. BMC Cancer 2024, 24, 83. [Google Scholar] [CrossRef] [PubMed]
- Colopi, A.; Fuda, S.; Santi, S.; Onorato, A.; Cesarini, V.; Salvati, M.; Balistreri, C.R.; Dolci, S.; Guida, E. Impact of age and gender on glioblastoma onset, progression, and management. Mech. Ageing Dev. 2023, 211, 111801. [Google Scholar] [CrossRef]
- Angom, R.S.; Nakka, N.M.R.; Bhattacharya, S. Advances in Glioblastoma Therapy: An Update on Current Approaches. Brain Sci. 2023, 13, 1536. [Google Scholar] [CrossRef]
- Sahoo, O.S.; Mitra, R.; Nagaiah, N.K.H. The hidden architects of glioblastoma multiforme: Glioma stem cells. MedComm—Oncol. 2024, 3, e66. [Google Scholar] [CrossRef]
- Rominiyi, O.; Collis, S.J. DDRugging glioblastoma: Understanding and targeting the DNA damage response to improve future therapies. Mol. Oncol. 2022, 16, 11–41. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Canney, M.; Carpentier, A.; Idbaih, A. Overcoming the blood brain barrier in glioblastoma: Status and future perspective. Rev. Neurol. 2023, 179, 430–436. [Google Scholar] [CrossRef]
- Chancellor, D.; Barrett, D.; Nguyen-Jatkoe, L.; Millington, S.; Eckhardt, F. The state of cell and gene therapy in 2023. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 3376–3388. [Google Scholar] [CrossRef]
- Cesur-Ergün, B.; Demir-Dora, D. Gene therapy in cancer. J. Gene Med. 2023, 25, e3550. [Google Scholar] [CrossRef] [PubMed]
- Volodina, O.; Smirnikhina, S. The Future of Gene Therapy: A Review of In Vivo and Ex Vivo Delivery Methods for Genome Editing-Based Therapies. Mol. Biotechnol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Sousa, A.; Faria, R.; Albuquerque, T.; Queiroz, J.A.; Costa, D. Cancer gene therapy mediated by RALA/plasmid DNA vectors: Nitrogen to phosphate groups ratio (N/P) as a tool for tunable transfection efficiency and apoptosis. Colloids Surf. B Biointerfaces 2020, 185, 110610. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Li, G.; Mei, X.; Ding, J.; Shen, H.; Zhu, D.; Wang, H. Co-Delivery of p53 Restored and E7 Targeted Nucleic Acids by Poly (Beta-Amino Ester) Complex Nanoparticles for the Treatment of HPV Related Cervical Lesions. Front. Pharmacol. 2022, 13, 826771. [Google Scholar] [CrossRef]
- Fang, Z.; Zhang, M.; Kang, R.; Cui, M.; Song, M.; Liu, K. A cancer cell membrane coated nanoparticles-based gene delivery system for enhancing cancer therapy. Int. J. Pharm. 2022, 629, 122415. [Google Scholar] [CrossRef]
- Kamath, D.; Iwakuma, T.; Bossmann, S.H. Therapeutic potential of combating cancer by restoring wild-type p53 through mRNA nanodelivery. Nanomed. Nanotechnol. Biol. Med. 2024, 56, 102732. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- D’Orazi, G. p53 Function and Dysfunction in Human Health and Diseases. Biomolecules 2023, 13, 506. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Li, H.; Pan, X.; Wang, Y. New Insights into the Roles of p53 in Central Nervous System Diseases. Int. J. Neuropsychopharmacol. 2023, 26, 465–473. [Google Scholar] [CrossRef]
- Hu, X.; Xu, W.; Ren, Y.; Wang, Z.; He, X.; Huang, R.; Ma, B.; Zhao, J.; Zhu, R.; Cheng, L. Spinal cord injury: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 245. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed]
- Borowczak, J.; Szczerbowski, K.; Maniewski, M.; Zdrenka, M.; Słupski, P.; Andrusewicz, H.; Łysik-Miśkurka, J.; Rutkiewicz, P.; Bodnar, M.; Szylberg, Ł. The prognostic role of p53 and its correlation with CDK9 in urothelial carcinoma. Clin. Transl. Oncol. 2023, 25, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, D.; Song, N.; Chen, X.; Zhang, X.; Zheng, W.; Yu, Y.; Han, C. Mutant p53 in head and neck squamous cell carcinoma: Molecular mechanism of gain-of-function and targeting therapy (Review). Oncol. Rep. 2023, 50, 162. [Google Scholar] [CrossRef] [PubMed]
- Marvalim, C.; Datta, A.; Lee, S.C. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics 2023, 13, 1421–1442. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Forte, I.M.; Indovina, P.; Iannuzzi, C.A.; Cirillo, D.; Di Marzo, D.; Barone, D.; Capone, F.; Pentimalli, F.; Giordano, A. Targeted therapy based on p53 reactivation reduces both glioblastoma cell growth and resistance to temozolomide. Int. J. Oncol. 2019, 54, 2189–2199. [Google Scholar] [CrossRef]
- Chen, Y.; Qu, W.; Tu, J.; Qi, H. Implications of Advances in Studies of O6-Methylguanine-DNA- Methyltransferase for Tumor Prognosis and Treatment. FBL 2023, 28, 197. [Google Scholar] [CrossRef]
- Sandoval, J.E.; Reich, N.O. p53 and TDG are dominant in regulating the activity of the human de novo DNA methyltransferase DNMT3A on nucleosomes. J. Biol. Chem. 2021, 296, 100058. [Google Scholar] [CrossRef]
- Szewczyk-Roszczenko, O.; Barlev, N.A. The Role of p53 in Nanoparticle-Based Therapy for Cancer. Cells 2023, 12, 2803. [Google Scholar] [CrossRef]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical-Physical Applications to Nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef]
- Malik, S.; Muhammad, K.; Waheed, Y. Nanotechnology: A Revolution in Modern Industry. Molecules 2023, 28, 661. [Google Scholar] [CrossRef] [PubMed]
- Erel-Akbaba, G.; Carvalho, L.A.; Tian, T.; Zinter, M.; Akbaba, H.; Obeid, P.J.; Chiocca, E.A.; Weissleder, R.; Kantarci, A.G.; Tannous, B.A. Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano 2019, 13, 4028–4040. [Google Scholar] [CrossRef]
- Wang, M.; Wang, C.; Ren, S.; Pan, J.; Wang, Y.; Shen, Y.; Zeng, Z.; Cui, H.; Zhao, X. Versatile Oral Insulin Delivery Nanosystems: From Materials to Nanostructures. Int. J. Mol. Sci. 2022, 23, 3362. [Google Scholar] [CrossRef] [PubMed]
- Kunene, S.; Lin, K.-S.; Weng, M.-T.; Carrera Espinoza, M.; Lin, Y.-S.; Lin, Y.-T. Design of biomimetic targeting nanoclusters for enhanced doxorubicin delivery to liver cancer. Eur. Polym. J. 2023, 186, 111861. [Google Scholar] [CrossRef]
- Albuquerque, T.; Faria, R.; Sousa, Â.; Neves, A.R.; Queiroz, J.; Costa, D. Methotrexate-plasmid DNA polyplexes for cancer therapy: Characterization, cancer cell targeting ability and tuned in vitro transfection. J. Mol. Liq. 2019, 292, 111391. [Google Scholar] [CrossRef]
- Song, M.; Fang, Z.; Wang, J.; Liu, K. A nano-targeted co-delivery system based on gene regulation and molecular blocking strategy for synergistic enhancement of platinum chemotherapy sensitivity in ovarian cancer. Int. J. Pharm. 2023, 640, 123022. [Google Scholar] [CrossRef]
- Priya, S.S.; Rekha, M.R. Synergistic effect of p53 gene/DOX intracellular delivery and P-gp inhibition by pullulan thiomers on cancer cells: In vitro and in vivo evaluations. J. Mater. Chem. B 2023, 11, 1365–1377. [Google Scholar] [CrossRef]
- Boisguérin, P.; Konate, K.; Josse, E.; Vivès, E.; Deshayes, S. Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery. Biomedicines 2021, 9, 583. [Google Scholar] [CrossRef]
- Deshayes, S.; Konate, K.; Vivès, E.; Boisguérin, P. Tips and Tools to Understand Direct Membrane Translocation of siRNA-Loaded WRAP-Based Nanoparticles. Methods Mol. Biol. 2022, 2383, 475–490. [Google Scholar] [CrossRef]
- Faria, R.; Paul, M.; Biswas, S.; Vivès, E.; Boisguérin, P.; Sousa, Â.; Costa, D. Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy. Pharmaceutics 2022, 14, 757. [Google Scholar] [CrossRef]
- Lemech, C.R.; Kichenadasse, G.; Marschner, J.-P.; Alevizopoulos, K.; Otterlei, M.; Millward, M. ATX-101, a cell-penetrating protein targeting PCNA, can be safely administered as intravenous infusion in patients and shows clinical activity in a Phase 1 study. Oncogene 2023, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi, B.; Tanwar, S.; Singh, A.; Arifin, D.R.; McMahon, M.T.; Barman, I.; Bulte, J.W.M. Cell-Penetrating and Enzyme-Responsive Peptides for Targeted Cancer Therapy: Role of Arginine Residue Length on Cell Penetration and In Vivo Systemic Toxicity. ACS Appl. Mater. Interfaces 2024, 16, 11159–11171. [Google Scholar] [CrossRef] [PubMed]
- Mander, S.; Naffouje, S.A.; Gao, J.; Li, W.; Christov, K.; Green, A.; Bongarzone, E.R.; Das Gupta, T.K.; Yamada, T. Tumor-targeting cell-penetrating peptide, p28, for glioblastoma imaging and therapy. Front. Oncol. 2022, 12, 940001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Kong, J.; Huang, X.; Zeng, J.; Du, Q.; Yang, T.; Yue, H.; Bao, Q.; Miao, Y.; Xu, Y.; et al. Targeted glioblastoma therapy by integrating brain-targeting peptides and corn-derived cancer cell-penetrating proteins into nanoparticles to cross blood-brain tumor barriers. Mater. Today Nano 2023, 23, 100347. [Google Scholar] [CrossRef]
- Neves, A.R.; Albuquerque, T.; Faria, R.; Gonçalves, A.M.; Santos, C.; Vivès, E.; Boisguérin, P.; Passarinha, L.A.; Sousa, Â.; Costa, D. Development of WRAP5 Peptide Complexes for Targeted Drug/Gene Co-Delivery toward Glioblastoma Therapy. Pharmaceutics 2022, 14, 2213. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Yang, X.; Zhang, S.; Cui, C. Preparation Optimization of Bovine Serum Albumin Nanoparticles and Its Application for siRNA Delivery. Drug Des. Dev. Ther. 2021, 15, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Zhu, H.; Qin, Y.; Zhang, R.; Wang, L.; Zhang, E.; Zhou, X.; Meng, R. GP60 and SPARC as albumin receptors: Key targeted sites for the delivery of antitumor drugs. Front. Pharmacol. 2024, 15, 1329636. [Google Scholar] [CrossRef]
- Hashiba, A.; Toyooka, M.; Sato, Y.; Maeki, M.; Tokeshi, M.; Harashima, H. The use of design of experiments with multiple responses to determine optimal formulations for in vivo hepatic mRNA delivery. J. Control. Release 2020, 327, 467–476. [Google Scholar] [CrossRef]
- Rampado, R.; Peer, D. Design of experiments in the optimization of nanoparticle-based drug delivery systems. J. Control Release 2023, 358, 398–419. [Google Scholar] [CrossRef]
- Karimi, M.; Bahrami, S.; Ravari, S.B.; Zangabad, P.S.; Mirshekari, H.; Bozorgomid, M.; Shahreza, S.; Sori, M.; Hamblin, M.R. Albumin nanostructures as advanced drug delivery systems. Expert Opin. Drug Deliv. 2016, 13, 1609–1623. [Google Scholar] [CrossRef]
- Babcock, J.J.; Brancaleon, L. Bovine serum albumin oligomers in the E- and B-forms at low protein concentration and ionic strength. Int. J. Biol. Macromol. 2013, 53, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Li, X.; Oh, J.S.; Lee, Y.; Lee, E.C.; Yang, M.; Kwon, N.; Ha, T.W.; Hong, D.Y.; Song, Y.; Kim, H.K.; et al. Albumin-binding photosensitizer capable of targeting glioma via the SPARC pathway. Biomater. Res. 2023, 27, 23. [Google Scholar] [CrossRef] [PubMed]
- Solanki, R.; Rostamabadi, H.; Patel, S.; Jafari, S.M. Anticancer nano-delivery systems based on bovine serum albumin nanoparticles: A critical review. Int. J. Biol. Macromol. 2021, 193, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, H.; Sefidi, N.; Sharafi, A.; Danafar, H.; Kheiri Manjili, H. Bovine Serum Albumin (BSA) coated iron oxide magnetic nanoparticles as biocompatible carriers for curcumin-anticancer drug. Bioorg. Chem. 2018, 76, 501–509. [Google Scholar] [CrossRef]
- Al-Jawad, S.M.H.; Taha, A.A.; Al-Halbosiy, M.M.F.; Al-Barram, L.F.A. Synthesis and characterization of small-sized gold nanoparticles coated by bovine serum albumin (BSA) for cancer photothermal therapy. Photodiagnosis Photodyn. Ther. 2018, 21, 201–210. [Google Scholar] [CrossRef]
- Fan, D.; Cao, Y.; Cao, M.; Wang, Y.; Cao, Y.; Gong, T. Nanomedicine in cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 293. [Google Scholar] [CrossRef]
- Jin, C.; Wang, K.; Oppong-Gyebi, A.; Hu, J. Application of Nanotechnology in Cancer Diagnosis and Therapy—A Mini-Review. Int. J. Med. Sci. 2020, 17, 2964–2973. [Google Scholar] [CrossRef]
- Kemp, J.A.; Kwon, Y.J. Cancer nanotechnology: Current status and perspectives. Nano Converg. 2021, 8, 34. [Google Scholar] [CrossRef]
- Tavares Luiz, M.; Santos Rosa Viegas, J.; Palma Abriata, J.; Viegas, F.; Testa Moura de Carvalho Vicentini, F.; Lopes Badra Bentley, M.V.; Chorilli, M.; Maldonado Marchetti, J.; Tapia-Blácido, D.R. Design of experiments (DoE) to develop and to optimize nanoparticles as drug delivery systems. Eur. J. Pharm. Biopharm. 2021, 165, 127–148. [Google Scholar] [CrossRef]
- Ben Ali, R.; Ben Ouada, S.; Leboulanger, C.; Ammar, J.; Sayadi, S.; Ben Ouada, H. Bisphenol A removal by the Chlorophyta Picocystis sp.: Optimization and kinetic study. Int. J. Phytoremed. 2021, 23, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Sankha, B. Central Composite Design for Response Surface Methodology and Its Application in Pharmacy. In Response Surface Methodology in Engineering Science; Palanikumar, K., Ed.; IntechOpen: Rijeka, Croatia, 2021; p. 5. [Google Scholar]
- Patil, S.A.; Surwase, S.N.; Jadhav, S.B.; Jadhav, J.P. Optimization of medium using response surface methodology for l-DOPA production by Pseudomonas sp. SSA. Biochem. Eng. J. 2013, 74, 36–45. [Google Scholar] [CrossRef]
- Amighi, F.; Emam-Djomeh, Z.; Labbafi-Mazraeh-Shahi, M. Effect of different cross-linking agents on the preparation of bovine serum albumin nanoparticles. J. Iran. Chem. Soc. 2020, 17, 1223–1235. [Google Scholar] [CrossRef]
- Pirkhezranian, Z.; Tahmoorespur, M.; Daura, X.; Monhemi, H.; Sekhavati, M.H. Interaction of camel Lactoferrin derived peptides with DNA: A molecular dynamics study. BMC Genom. 2020, 21, 60. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.C.; Posey, N.D.; Tew, G.N. Protein Binding and Release by Polymeric Cell-Penetrating Peptide Mimics. Biomacromolecules 2022, 23, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Aramesh-Boroujeni, Z.; Jahani, S.; Khorasani-Motlagh, M.; Kerman, K.; Noroozifar, M. Evaluation of parent and nano-encapsulated terbium(III) complex toward its photoluminescence properties, FS-DNA, BSA binding affinity, and biological applications. J. Trace Elem. Med. Biol. 2020, 61, 126564. [Google Scholar] [CrossRef]
- Galdino, F.E.; Picco, A.S.; Sforca, M.L.; Cardoso, M.B.; Loh, W. Effect of particle functionalization and solution properties on the adsorption of bovine serum albumin and lysozyme onto silica nanoparticles. Colloids Surf. B Biointerfaces 2020, 186, 110677. [Google Scholar] [CrossRef]
- Srivastava, A.; Prajapati, A. Albumin and functionalized albumin nanoparticles: Production strategies, characterization, and target indications. Asian Biomed. 2020, 14, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Luis de Redín, I.; Boiero, C.; Martínez-Ohárriz, M.C.; Agüeros, M.; Ramos, R.; Peñuelas, I.; Allemandi, D.; Llabot, J.M.; Irache, J.M. Human serum albumin nanoparticles for ocular delivery of bevacizumab. Int. J. Pharm. 2018, 541, 214–223. [Google Scholar] [CrossRef]
- Bukackova, M.; Marsalek, R. Interaction of BSA with ZnO, TiO2, and CeO2 nanoparticles. Biophys. Chem. 2020, 267, 106475. [Google Scholar] [CrossRef]
- Li, Z.; Lu, D.; Gao, X. Optimization of mixture proportions by statistical experimental design using response surface method—A review. J. Build. Eng. 2021, 36, 102101. [Google Scholar] [CrossRef]
- Lakka, N.S.; Kuppan, C.; Vadagam, N.; Reddamoni, S.Y.; Muthusamy, C. Degradation pathways and impurity profiling of the anticancer drug apalutamide by HPLC and LC–MS/MS and separation of impurities using Design of Experiments. Biomed. Chromatogr. 2023, 37, e5549. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, A.; Chaudhary, G.; Goia, F. Designing the design of experiments (DOE)—An investigation on the influence of different factorial designs on the characterization of complex systems. Energy Build. 2021, 250, 111298. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Pedersen, J.N.; Marie, R. Size and surface charge characterization of nanoparticles with a salt gradient. Nat. Commun. 2020, 11, 2337. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]
- Filippov, S.K.; Khusnutdinov, R.; Murmiliuk, A.; Inam, W.; Zakharova, L.Y.; Zhang, H.; Khutoryanskiy, V.V. Dynamic light scattering and transmission electron microscopy in drug delivery: A roadmap for correct characterization of nanoparticles and interpretation of results. Mater. Horiz. 2023, 10, 5354–5370. [Google Scholar] [CrossRef]
- Farkas, N.; Kramar, J.A. Dynamic light scattering distributions by any means. J. Nanopart. Res. 2021, 23, 120. [Google Scholar] [CrossRef]
- ISO 10993-5: 2009; Biological Evaluation of Medical Devices. Part 5: Tests for In Vitro Cytotoxicity. ISO: Geneva, Switzerland, 2009.
- Valente, J.F.A.; Sousa, A.; Azevedo, G.A.; Queiroz, J.A.; Sousa, F. Purification of supercoiled p53-encoding plasmid using an arginine-modified macroporous support. J. Chromatogr. A 2020, 1618, 460890. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Seo, H.W.; Baek, J.-H.; Lim, S.H.; Hwang, S.-G.; Kim, E.H. Gene expression profiling of glioblastoma cell lines depending on TP53 status after tumor-treating fields (TTFields) treatment. Sci. Rep. 2020, 10, 12272. [Google Scholar] [CrossRef]

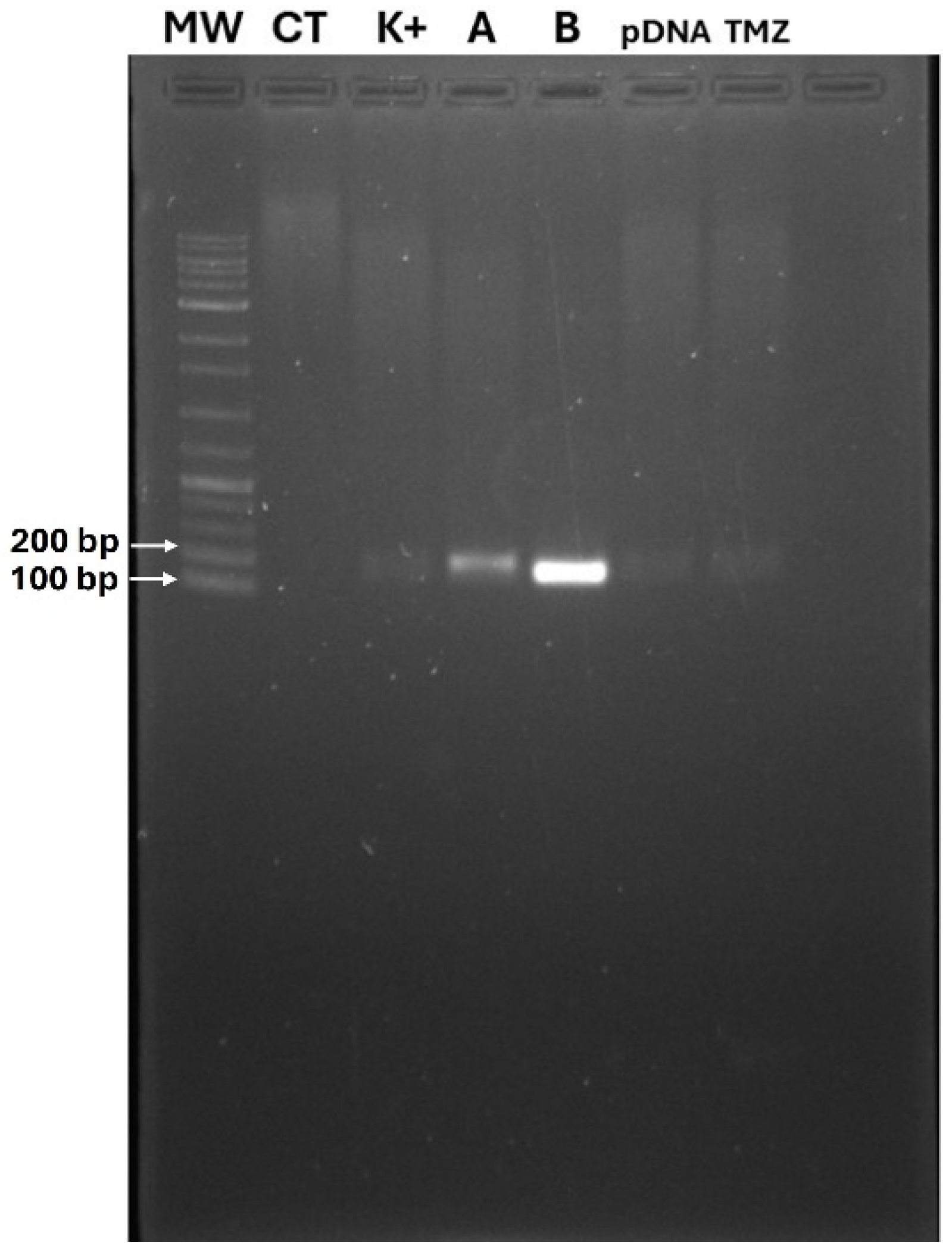
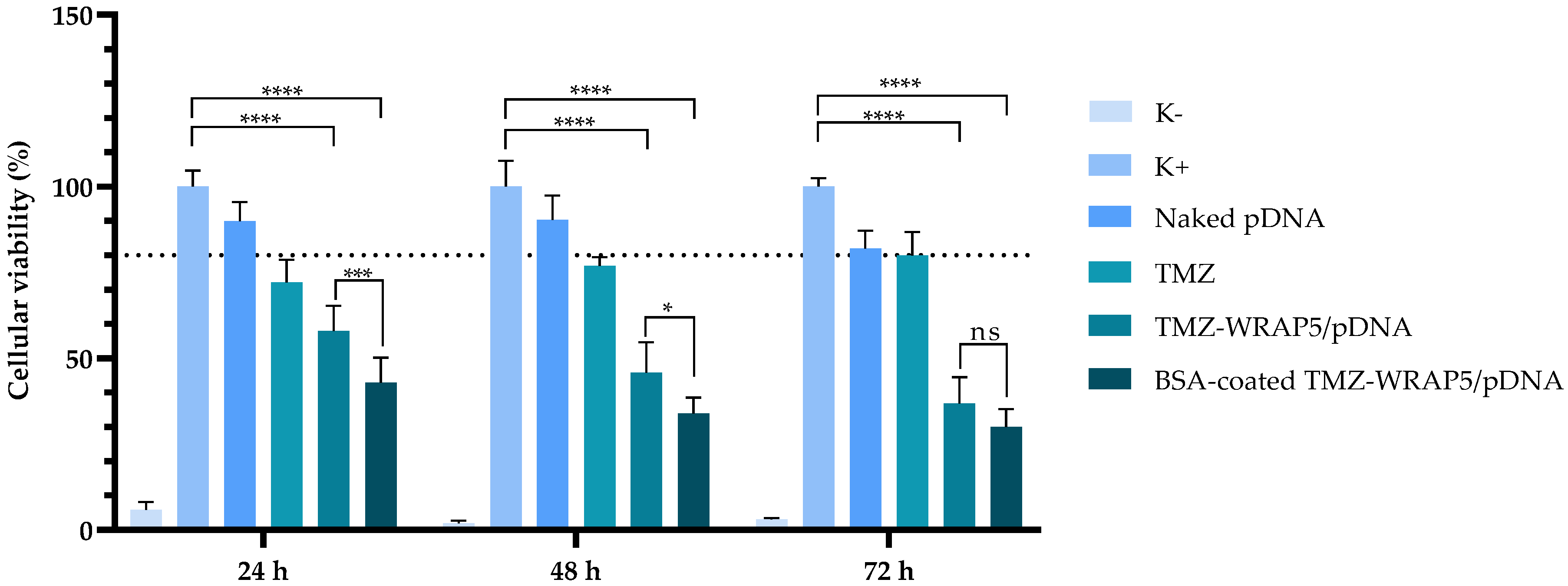

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inputs | Outputs | |||||
|---|---|---|---|---|---|---|
| Time (min) | N/P Ratio | BSA (%) | Charge (mV) | Size (nm) | PdI | pDNA CC (%) |
| 0 | 0.5 | 0.08 | −17.3 | 359.20 | 0.580 | 35.5 |
| 0.5 | 0.18 | −17.7 | 358.00 | 0.490 | 26.8 | |
| 0.5 | 0.28 | −31.6 | 431.84 | 0.483 | 25.8 | |
| 1.0 | 0.08 | 3.8 | 250.82 | 0.378 | 86.4 | |
| 1.0 | 0.18 | −1.0 | 307.67 | 0.361 | 80.3 | |
| 1.0 | 0.18 | 0.9 | 299.44 | 0.344 | 78.3 | |
| 1.0 | 0.18 | 1.2 | 290.97 | 0.381 | 76.7 | |
| 1.0 | 0.28 | −3.4 | 308.2 | 0.480 | 86.9 | |
| 1.5 | 0.08 | 1.9 | 217.94 | 0.370 | 91.0 | |
| 1.5 | 0.18 | 2.6 | 243.96 | 0.310 | 89.0 | |
| 1.5 | 0.28 | 1.1 | 280.60 | 0.331 | 91.7 | |
| 25 | 0.5 | 0.08 | −12.4 | 311.25 | 0.391 | 51.2 |
| 0.5 | 0.18 | −18.3 | 324.99 | 0.309 | 39.4 | |
| 0.5 | 0.28 | −25.7 | 350.13 | 0.276 | 27.0 | |
| 1.0 | 0.08 | 9.0 | 182.84 | 0.281 | 97.1 | |
| 1.0 | 0.18 | 5.0 | 211.47 | 0.214 | 91.4 | |
| 1.0 | 0.18 | 3.2 | 203.47 | 0.242 | 90.9 | |
| 1.0 | 0.18 | 4.5 | 207.74 | 0.234 | 87.2 | |
| 1.0 | 0.28 | 0.0 | 218.90 | 0.290 | 91.8 | |
| 1.5 | 0.08 | 11.6 | 134.09 | 0.223 | 95.3 | |
| 1.5 | 0.18 | 8.6 | 175.37 | 0.224 | 91.7 | |
| 1.5 | 0.28 | 1.3 | 193.59 | 0.228 | 95.8 |
| Output | Multiple Regression Equations | Model | R2 | R2 Adjusted | R2 Predicted | Adequate Precision |
|---|---|---|---|---|---|---|
| Charge | +2.88 + 12.51 A − 4.58 B + 2.10 C + 2.06 AB + 0.47 AC − 0.86 BC − 9.94 A2 − 1.39 B2 | Quadratic | 0.9841 | 0.9744 | 0.9317 | 31.863 |
| Size | +248.72 − 74.15 A + 27.26 B − 37.95 C + 1.33 AB − 6.40 AC − 4.85 BC + 33.97 A2 − 1.42 B2 | Quadratic | 0.9786 | 0.9654 | 0.9371 | 31.522 |
| PdI | +0.30 − 0.070 A − 0.011 B − 0.073 C + 0.022 AB + 0.020 AC − 0.0056 BC + 0.019 A2 + 0.043 B2 | Quadratic | 0.9296 | 0.8863 | 0.7335 | 16.326 |
| pDNA CC | +78.39 + 28.06 A − 3.13 B + 1.84 C + 4.39 AB − 1.53 AC − 1.71 BC − 20.55 A2 + 8.27 B2 | Quadratic | 0.8436 | 0.7474 | 0.7292 | 8.405 |
| Source | Charge | Size | PdI | pDNA CC |
|---|---|---|---|---|
| Model | <0.0001 | <0.0001 | <0.0001 | 0.0004 |
| A | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| B | <0.0001 | <0.0001 | 0.2746 | 0.4358 |
| C | 0.0002 | <0.0001 | <0.0001 | 0.5340 |
| AB | 0.0085 | 0.7898 | 0.0884 | 0.3734 |
| AC | 0.3981 | 0.1329 | 0.0626 | 0.7003 |
| BC | 0.1383 | 0.2456 | 0.5809 | 0.6682 |
| A2 | <0.0001 | <0.0001 | 0.2275 | 0.0044 |
| B2 | 0.1203 | 0.8211 | 0.0137 | 0.1903 |
| Lack of fit | 0.0961 | 0.0501 | 0.0550 | 0.9982 |
| Predicted Input | Output | Predicted Mean | 95% CI Low for Mean | 95% CI High for Mean | Obtained Mean |
|---|---|---|---|---|---|
| Charge | 9.7 | 7.2 | 12.2 | 9.8 |
| Size | 181.5 | 163.0 | 200.0 | 182.3 | |
| PdI | 0.286 | 0.240 | 0.332 | 0.248 | |
| pDNA CC | 94.8 | 76.8 | 112.8 | 96.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, I.; Neves, A.R.; Eusébio, D.; Albuquerque, T.; Vivès, E.; Boisguérin, P.; Santos, A.O.; Sousa, Â.; Costa, D. Design of Experiments to Tailor the Potential of BSA-Coated Peptide Nanocomplexes for Temozolomide/p53 Gene Co-Delivery. Pharmaceutics 2024, 16, 1389. https://doi.org/10.3390/pharmaceutics16111389
Afonso I, Neves AR, Eusébio D, Albuquerque T, Vivès E, Boisguérin P, Santos AO, Sousa Â, Costa D. Design of Experiments to Tailor the Potential of BSA-Coated Peptide Nanocomplexes for Temozolomide/p53 Gene Co-Delivery. Pharmaceutics. 2024; 16(11):1389. https://doi.org/10.3390/pharmaceutics16111389
Chicago/Turabian StyleAfonso, Inês, Ana R. Neves, Dalinda Eusébio, Tânia Albuquerque, Eric Vivès, Prisca Boisguérin, Adriana O. Santos, Ângela Sousa, and Diana Costa. 2024. "Design of Experiments to Tailor the Potential of BSA-Coated Peptide Nanocomplexes for Temozolomide/p53 Gene Co-Delivery" Pharmaceutics 16, no. 11: 1389. https://doi.org/10.3390/pharmaceutics16111389
APA StyleAfonso, I., Neves, A. R., Eusébio, D., Albuquerque, T., Vivès, E., Boisguérin, P., Santos, A. O., Sousa, Â., & Costa, D. (2024). Design of Experiments to Tailor the Potential of BSA-Coated Peptide Nanocomplexes for Temozolomide/p53 Gene Co-Delivery. Pharmaceutics, 16(11), 1389. https://doi.org/10.3390/pharmaceutics16111389