


Burst Release from In Situ Forming PLGA-Based Implants: 12 Effectors and Ways of Correction

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Base PLGA Characteristics
- Hydration: water penetrates the amorphous region and destroys the van der Waals interaction forces and hydrogen bonds, causing a decrease in the glass transition temperature;
- Initial degradation: breakage of covalent bonds with a decrease in molecular weight;
- Permanent degradation: carboxyl end groups autocatalyze the degradation process, and mass loss begins due to massive breakage of main chain covalent bonds, resulting in loss of integrity;
- Solubilization: the fragments are further degraded to aqueous soluble molecules [12].

- Molecular weight: when the molecular weight of conventional PLGA is increased from 10–20 to 100 kDa, the degradation rate is reported to range from several weeks to several months;
- GA to LA ratio: PLGAs with higher LA content are less hydrophilic, absorb less water, and subsequently degrade more slowly due to the presence of methyl side groups in PLA, making it more hydrophobic than PGA. The exception to this rule is the 50:50 copolymer, which degrades faster [13];
- Stereochemistry: mixtures of lactic acid monomers D and L are most commonly used to make PLGA because the rate of water penetration is higher in the amorphous regions of D and L, leading to accelerated degradation of poly-lactic-co-glycolic acid [13];
- Modification of end groups: polymers that are capped by esters (as opposed to free carboxylic acid) have longer half-lives [14]. Moreover, the shape of the polymer chain strongly influences the degradation behaviour of PLGA depending on water availability. In addition, low pH accelerates the degradation of PLGA due to the autocatalysis of the reaction [15].
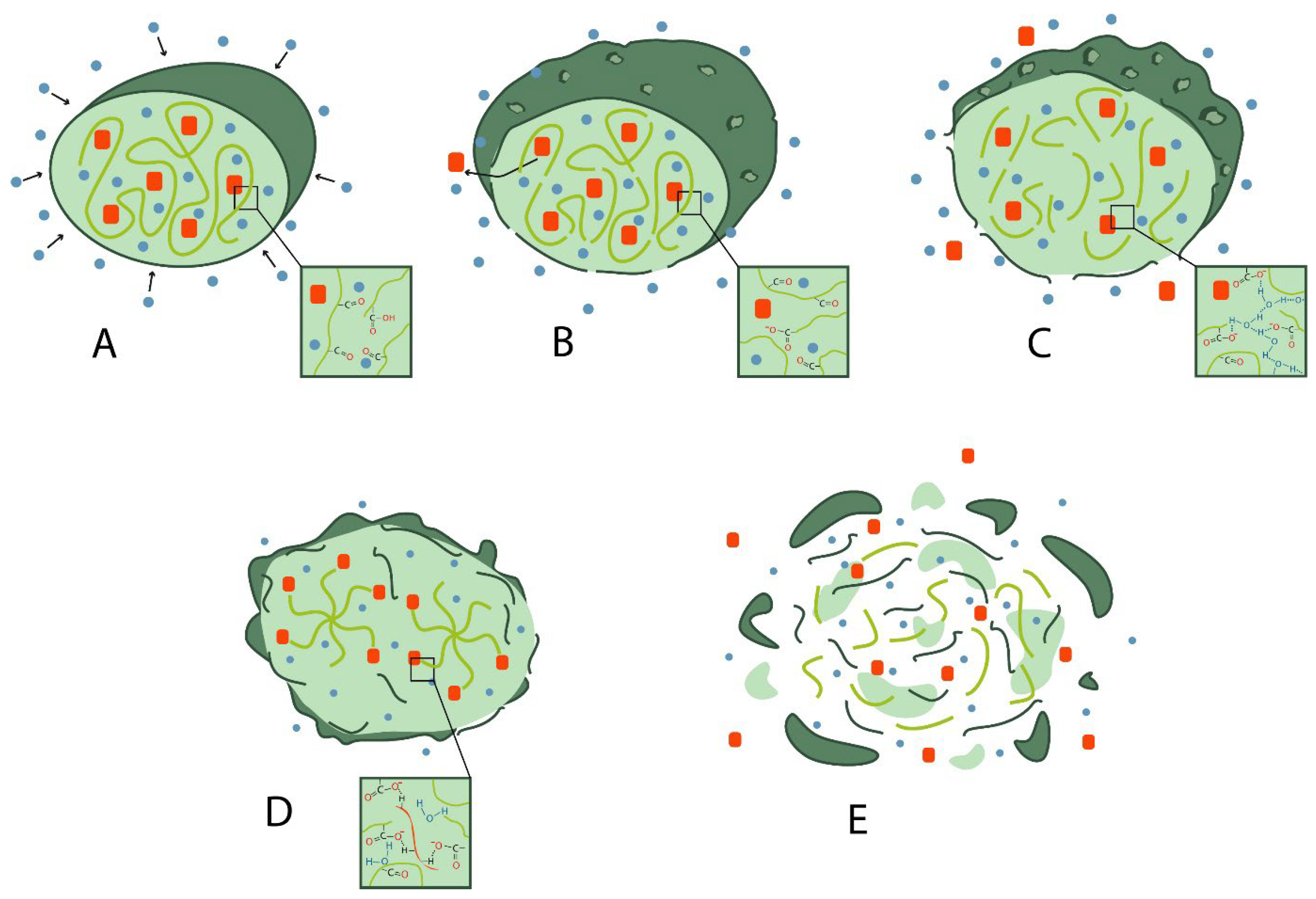
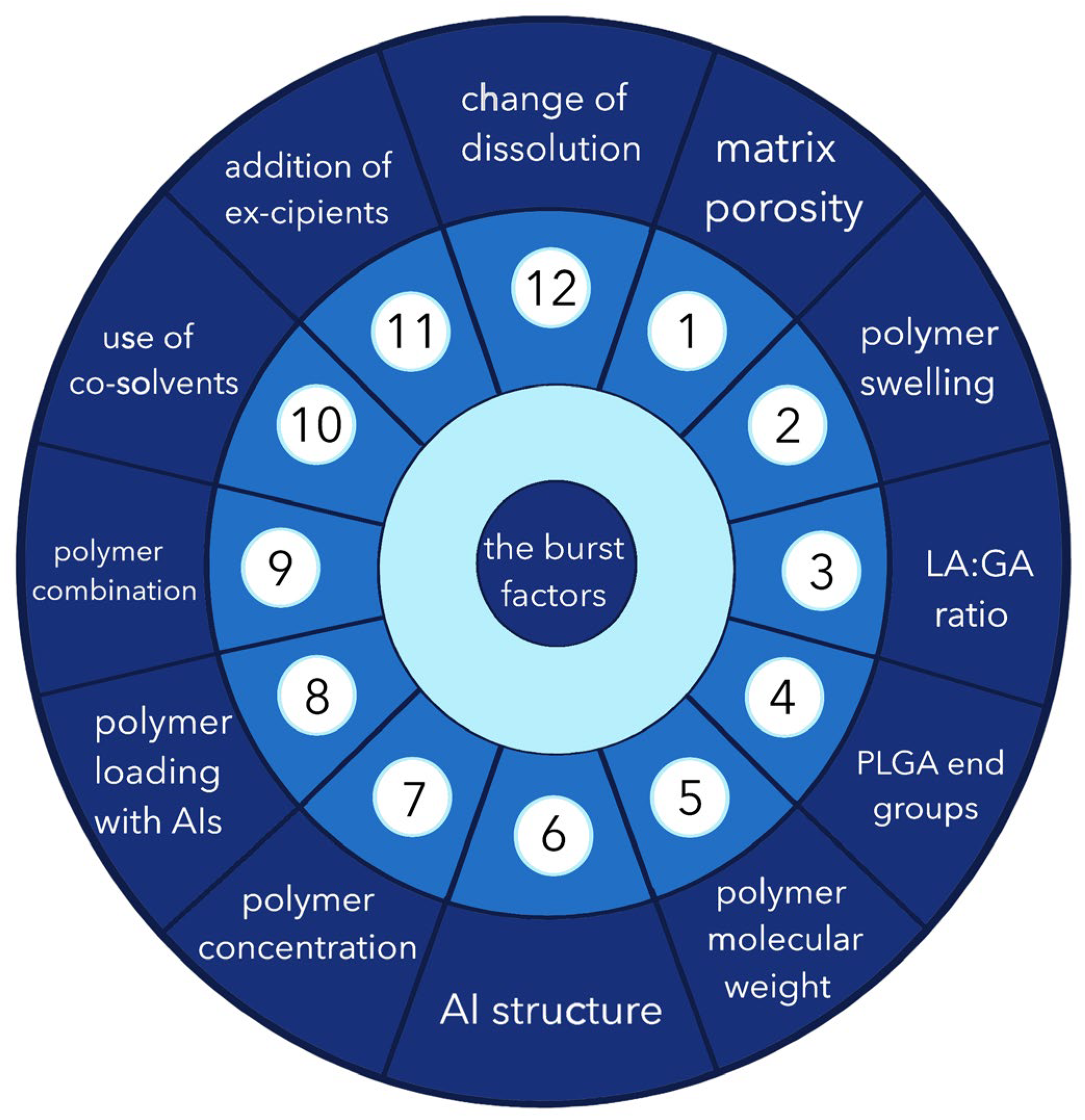
3. Possible Causes of the “Burst Release” Problem
3.1. Porosity
3.2. Swelling of the Polymer
3.3. LA/GA Ratio
3.4. Impact of End Groups
3.5. Molecular Weight of the Polymer
3.6. Structure of the Active Molecule
3.7. Polymer Concentration
3.8. Polymer Loading with Active Ingredient
3.9. Combination of Composition
3.10. Use of Cosolvents
3.11. Inclusion of Excipients
3.12. Release Environment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kempe, S.; Mader, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.S.; Kastellorizios, M.; Tipnis, N.; Zou, Y.; Wang, Y.; Choi, S.; Burgess, D.J. Effect of implant formation on drug release kinetics of in situ forming implants. Int. J. Pharm. 2021, 592, 120105. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jensen, H.; Petersen, N.J.; Larsen, S.W.; Ostergaard, J. Concomitant monitoring of implant formation and drug release of in situ forming poly (lactide-co-glycolide acid) implants in a hydrogel matrix mimicking the subcutis using UV-vis imaging. J. Pharm. Biomed. Anal. 2018, 150, 95–106. [Google Scholar] [CrossRef]
- Thakur, R.R.; McMillan, H.L.; Jones, D.S. Solvent induced phase inversion-based in situ forming controlled release drug delivery implants. J. Control. Release 2014, 176, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.N.; Bhatia, A.; Kaur, R.; Sharma, R.; Kaur, G.; Dhawan, S. PLGA: A unique polymer for drug delivery. Ther. Deliv. 2015, 6, 41–58. [Google Scholar] [CrossRef]
- Kanwar, N.; Sinha, V.R. In situ Forming Depot as Sustained-Release Drug Delivery Systems. Crit. Rev. Ther. Drug Carrier Syst. 2019, 36, 93–136. [Google Scholar] [CrossRef] [PubMed]
- Paillard-Giteau, A.; Tran, V.; Thomas, O.; Garric, X.; Coudane, J.; Marchal, S.; Chourpa, I.; Benoît, J.; Montero-Menei, C.; Venier-Julienne, M. Effect of various additives and polymers on lysozyme release from PLGA microspheres prepared by an s/o/w emulsion technique. Eur. J. Pharm. Biopharm. 2010, 75, 128–136. [Google Scholar] [CrossRef]
- Allison, S.D. Analysis of initial burst in PLGA microparticles. Expert Opin. Drug Deliv. 2008, 5, 615–628. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Park, T.G. Degradation of poly (lactic-co-glycolic acid) microspheres: Effect of copolymer composition. Biomaterials 1995, 16, 1123–1130. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, J.; Jing, D.; Ding, J. “Wet-state” mechanical properties of three-dimensional polyester porous scaffolds. J. Biomed. Mater. Res. A 2006, 76, 264–271. [Google Scholar] [CrossRef]
- Engineer, C.; Parikh, J.; Raval, A. Review on hydrolytic degradation behavior of biodegradable polymers from controlled drug delivery system. Trends Biomater. Artif. Organs 2011, 2, 79–85. [Google Scholar]
- Wang, M.; Zhan, J.; Xu, L.; Wang, Y.; Lu, D.; Li, Z.; Li, J.; Luo, F.; Tan, H. Synthesis and characterization of PLGA-PEG-PLGA based thermosensitive polyurethane micelles for potential drug delivery. J. Biomater. Sci. Polym. Ed. 2021, 32, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.S.; Wang, N. Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers. Part II: Biodegradation. J. Biomater. Sci. Polym. Ed. 2001, 12, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Holy, C.E.; Dang, S.M.; Davies, J.E.; Shoichet, M.S. In vitro degradation of a novel poly(lactide-co-glycolide) 75/25 foam. Biomaterials 1999, 20, 1177–1185. [Google Scholar] [CrossRef]
- Wang, X.; Burgess, D.J. Drug release from in situ forming implants and advances in release testing. Adv. Drug Deliv. Rev. 2021, 178, 113912. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Tan, W.S.; Ho, K.L.; Mariatulqabtiah, A.R.; Abu Kasim, N.H.; Rahman, N.A.; Wong, T.W.; Chee, C.F. Challenges and Complications of Poly(lactic-co-glycolic acid)-Based Long-Acting Drug Product Development. Pharmaceutics 2022, 14, 614. [Google Scholar] [CrossRef]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef]
- Wan, B.; Bao, Q.; Burgess, D.J. In vitro-in vivo correlation of PLGA microspheres: Effect of polymer source variation and temperature. J. Control. Release 2022, 347, 347–355. [Google Scholar] [CrossRef]
- Thalhauser, S.; Peterhoff, D.; Wagner, R.; Breunig, M. Silica particles incorporated into PLGA-based in situ-forming implants exploit the dual advantage of sustained release and particulate delivery. Eur. J. Pharm. Biopharm. 2020, 156, 1–10. [Google Scholar] [CrossRef]
- Brodbeck, K.J.; DesNoyer, J.R.; McHugh, A.J. Phase inversion dynamics of PLGA solutions related to drug delivery. Part II. The role of solution thermodynamics and bath-side mass transfer. J. Control. Release 1999, 62, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mishra, B. An overview of recent patents on oral osmotic drug delivery systems. Recent Pat. Drug Deliv. Formul. 2007, 1, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Batycky, R.P.; Hanes, J.; Langer, R.; Edwards, D.A. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J. Pharm. Sci. 1997, 86, 1464–1477. [Google Scholar] [CrossRef] [PubMed]
- Joiner, J.B.; Prasher, A.; Young, I.C.; Kim, J.; Shrivastava, R.; Maturavongsadit, P.; Benhabbour, S.R. Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics. Pharmaceutics 2022, 14, 1188. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Matsukawa, Y.; Suzuki, T.; Yoshino, H. Drug release characteristics of multi-reservoir type microspheres with poly(dl-lactide-co-glycolide) and poly(dl-lactide). J. Control. Release 2005, 106, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F. Factors affecting the degradation and drug-release mechanism of poly (lactic acid) and poly[(lactic acid)-co-(glycolic acid)]. Polym. Int. 2004, 54, 36–46. [Google Scholar] [CrossRef]
- Fredenberg, S.; Reslow, M.; Axelsson, A. Encapsulated zinc salt increases the diffusion of protein through PLG films. Int. J. Pharm. 2009, 370, 47–53. [Google Scholar] [CrossRef]
- Siepmann, J.; Elkharraz, K.; Siepmann, F.; Klose, D. How autocatalysis accelerates drug release from PLGA-based microparticles: A quantitative treatment. Biomacromolecules 2005, 6, 2312–2319. [Google Scholar] [CrossRef]
- Ford Versypt, A.N.; Pack, D.W.; Braatz, R.D. Mathematical modeling of drug delivery from autocatalytically degradable PLGA microspheres—A review. J. Control. Release 2013, 165, 29–37. [Google Scholar] [CrossRef]
- Brunner, A.; Mader, K.; Gopferich, A. pH and osmotic pressure inside biodegradable microspheres during erosion. Pharm. Res. 1999, 16, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Schwendeman, S.P. Mapping neutral microclimate pH in PLGA microspheres. J. Control. Release 2005, 101, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Klose, D.; Siepmann, F.; Elkharraz, K.; Krenzlin, S.; Siepmann, J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int. J. Pharm. 2006, 314, 198–206. [Google Scholar] [CrossRef]
- Dunne, M.; Corrigan, I.; Ramtoola, Z. Influence of particle size and dissolution conditions on the degradation properties of polylactide-co-glycolide particles. Biomaterials 2000, 21, 1659–1668. [Google Scholar] [CrossRef]
- Kang, J.; Schwendeman, S.P. Comparison of the effects of Mg(OH)2 and sucrose on the stability of bovine serum albumin encapsulated in injectable poly(D,L-lactide-co-glycolide) implants. Biomaterials 2002, 23, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Qiu, X.; Jin, R.; Bai, Y.; Liu, S.; Chen, X. One-pot preparation of polymer microspheres with different porous structures to sequentially release bio-molecules for cutaneous regeneration. Biomater. Sci. 2018, 6, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Bassand, C.; Verin, J.; Lamatsch, M.; Siepmann, F.; Siepmann, J. How agarose gels surrounding PLGA implants limit swelling and slow down drug release. J. Control. Release 2022, 343, 255–266. [Google Scholar] [CrossRef]
- Gasmi, H.; Danede, F.; Siepmann, J.; Siepmann, F. Does PLGA microparticle swelling control drug release? New insight based on single particle swelling studies. J. Control. Release 2015, 213, 120–127. [Google Scholar] [CrossRef]
- Saini, V.; Jain, V.; Sudheesh, M.S.; Jaganathan, K.S.; Murthy, P.K.; Kohli, D.V. Comparison of humoral and cell-mediated immune responses to cationic PLGA microspheres containing recombinant hepatitis B antigen. Int. J. Pharm. 2011, 408, 50–57. [Google Scholar] [CrossRef]
- Nasonova, M.V.; Hodyrevskaya, Y.I.; Nemoykina, A.L.; Mikhaylenko, M.Y.; Kudryavtseva, Y.A. Optimization of physical, mechanical and degradation properties for biodegradable anti-adhesive membranes. SibScript 2015, 1, 65–69. Available online: https://cyberleninka.ru/article/n/optimizatsiya-srokov-degradatsii-i-fiziko-mehanicheskih-svoystv-protivospaechnyh-membran-na-osnove-biodegradiruemyh-polimerov (accessed on 1 October 2023). (In Russian).
- Vey, E.; Rodger, C.; Booth, J.; Claybourn, M.; Miller, A.F.; Saiani, A. Degradation kinetics of poly(lactic-co-glycolic) acid block copolymer cast films in phosphate buffer solution as revealed by infrared and Raman spectroscopies. Polym. Degrad. Stab. 2011, 96, 1882–1889. [Google Scholar] [CrossRef]
- Sakharova, P.S.; Pyzhov, V.S.; Bakhrushina, E.O. Poly(l-lactide-co-glycolide) and shellac in the development of phase-sensitive in situ implants. Aspir. Vestn. Povolzhiya 2022, 22, 51–57. [Google Scholar] [CrossRef]
- Sakharova, P.S.; Bakhrushina, E.O.; Krasnyuk, I.I. In Vitro Modeling for the Evaluation of Biopharmaceutical Parameters of Phase Inversion-Based Dental In Situ Implants. Med. Pharm. J. PULSE 2022, 24, 31–35. [Google Scholar] [CrossRef]
- Huang, C.L.; Kumar, S.; Tan, J.J.; Boey, F.Y.; Venkatraman, S.S.; Steele, T.W.; Loo, J.S. Modulating drug release from poly (lactic-co-glycolic acid) thin films through terminal end-groups and molecular weight. Polym. Degrad. Stab. 2013, 98, 619–626. [Google Scholar] [CrossRef]
- Wang, J.; Helder, L.; Shao, J.; Jansen, J.A.; Yang, M.; Yang, F. Encapsulation and release of doxycycline from electrospray-generated PLGA microspheres: Effect of polymer end groups. Int. J. Pharm. 2019, 564, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Kranz, H.; Fivez, A.; Siepmann, F.; Siepmann, J. Often neglected: PLGA/PLA swelling orchestrates drug release: HME implants. J. Control. Release 2019, 306, 97–107. [Google Scholar] [CrossRef]
- Lima, K.M.; Rodrigues Junior, J.M. Poly-DL-lactide-co-glycolide microspheres as a controlled release antigen delivery system. Braz. J. Med. Biol. Res. 1999, 32, 171–180. [Google Scholar] [CrossRef][Green Version]
- Patel, R.B.; Solorio, L.; Wu, H.; Krupka, T.; Exner, A.A. Effect of injection site on in situ implant formation and drug release in vivo. J. Control. Release 2010, 147, 350–358. [Google Scholar] [CrossRef]
- Yewey, G.L.; Duysen, E.G.; Cox, S.M.; Dunn, R.L. Delivery of proteins from a controlled release injectable implant. Pharm. Biotechnol. 1997, 10, 93–117. [Google Scholar] [CrossRef]
- Luan, X.; Bodmeier, R. Influence of the poly(lactide-co-glycolide) type on the leuprolide release from in situ forming microparticle systems. J. Control. Release 2006, 110, 266–272. [Google Scholar] [CrossRef]
- Astaneh, R.; Erfan, M.; Moghimi, H.; Mobedi, H. Changes in morphology of in situ forming PLGA implant prepared by different polymer molecular weight and its effect on release behavior. J. Pharm. Sci. 2009, 98, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Simo, C.; Gallardo, A.; Parejo, C.; San Roman, J.; Barbas, C.; Cifuentes, A. Monitoring ibuprofen enantiomers released from polymeric systems. Eur. J. Pharm. Sci. 2002, 16, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Liang, Z.H.; Zeng, S. Monitoring release of ketoprofen enantiomers from biodegradable poly(D,L-lactide-co-glycolide) injectable implants. Int. J. Pharm. 2007, 337, 102–108. [Google Scholar] [CrossRef]
- Quan, P.; Guo, W.; Lin, Y.; Cun, D.; Yang, M. Donepezil accelerates the release of PLGA microparticles via catalyzing the polymer degradation regardless of the end groups and molecular weights. Int. J. Pharm. 2023, 632, 122566. [Google Scholar] [CrossRef] [PubMed]
- Santhosh, J.K.; Santosh, V.; Raju, T.; Aravind, G.; Babu, D.S. Formulation and development of in-situ implant of Cytarabine. Int. J. Pharm. Pharm. Sci. 2012, 4, 412–420. [Google Scholar]
- Bode, C.; Kranz, H.; Siepmann, F.; Siepmann, J. Coloring of PLGA implants to better understand the underlying drug release mechanisms. Int. J. Pharm. 2019, 569, 118563. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.A.; Liu, J.; Chen, B.; Wang, Y.; Qin, B.; Xu, X.; Li, Q.; Lynd, N.A.; Zhang, F. Drug release mechanisms of high-drug-load, melt-extruded dexamethasone intravitreal implants. Eur. J. Pharm. Biopharm. 2023, 187, 46–56. [Google Scholar] [CrossRef]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Controlling the in vitro release profiles for a system of haloperidol-loaded PLGA nanoparticles. Int. J. Pharm. 2008, 346, 151–159. [Google Scholar] [CrossRef]
- Karp, F.; Turino, L.N.; Helbling, I.M.; Islan, G.A.; Luna, J.A.; Estenoz, D.A. In situ Formed Implants, Based on PLGA and Eudragit Blends, for Novel Florfenicol Controlled Release Formulations. J. Pharm. Sci. 2021, 110, 1270–1278. [Google Scholar] [CrossRef]
- Cao, Z.; Tang, X.; Zhang, Y.; Yin, T.; Gou, J.; Wang, Y.; He, H. Novel injectable progesterone-loaded nanoparticles embedded in SAIB-PLGA in situ depot system for sustained drug release. Int. J. Pharm. 2021, 607, 121021. [Google Scholar] [CrossRef]
- Li, F.; Wen, Y.; Zhang, Y.; Zheng, K.; Ban, J.; Xie, Q.; Wen, Y.; Liu, Q.; Chen, F.; Mo, Z.; et al. Characterisation of 2-HP-β-cyclodextrin-PLGA nanoparticle complexes for potential use as ocular drug delivery vehicles. Artif. Cells Nanomed. Biotechnol. 2019, 47, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Huang, Z.; Huang, J.; Liu, X.; Ban, J.; Huang, X.; Luo, H.; Chen, Z.; Xie, Q.; Chen, Y.; et al. Effect of a 2-HP-β-Cyclodextrin Formulation on the Biological Transport and Delivery of Chemotherapeutic PLGA Nanoparticles. Drug Des. Dev. Ther. 2021, 15, 2605–2618. [Google Scholar] [CrossRef]
- Duque, L.; Korber, M.; Bodmeier, R. Improving release completeness from PLGA-based implants for the acid-labile model protein ovalbumin. Int. J. Pharm. 2018, 538, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.; Eissa, N.G.; Ibrahim, T.M.; El-Bassossy, H.M.; El-Nahas, H.M.; Ayoub, M.M. Development of depot PLGA-based in-situ implant of Linagliptin: Sustained release and glycemic control. Saudi Pharm. J. 2023, 31, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Venkatraman, S.S. Cosolvent effects on the drug release and depot swelling in injectable in situ depot-forming systems. J. Pharm. Sci. 2012, 101, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, H.; Zhou, G.; Xie, S.; Zou, H.; Yu, Y.; Li, G.; Sun, D.; Zhang, G.; Lu, Y.; et al. In vitro and in vivo study of thymosin alpha1 biodegradable in situ forming poly(lactide-co-glycolide) implants. Int. J. Pharm. 2010, 397, 122–129. [Google Scholar] [CrossRef]
- Phaechamud, T.; Mahadlek, J.; Tuntarawongsa, S. Peppermint oil/doxycycline hyclate-loaded Eudragit RS in situ forming gel for periodontitis treatment. J. Pharm. Investig. 2018, 48, 451–464. [Google Scholar] [CrossRef]
- Khaing, E.M.; Mahadlek, J.; Okonogi, S.; Phaechamud, T. Lime Peel Oil-Incorporated Rosin-Based Antimicrobial In situ Forming Gel. Gels 2022, 8, 169. [Google Scholar] [CrossRef]
- Parent, M.; Nouvel, C.; Koerber, M.; Sapin, A.; Maincent, P.; Boudier, A. PLGA in situ implants formed by phase inversion: Critical physicochemical parameters to modulate drug release. J. Control. Release 2013, 172, 292–304. [Google Scholar] [CrossRef]
- Jain, R.A.; Rhodes, C.T.; Railkar, A.M.; Malick, A.W.; Shah, N.H. Controlled release of drugs from injectable in situ formed biodegradable PLGA microspheres: Effect of various formulation variables. Eur. J. Pharm. Biopharm. 2000, 50, 257–262. [Google Scholar] [CrossRef]
- Graham, P.D.; Brodbeck, K.J.; McHugh, A.J. Phase inversion dynamics of PLGA solutions related to drug delivery. J. Control. Release 1999, 58, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Kamali, H.; Khodaverdi, E.; Hadizadeh, F.; Mohajeri, S.A.; Nazari, A.; Jafarian, A.H. Comparison of in-situ forming composite using PLGA-PEG-PLGA with in-situ forming implant using PLGA: In-vitro, ex-vivo, and in-vivo evaluation of naltrexone release. J. Drug Deliv. Sci. Technol. 2019, 50, 188–200. [Google Scholar] [CrossRef]
- Bakhshi, R.; Vasheghani-Farahani, E.; Mobedi, H.; Jamshidi, A.; Khakpour, M. The effect of additives on naltrexone hydrochloride release and solvent removal rate from an injectable in situ forming PLGA implant. Polym. Adv. Technol. 2006, 17, 354–359. [Google Scholar] [CrossRef]
- Dong, W.Y.; Korber, M.; Lopez Esguerra, V.; Bodmeier, R. Stability of poly(D,L-lactide-co-glycolide) and leuprolide acetate in in-situ forming drug delivery systems. J. Control. Release 2006, 115, 158–167. [Google Scholar] [CrossRef]
- Ali, A.; Madni, A.; Shah, H.; Jamshaid, T.; Jan, N.; Khan, S.; Khan, M.M.; Mahmood, M.A. Solid lipid-based nanoparticulate system for sustained release and enhanced in-vitro cytotoxic effect of 5-fluorouracil on skin Melanoma and squamous cell carcinoma. PLoS ONE 2023, 18, e0281004. [Google Scholar] [CrossRef]
- Abla, K.K.; Mehanna, M.M. Lipid-based nanocarriers challenging the ocular biological barriers: Current paradigm and future perspectives. J. Control. Release 2023, 362, 70–96. [Google Scholar] [CrossRef] [PubMed]
- Hamoudi-Ben Yelles, M.C.; Tran Tan, V.; Danede, F.; Willart, J.F.; Siepmann, J. PLGA implants: How Poloxamer/PEO addition slows down or accelerates polymer degradation and drug release. J. Control. Release 2017, 253, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Augusthy, A.R.; Chandran, S.; Vipin, K.V. Design and evaluation of an in situ forming implant system of an anti-inflammatory drug. Indo Am. J. Pharm. Res. 2017, 4, 983–994. [Google Scholar] [CrossRef]
- Silva, A.; Rosalia, R.; Sazak, A.; Carstens, M.; Ossendorp, F.; Oostendorp, J.; Jiskoot, W. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: Low-burst release is crucial for efficient CD8(+) T cell activation. Eur. J. Pharm. Biopharm. 2013, 83, 338–345. [Google Scholar] [CrossRef]
- Kang, J.; Schwendeman, S.P. Pore closing and opening in biodegradable polymers and their effect on the controlled release of proteins. Mol. Pharm. 2007, 4, 104–118. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Borcherding, K.; Marx, D.; Gätjen, L.; Bormann, N.; Wildemann, B.; Specht, U.; Salz, D.; Thiel, K.; Grunwald, I. Burst Release of Antibiotics Combined with Long-Term Release of Silver Targeting Implant-Associated Infections: Design, Characterization and in vitro Evaluation of Novel Implant Hybrid Surface. Materials 2019, 12, 3838. [Google Scholar] [CrossRef] [PubMed]
- Qnouch, A.; Solarczyk, V.; Verin, J.; Tourrel, G.; Stahl, P.; Danede, F.; Willart, J.; Lemesre, P.; Vincent, C.; Siepmann, J.; et al. Dexamethasone-loaded cochlear implants: How to provide a desired “burst release”. Int. J. Pharm. X 2021, 3, 100088. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, J.; Song, T.; Sun, Y.; Lu, X.; Li, N.; Sun, K. Effects of water-soluble additive on the release profile and pharmacodynamics of triptorelin loaded in PLGA microspheres. Drug Dev. Ind. Pharm. 2023, 49, 357–366. [Google Scholar] [CrossRef]
- Du, C.; Fikhman, D.A.; Persaud, D.; Monroe, M.B.B. Dual Burst and Sustained Release of p-Coumaric Acid from Shape Memory Polymer Foams for Polymicrobial Infection Prevention in Trauma-Related Hemorrhagic Wounds. ACS Appl. Mater. Interfaces 2023, 15, 24228–24243. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Garg, A.; Farooq, U.; Panda, A.K.; Mirza, M.A.; Noureldeen, A.; Darwish, H.; Iqbal, Z. Preparation and quality by design assisted (Qb-d) optimization of bioceramic loaded microspheres for periodontal delivery of doxycycline hyclate. Saudi J. Biol. Sci. 2021, 28, 2677–2685. [Google Scholar] [CrossRef]
- Mahendra, J.; Mahendra, L.; Mugri, M.H.; Sayed, M.E.; Bhandi, S.; Alshahrani, R.T.; Balaji, T.M.; Varadarajan, S.; Tanneeru, S.; P., A.N.R.; et al. Role of Periodontal Bacteria, Viruses, and Placental mir155 in Chronic Periodontitis and Preeclampsia-A Genetic Microbiological Study. Curr. Issues Mol. Biol. 2021, 43, 831–844. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakhrushina, E.O.; Sakharova, P.S.; Konogorova, P.D.; Pyzhov, V.S.; Kosenkova, S.I.; Bardakov, A.I.; Zubareva, I.M.; Krasnyuk, I.I.; Krasnyuk, I.I., Jr. Burst Release from In Situ Forming PLGA-Based Implants: 12 Effectors and Ways of Correction. Pharmaceutics 2024, 16, 115. https://doi.org/10.3390/pharmaceutics16010115
Bakhrushina EO, Sakharova PS, Konogorova PD, Pyzhov VS, Kosenkova SI, Bardakov AI, Zubareva IM, Krasnyuk II, Krasnyuk II Jr. Burst Release from In Situ Forming PLGA-Based Implants: 12 Effectors and Ways of Correction. Pharmaceutics. 2024; 16(1):115. https://doi.org/10.3390/pharmaceutics16010115
Chicago/Turabian StyleBakhrushina, Elena O., Polina S. Sakharova, Polina D. Konogorova, Victor S. Pyzhov, Svetlana I. Kosenkova, Alexander I. Bardakov, Irina M. Zubareva, Ivan I. Krasnyuk, and Ivan I. Krasnyuk, Jr. 2024. "Burst Release from In Situ Forming PLGA-Based Implants: 12 Effectors and Ways of Correction" Pharmaceutics 16, no. 1: 115. https://doi.org/10.3390/pharmaceutics16010115
APA StyleBakhrushina, E. O., Sakharova, P. S., Konogorova, P. D., Pyzhov, V. S., Kosenkova, S. I., Bardakov, A. I., Zubareva, I. M., Krasnyuk, I. I., & Krasnyuk, I. I., Jr. (2024). Burst Release from In Situ Forming PLGA-Based Implants: 12 Effectors and Ways of Correction. Pharmaceutics, 16(1), 115. https://doi.org/10.3390/pharmaceutics16010115