


One Step In Situ Co-Crystallization of Dapsone and Polyethylene Glycols during Fluidized Bed Granulation



Abstract
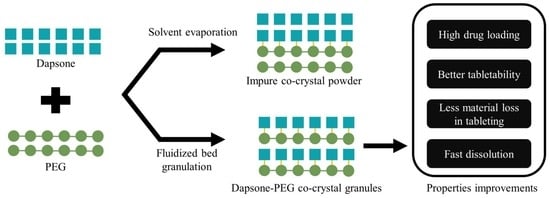
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation Methods
2.2.1. Solvent Evaporation (SE)
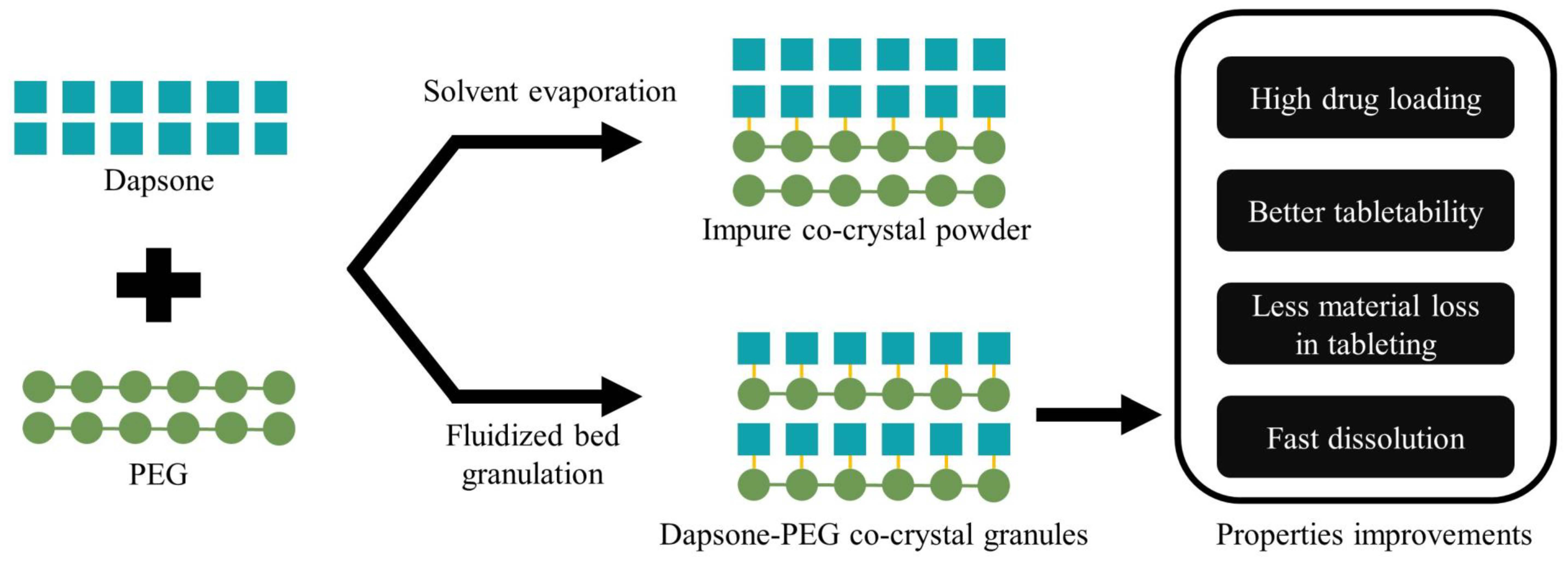
2.2.2. Fluidized Bed Granulation
2.3. Material Characterization
2.3.1. Powder X-ray Diffraction (PXRD)
2.3.2. Modulated Differential Scanning Calorimetry (mDSC)
2.3.3. Thermogravimetric Analysis (TGA)
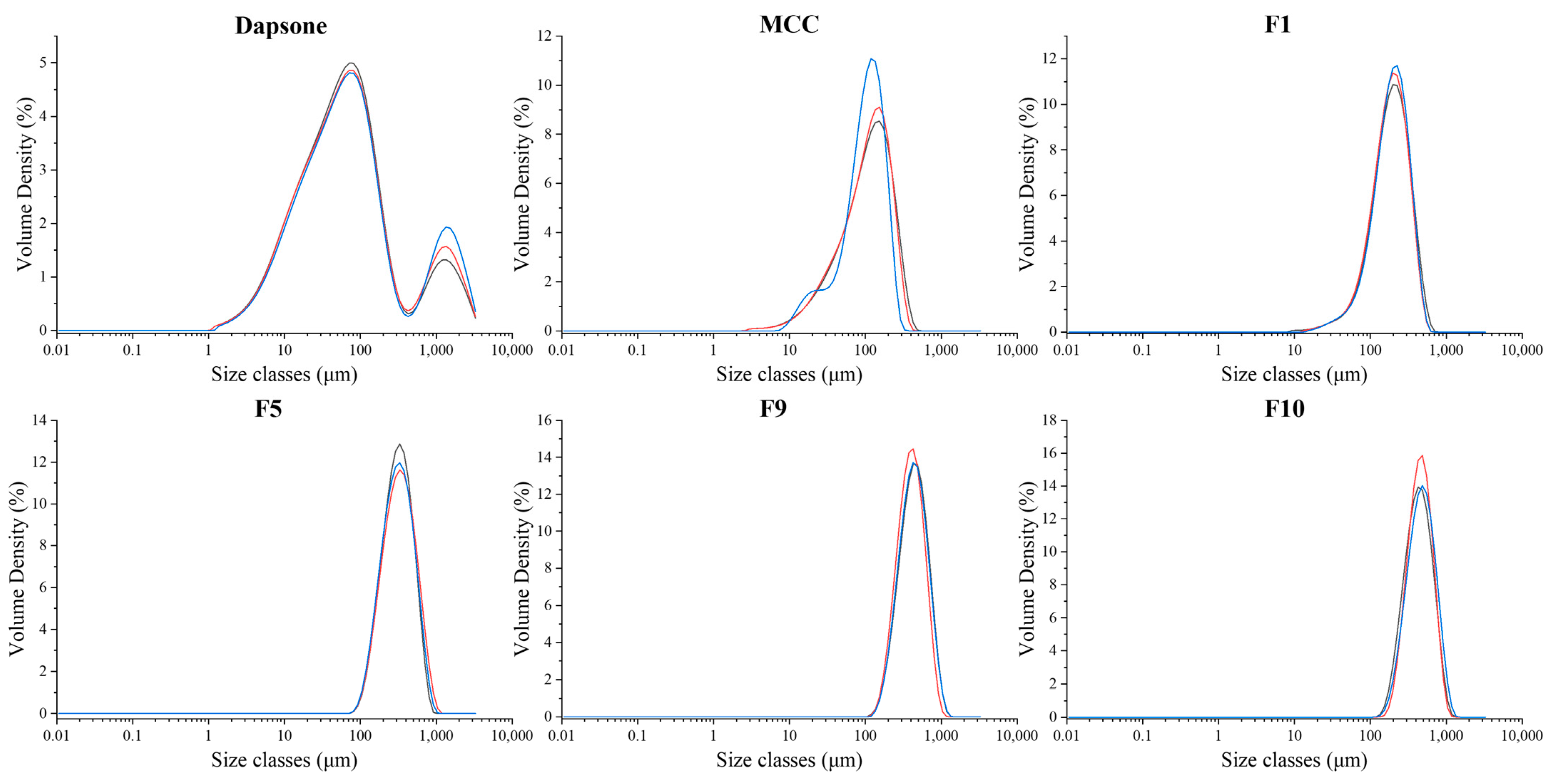
2.3.4. Particle Size Analysis
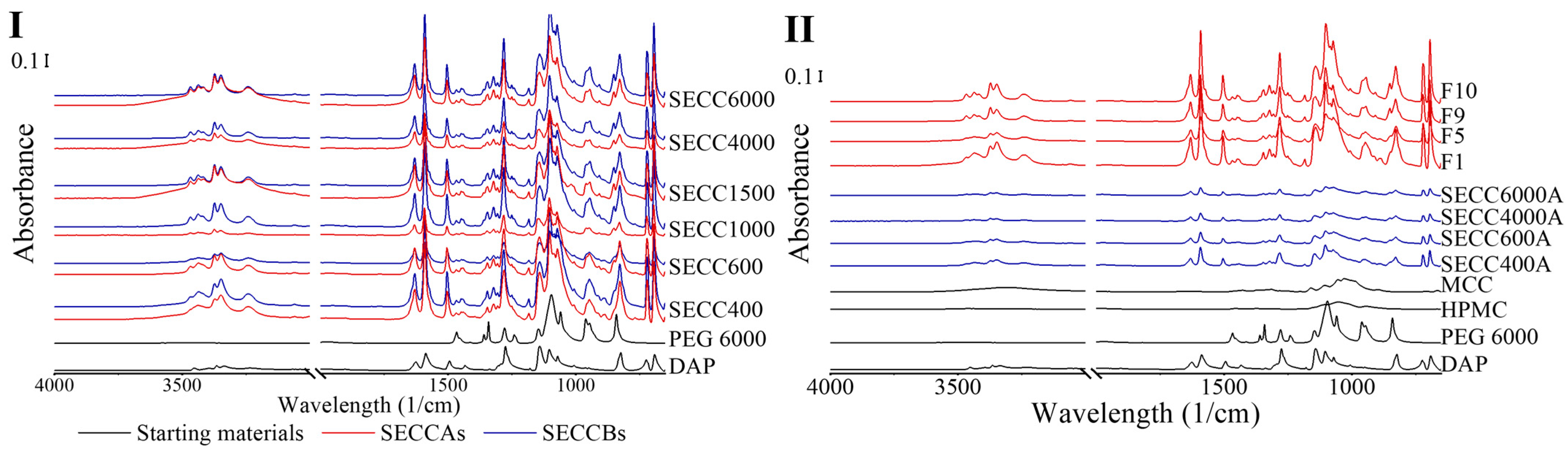
2.3.5. Fourier Transform Infrared Spectroscopy
2.3.6. Raman Spectroscopy
2.3.7. Scanning Electron Microscopy (SEM)
2.3.8. Imaging of Granules/Powder in Bulk
2.3.9. Simulated PXRD Patterns
2.3.10. Ultraviolet Spectroscopy (UV)
2.4. Micromeritic and Tableting Properties Characterization
2.4.1. Flowability Study of Granules/Powder
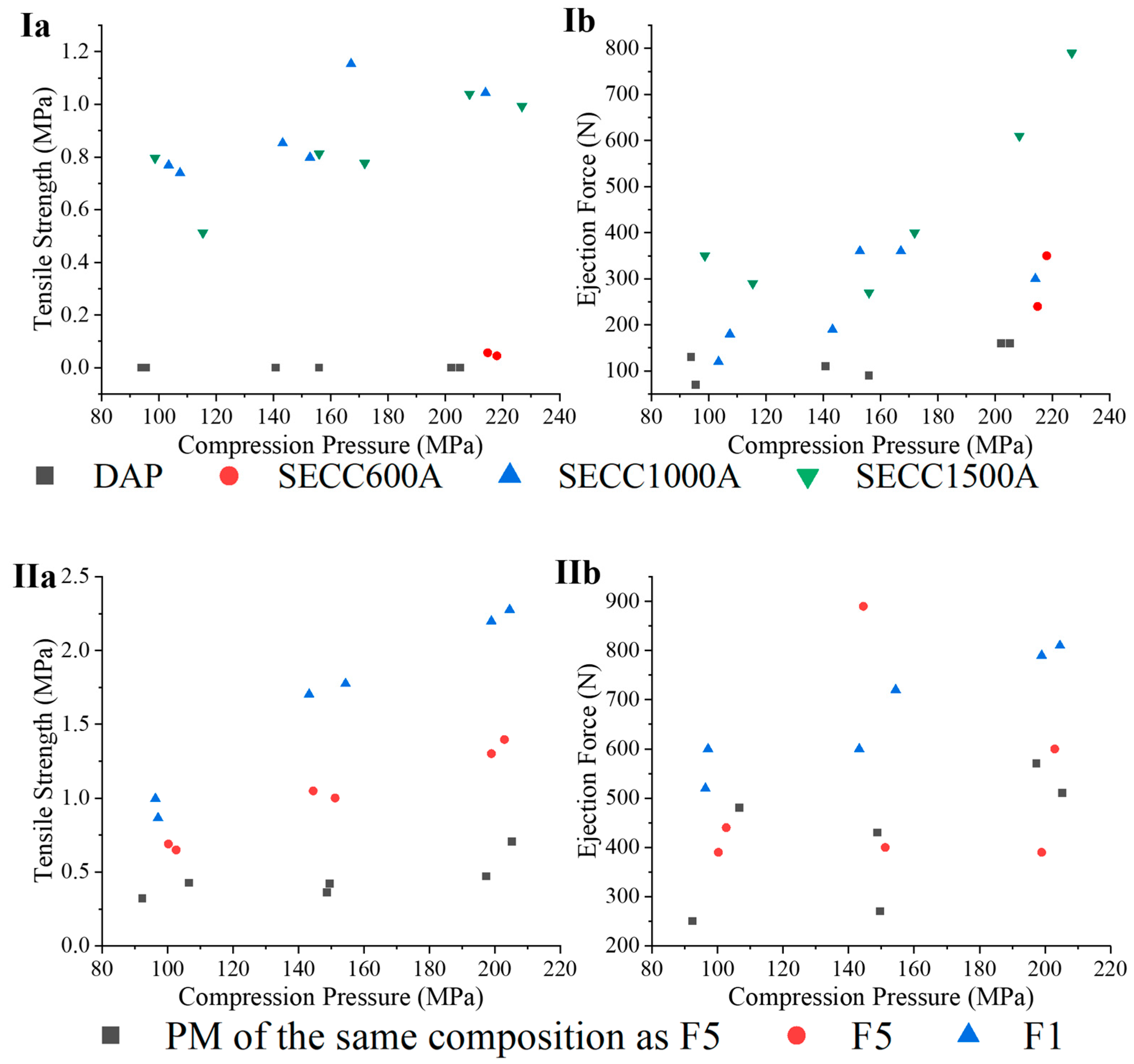
2.4.2. Tableting Study
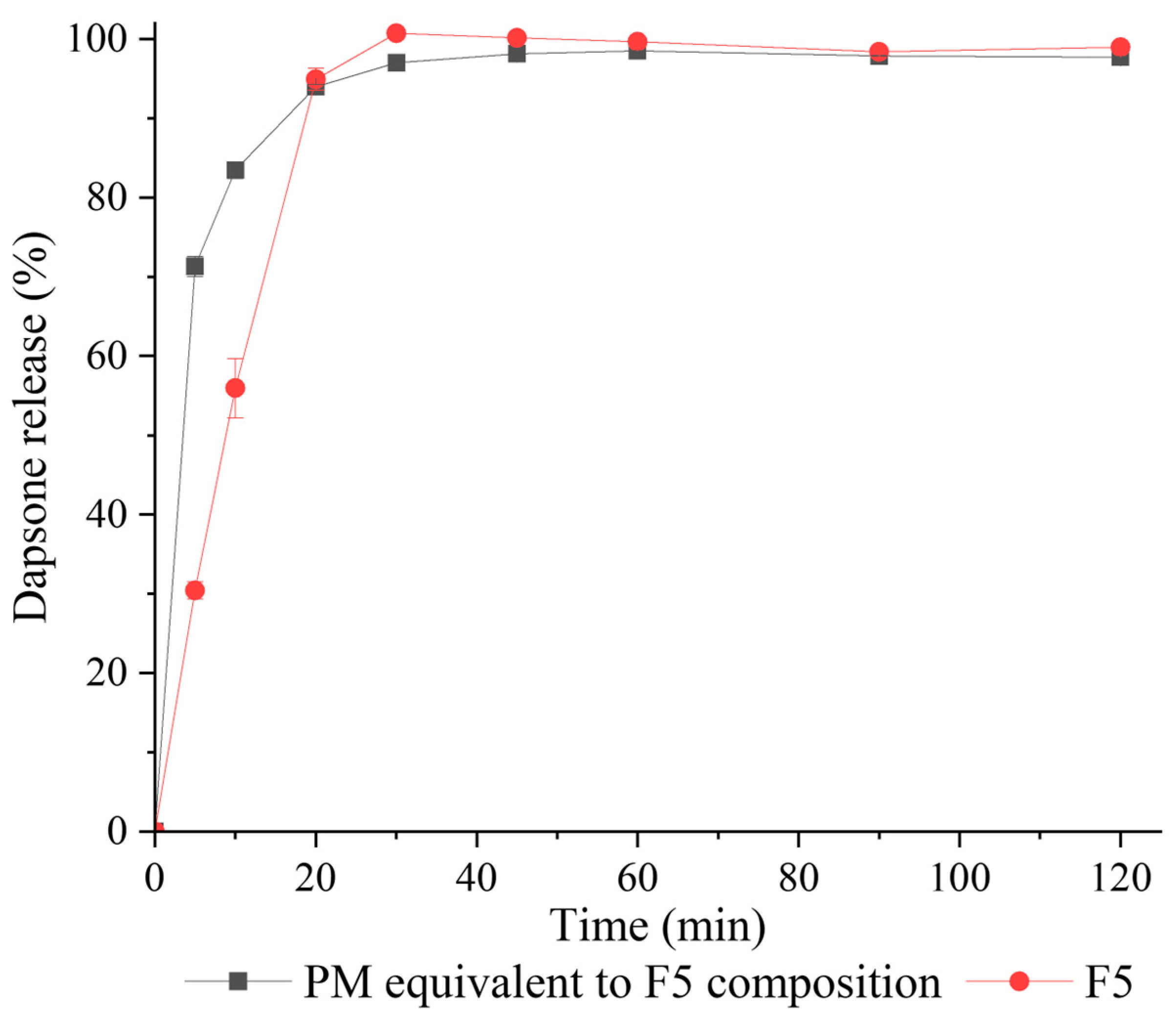
2.4.3. Dissolution Study
3. Results and Discussions
3.1. DAP-PEG Co-Crystals Generated by Solvent Evaporation
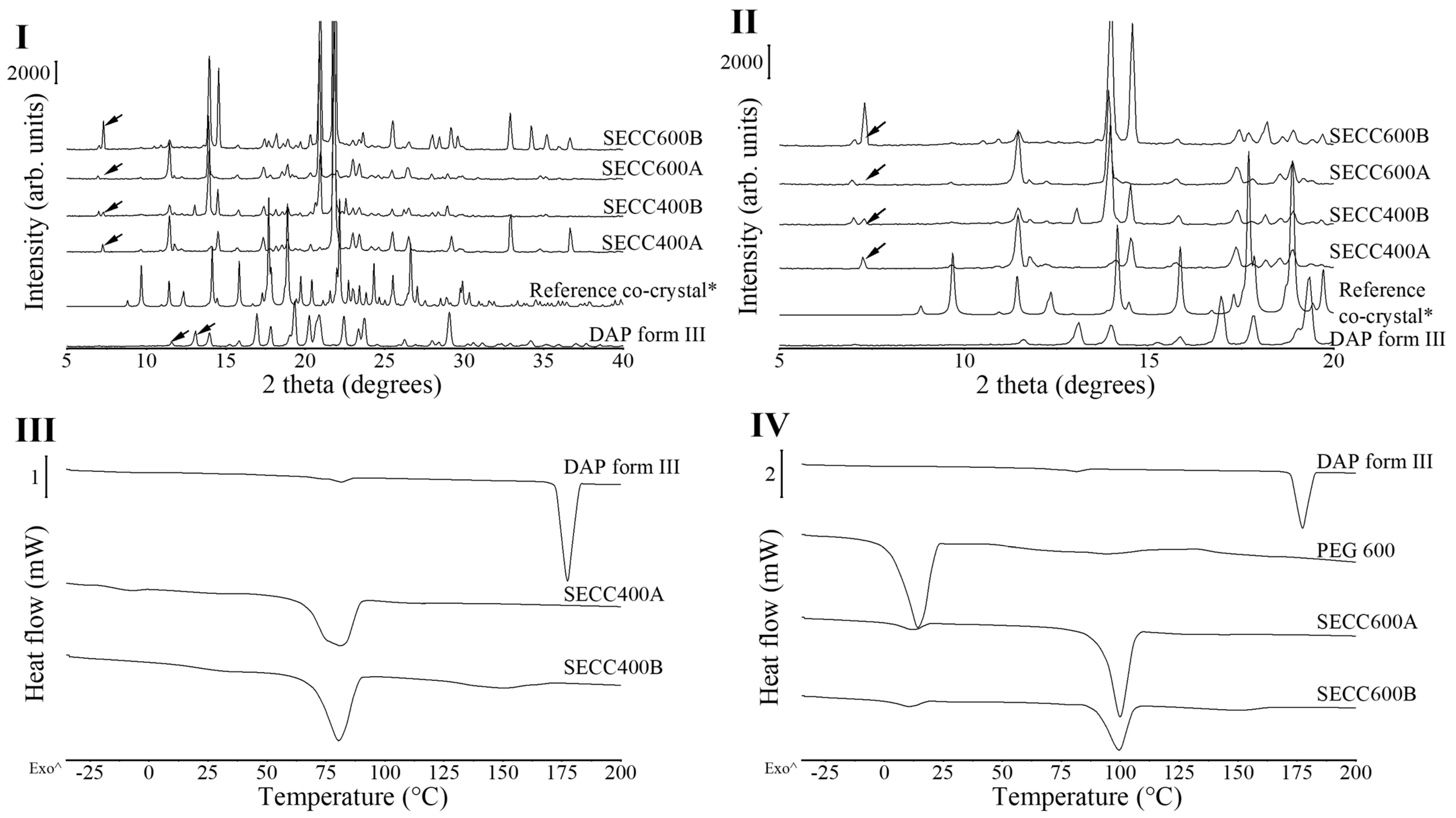
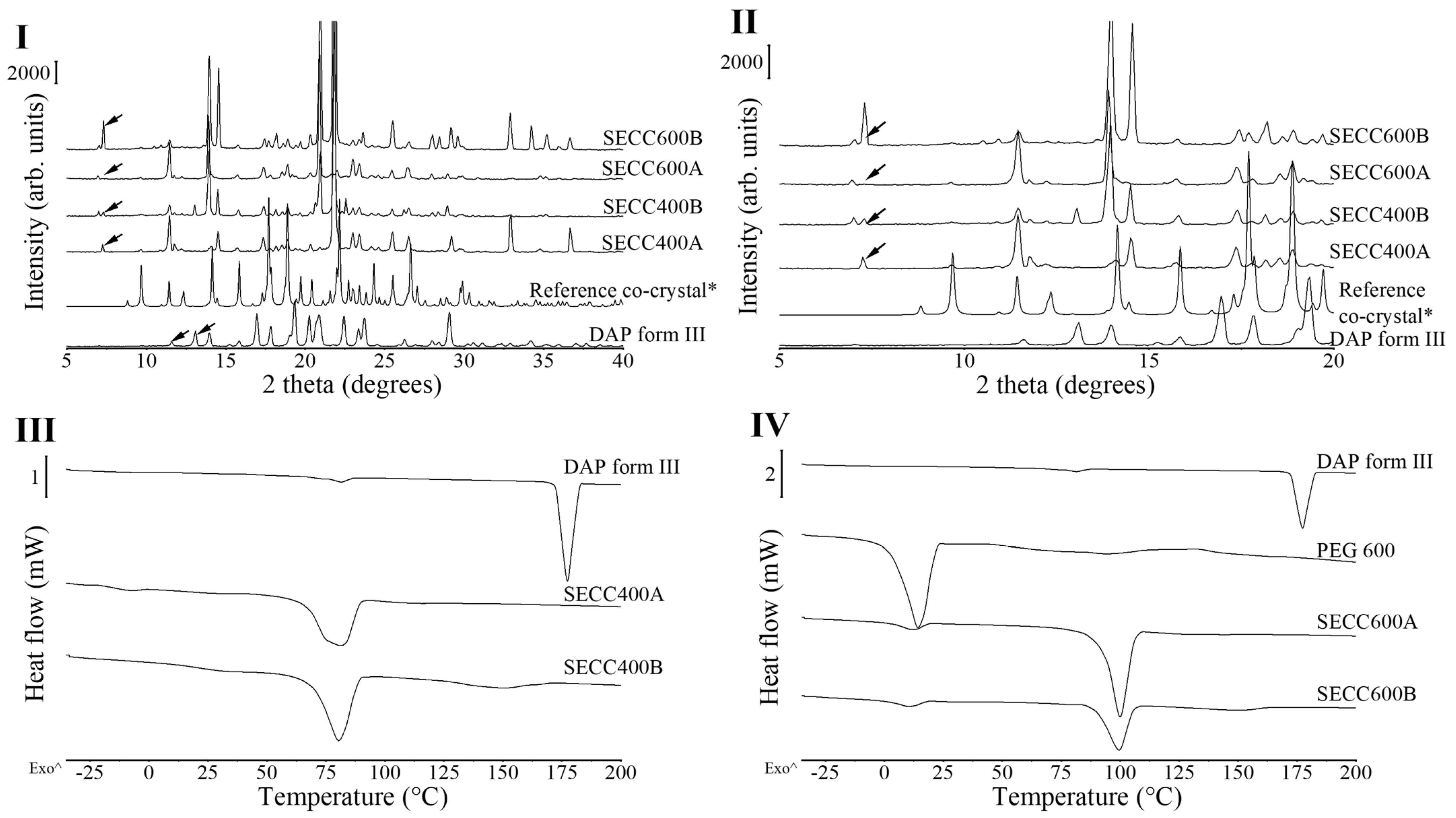
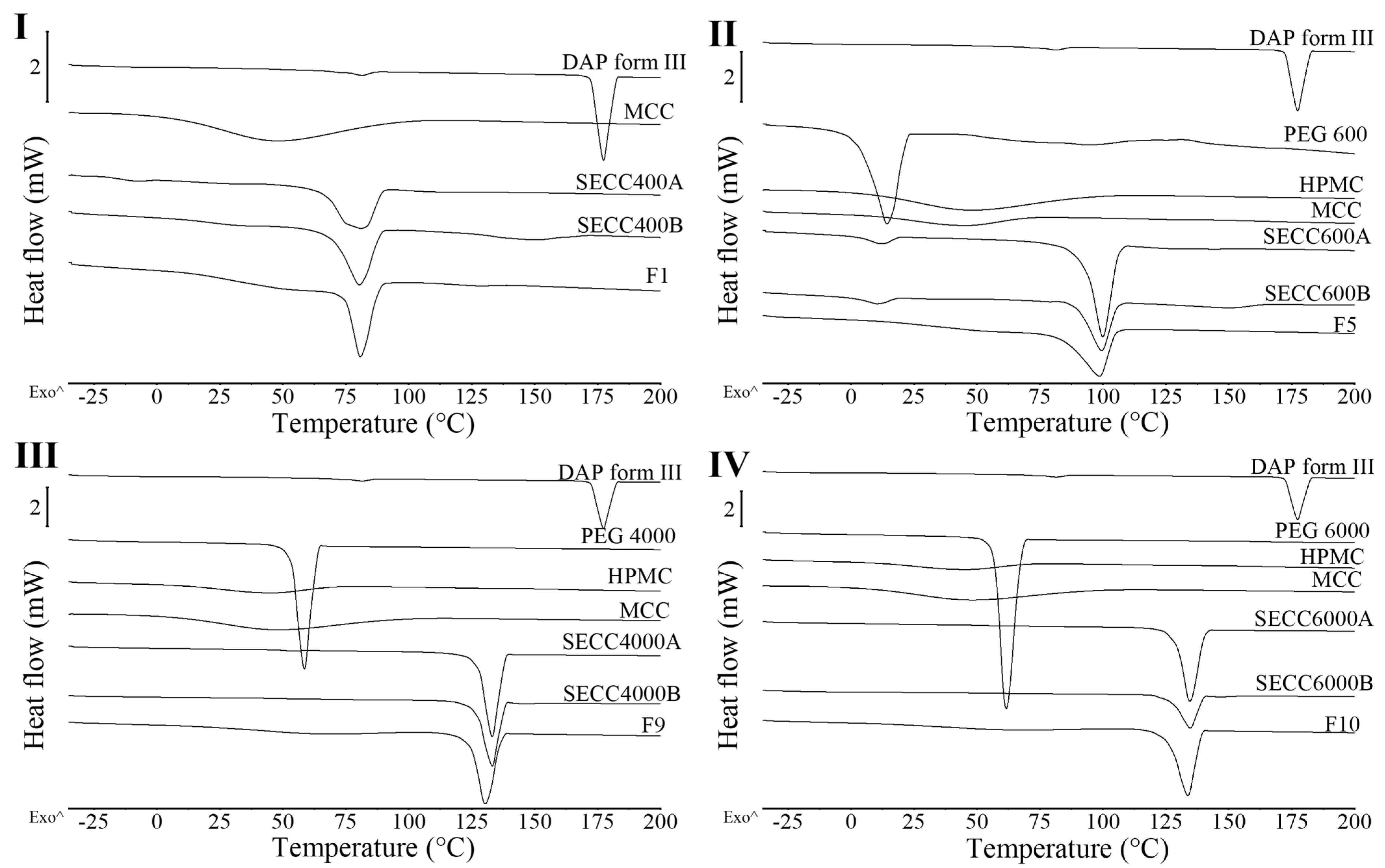
3.1.1. XRD and DSC Analysis of SECC400 and SECC600
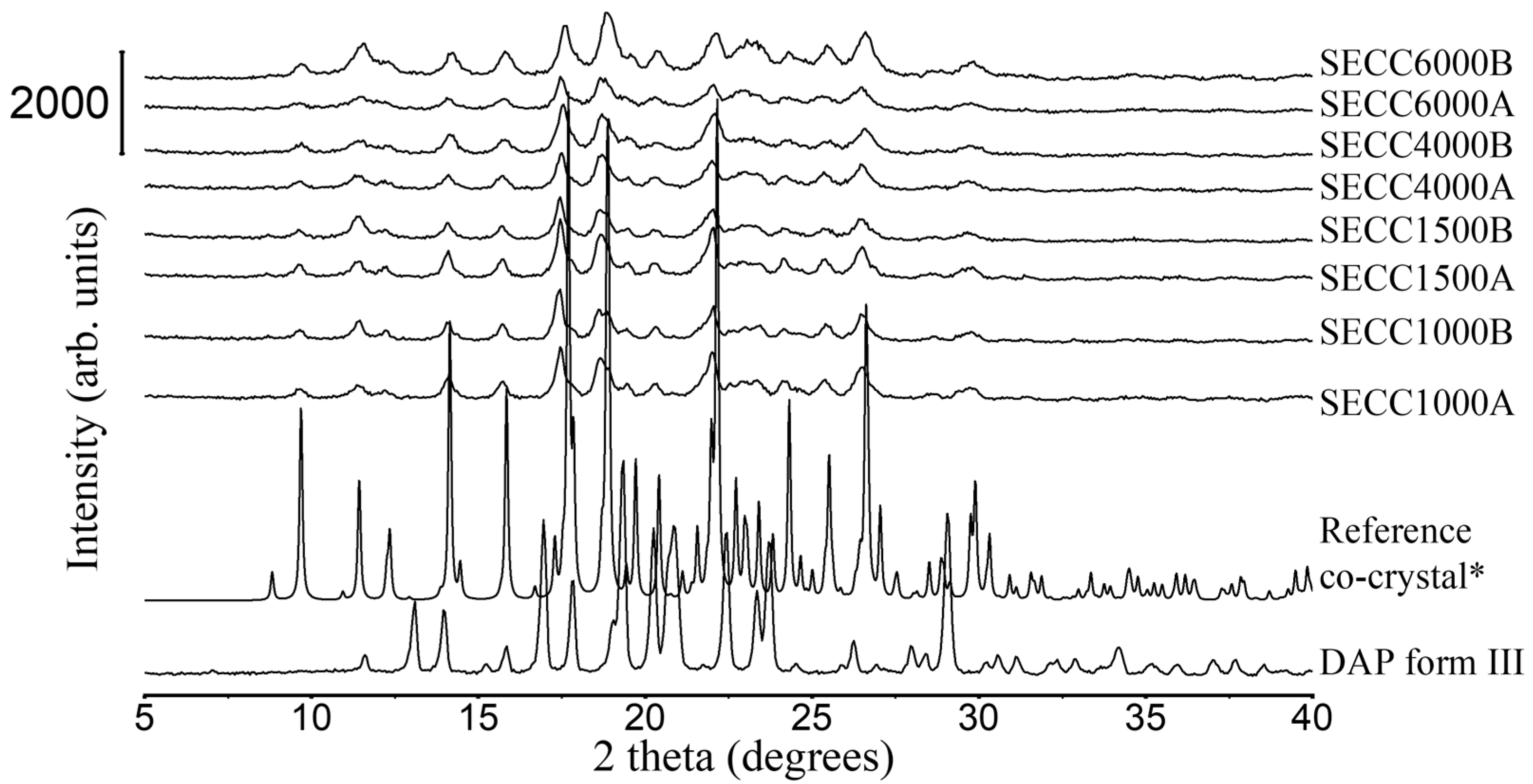
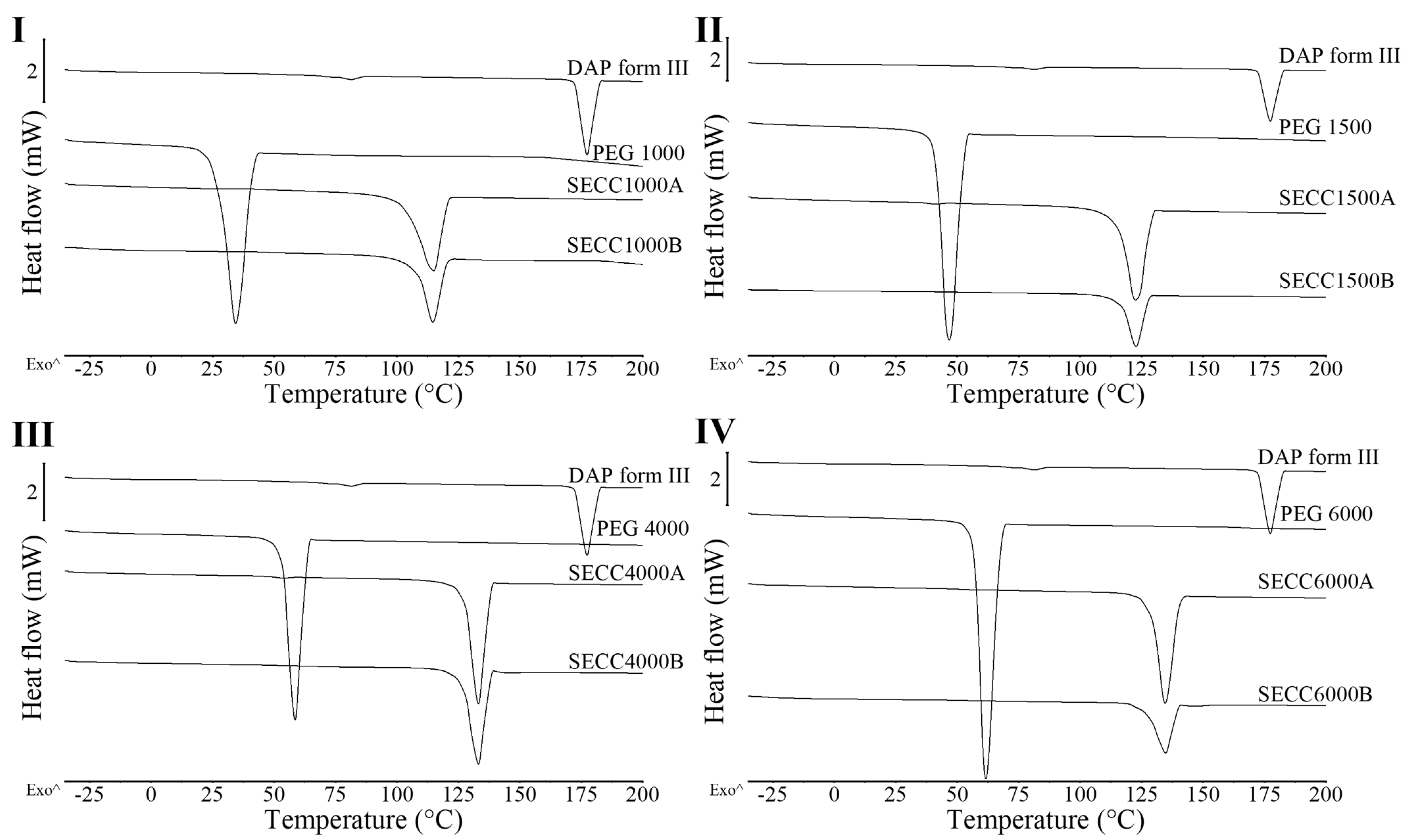
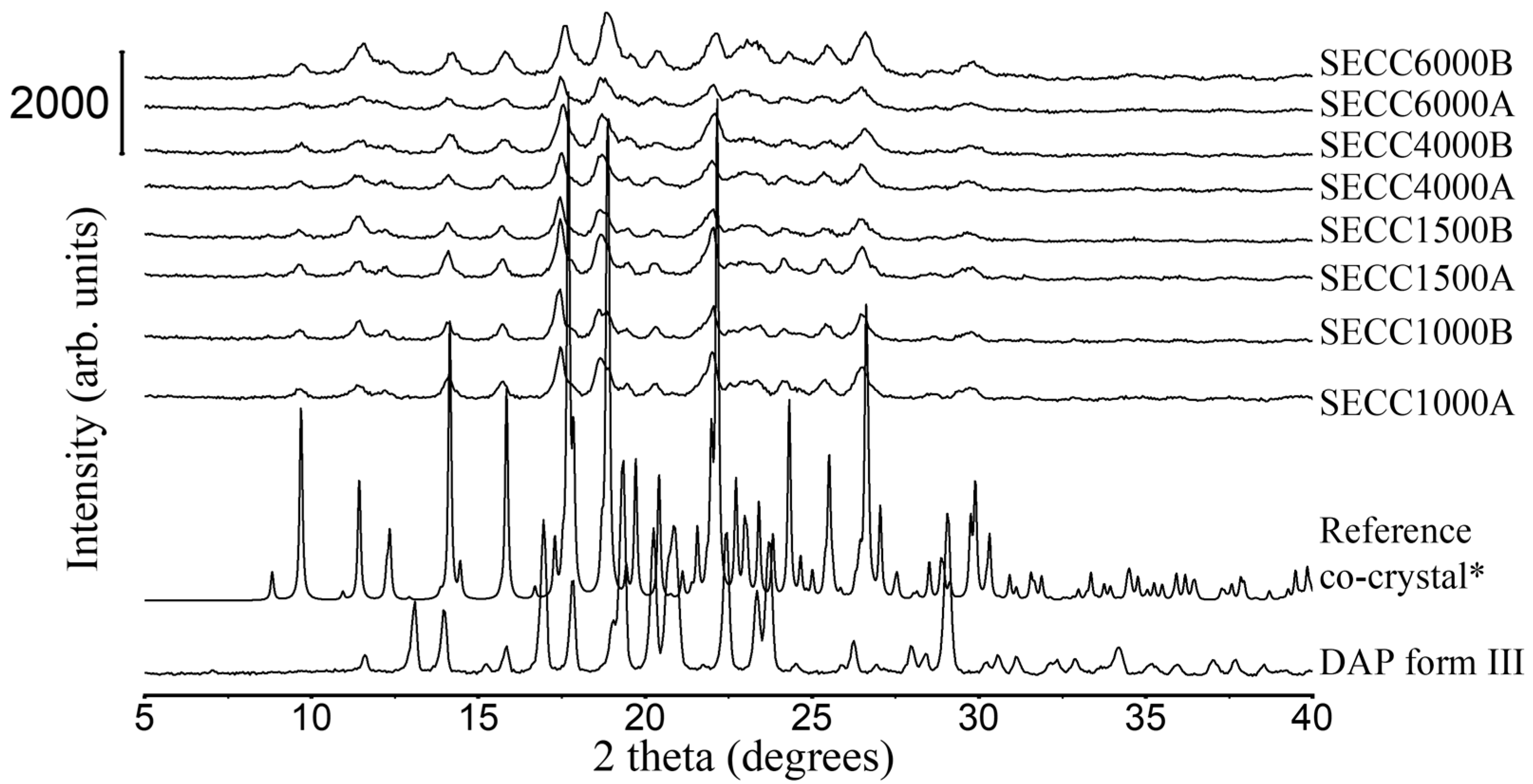
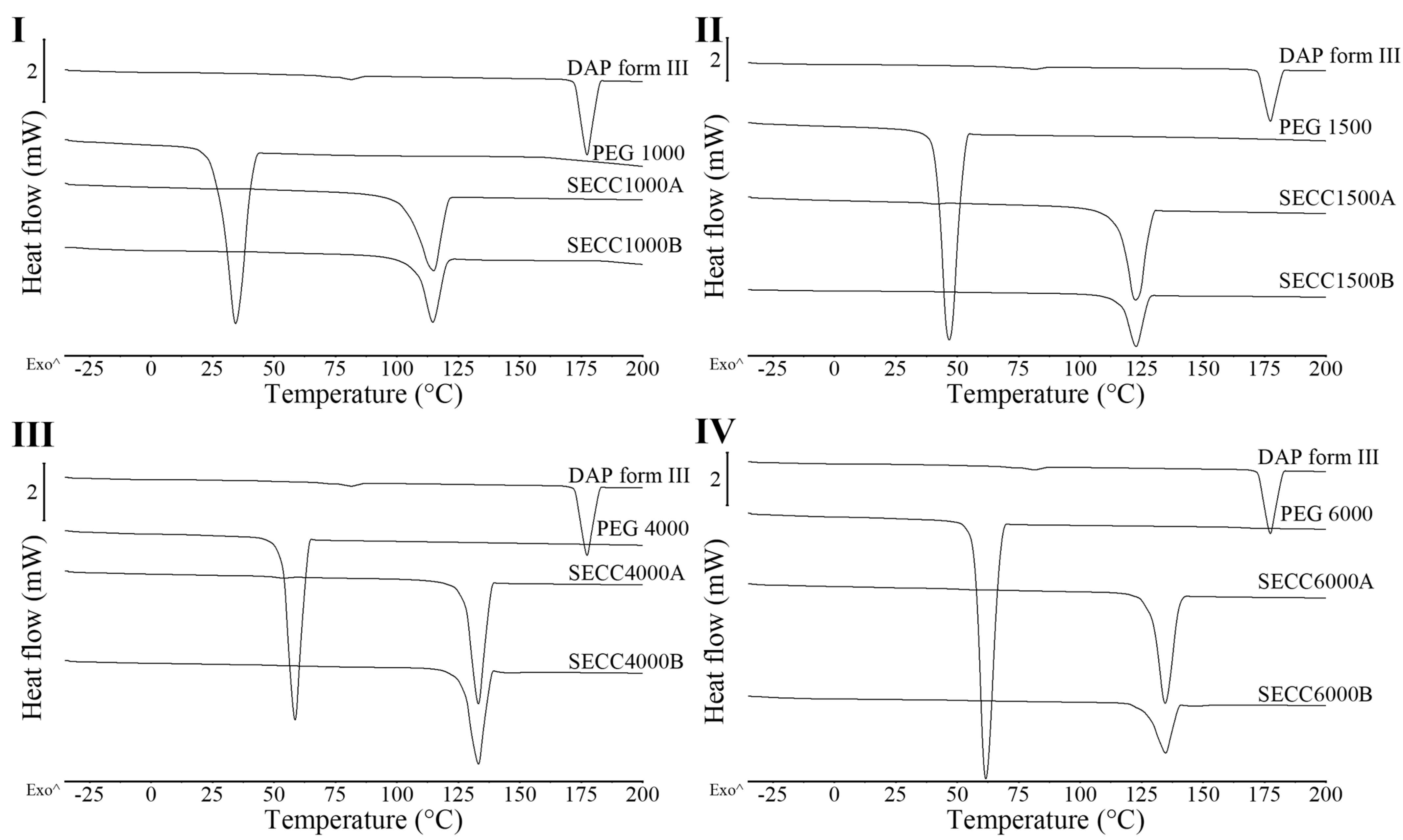
3.1.2. XRD and DSC Analysis of SECC1000, SECC1500, SECC4000, and SECC6000
3.1.3. Determination of DAP Weight Fraction in DAP-PEGs Co-Crystals
3.2. Solid-State Characterization Comparing SE Powder with FBG Granules
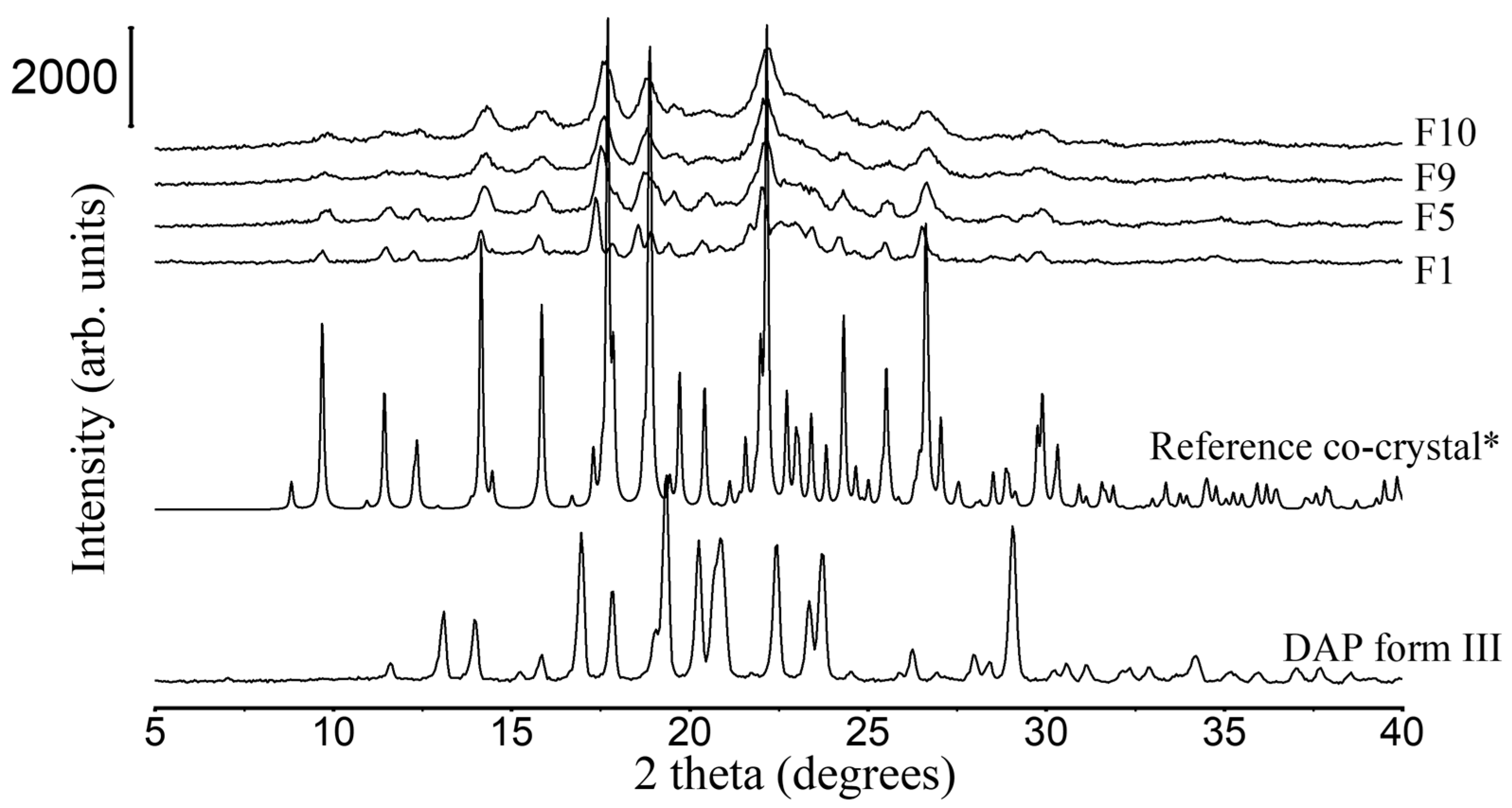
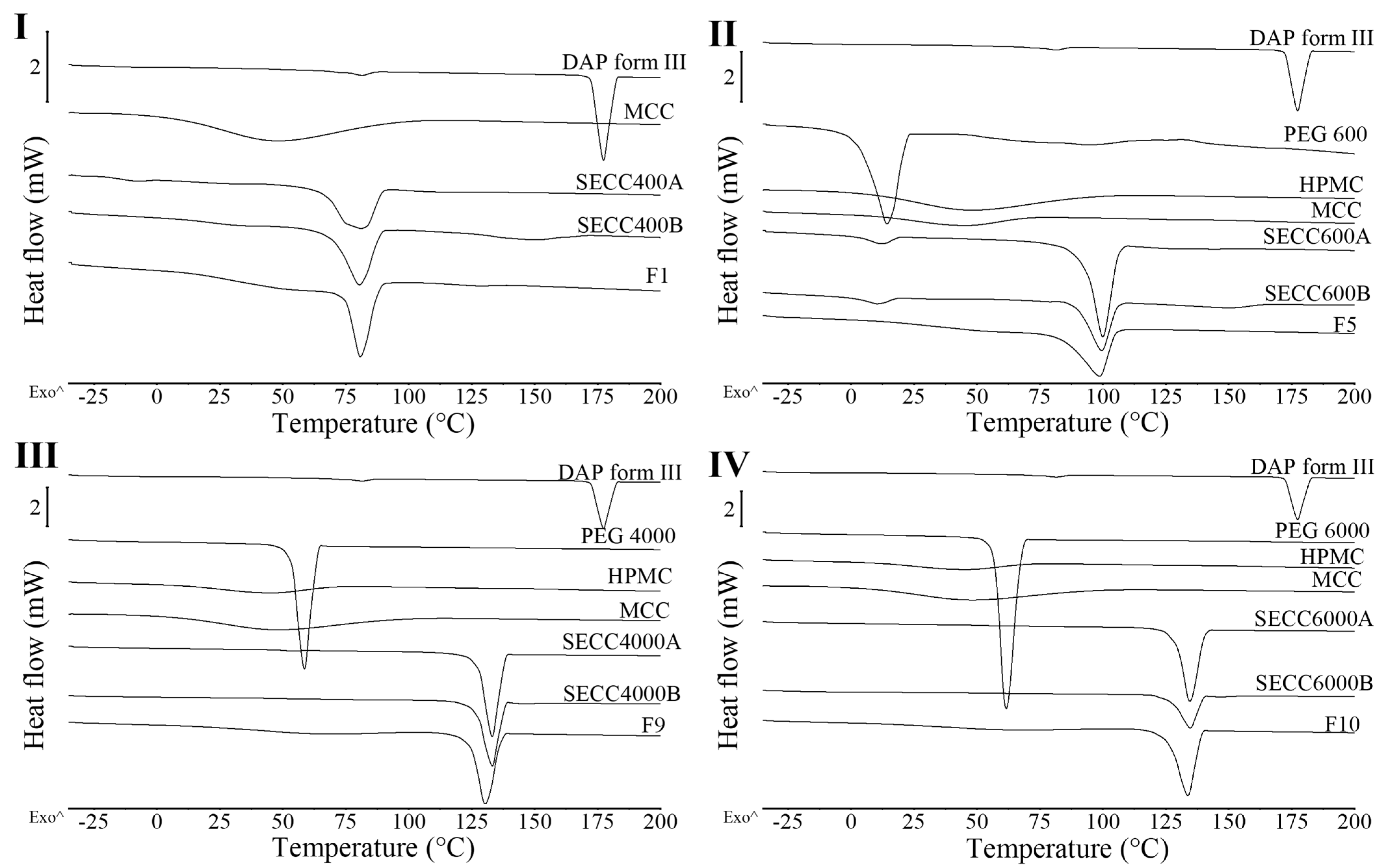
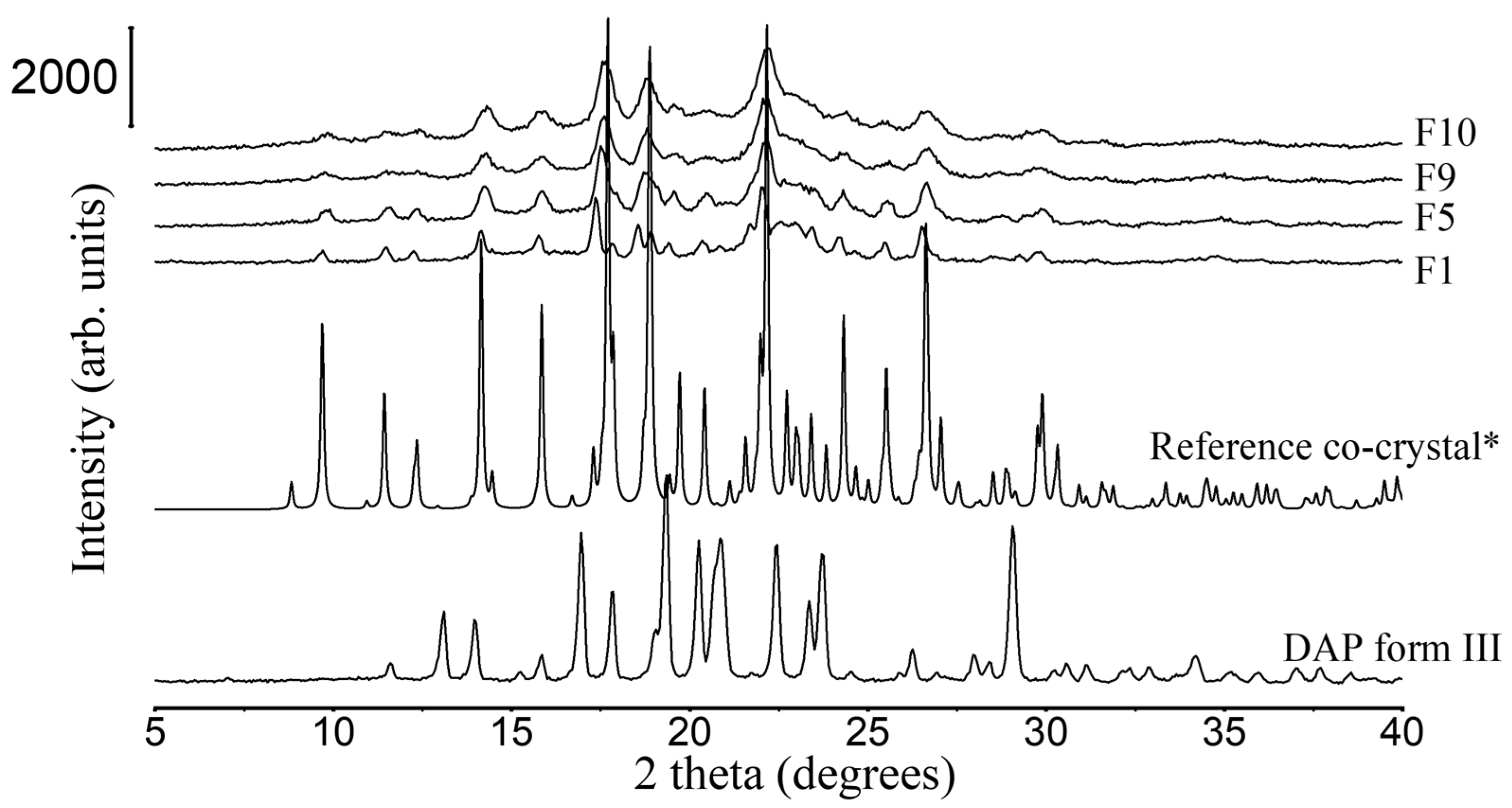
3.2.1. XRD and DSC Analysis
3.2.2. TGA Analysis
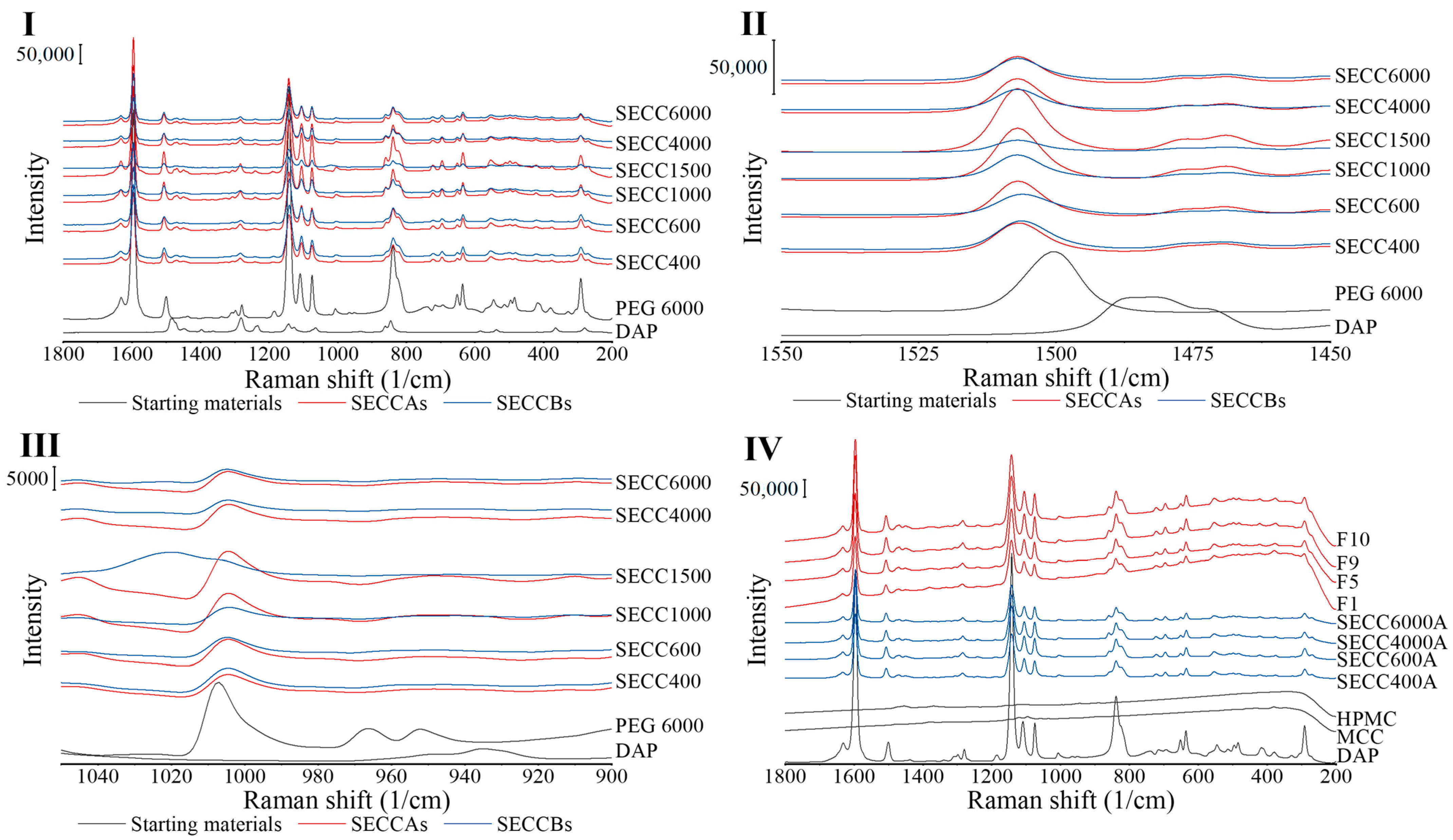
3.2.3. Raman Spectroscopy
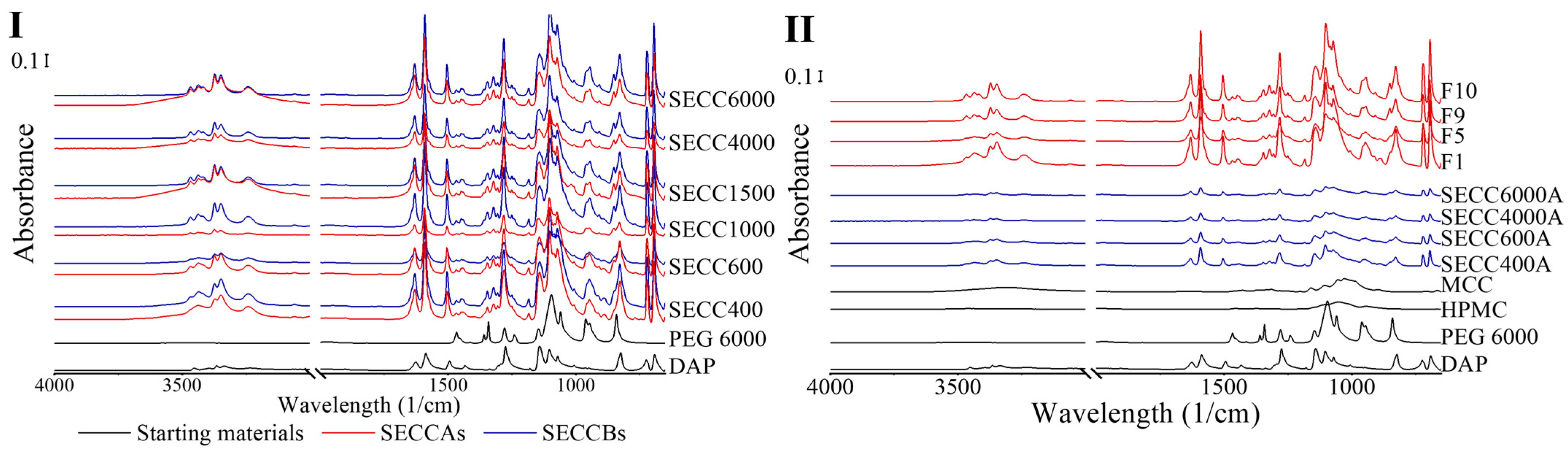
3.2.4. FTIR Spectroscopy
3.3. Pharmaceutical Properties of FBG Granules
3.3.1. Granule Drug Loading
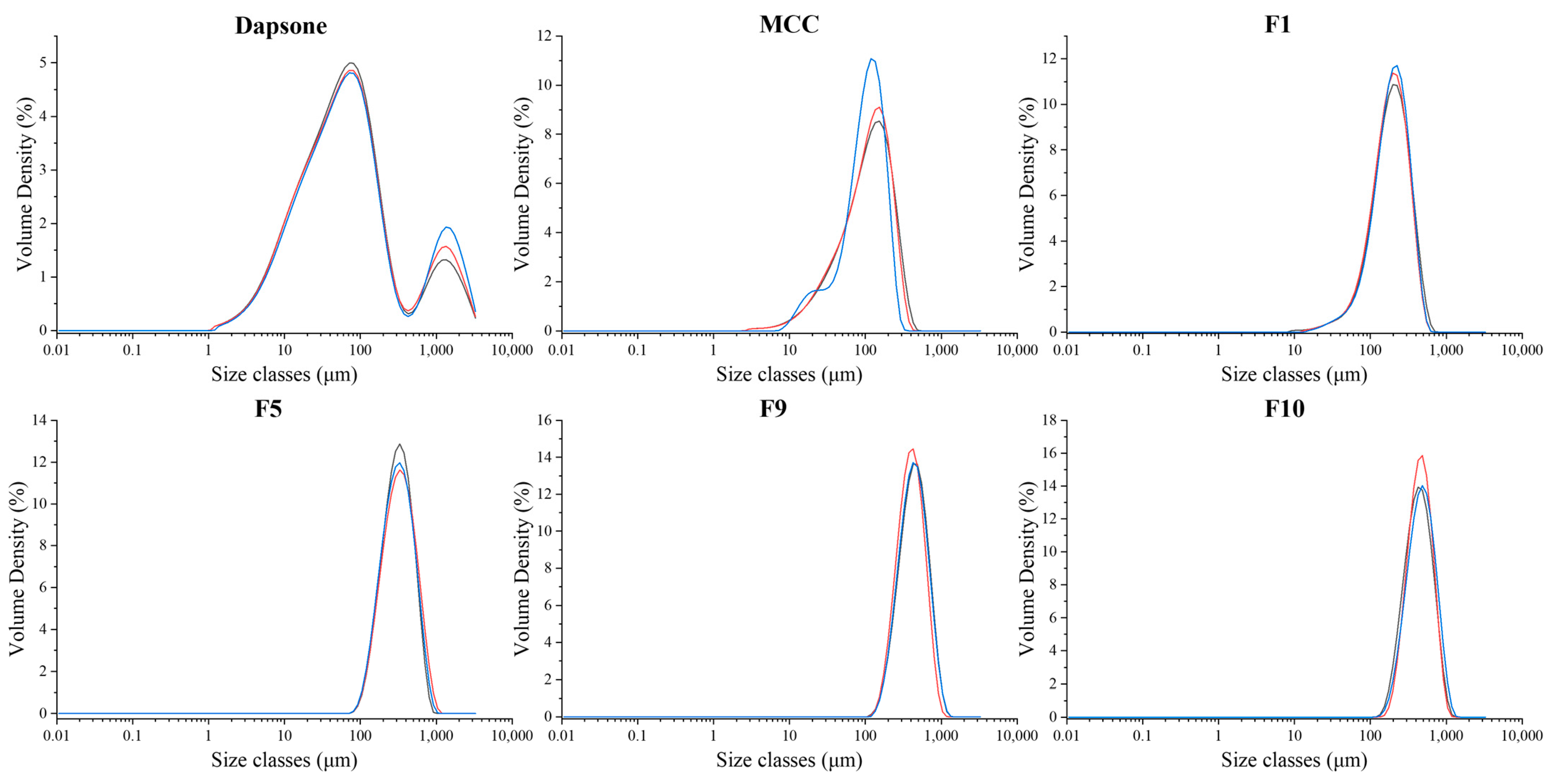
3.3.2. Particle Size Analysis
3.3.3. Imaging of Granules/Powder in Bulk
3.3.4. Morphology Study on Powder/Granules by SEM
3.3.5. Flowability of Powder/Granules
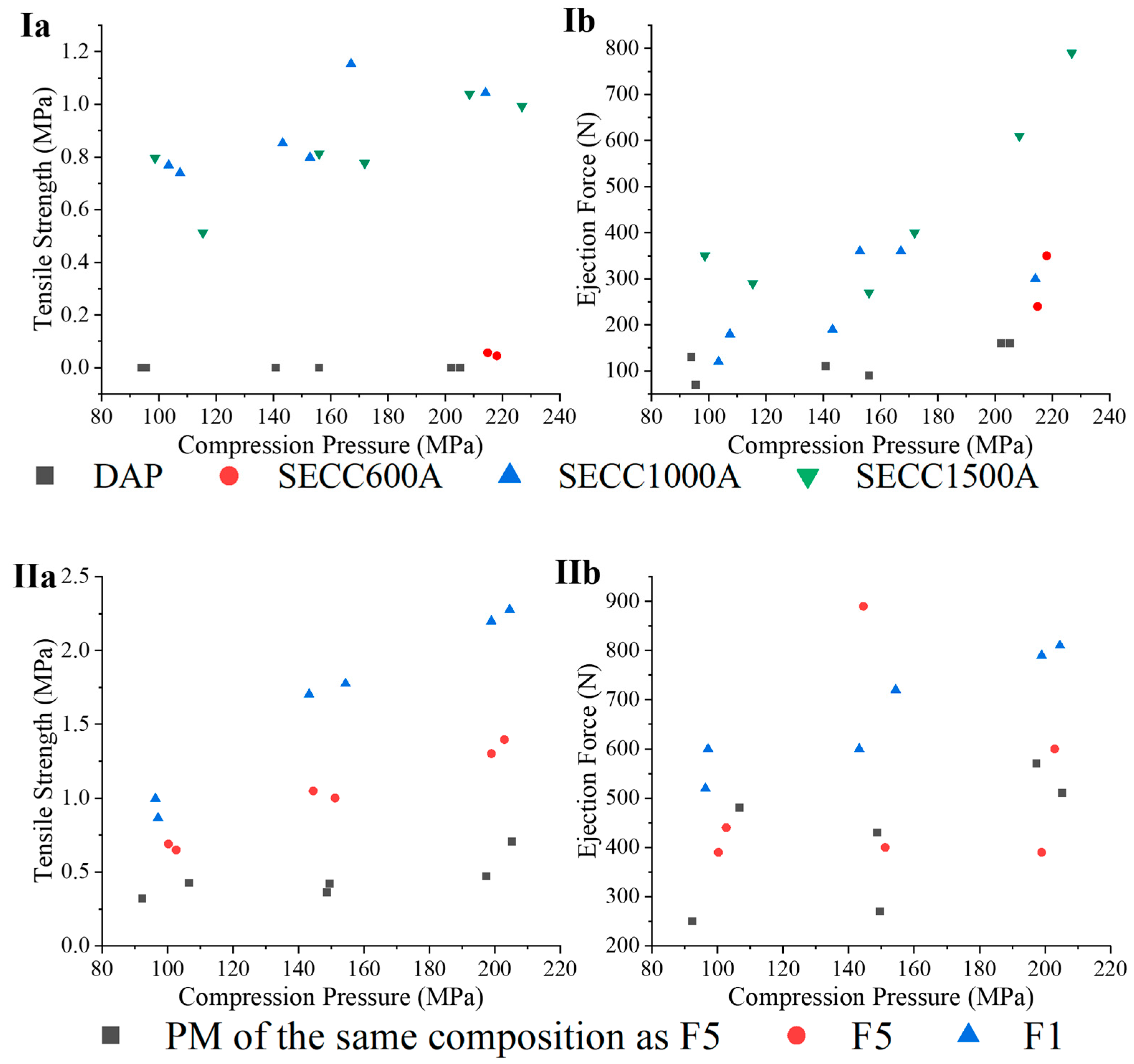
3.3.6. Tablet Mechanical Characteristics
3.3.7. Tablet Dissolution Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodríguez-Hornedo, N. Pharmaceutical Cocrystals and Poorly Soluble Drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.M.; Porter, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Trask, A.V.; Haynes, D.A.; Motherwell, W.D.S.; Jones, W. Screening for Crystalline Salts via Mechanochemistry. Chem. Commun. 2006, 1, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Das, S.; Lal, G.; Soni, S.R.; Ghosh, A.; Reddy, C.M.; Ghosh, S. Screening, Crystal Structures and Solubility Studies of a Series of Multidrug Salt Hydrates and Cocrystals of Fenamic Acids with Trimethoprim and Sulfamethazine. J. Mol. Struct. 2020, 1199, 127028. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Hu, Y.; Gniado, K.; Erxleben, A.; McArdle, P. Mechanochemical Reaction of Sulfathiazole with Carboxylic Acids: Formation of a Cocrystal, a Salt, and Coamorphous Solids. Cryst. Growth Des. 2014, 14, 803–813. [Google Scholar] [CrossRef]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical Solvates, Hydrates and Amorphous Forms: A Special Emphasis on Cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef]
- Regulatory Classification of Pharmaceutical Co-Crystals, Guidance for Industry; U.S. Department of Health & Human Services: Washington, DC, USA, 2018; pp. 1–4.
- Ngilirabanga, J.B.; Samsodien, H. Pharmaceutical Co-Crystal: An Alternative Strategy for Enhanced Physicochemical Properties and Drug Synergy. Nano Sel. 2021, 2, 512–526. [Google Scholar] [CrossRef]
- Wang, X.; Du, S.; Zhang, R.; Jia, X.; Yang, T.; Zhang, X. Drug-Drug Cocrystals: Opportunities and Challenges. Asian J. Pharm. Sci. 2021, 16, 307–317. [Google Scholar] [CrossRef]
- Zhong, Z.; Guo, C.; Chen, L.; Xu, J.; Huang, Y. Co-Crystal Formation between Poly (Ethylene Glycol) and a Small Molecular Drug Griseofulvin. Chem. Commun. 2014, 50, 6375–6378. [Google Scholar] [CrossRef]
- Sun, C.C. Novel Co-Crystals between Polyethylene Glycols and 5-Phenylpyrazolyl-1-Benzenesulfonamides. WO2006024930A1, 9 March 2006. [Google Scholar]
- Chappa, P.; Maruthapillai, A.; Voguri, R.; Dey, A.; Ghosal, S.; Basha, M.A. Drug–Polymer Co-Crystals of Dapsone and Polyethylene Glycol: An Emerging Subset in Pharmaceutical Co-Crystals. Cryst. Growth Des. 2018, 18, 7590–7598. [Google Scholar] [CrossRef]
- Ye, H.-M.; Song, Y.-Y.; Xu, J.; Guo, B.-H.; Zhou, Q. Melting Behavior of Inclusion Complex Formed between Polyethylene Glycol Oligomer and Urea. Polymer 2013, 54, 3385–3391. [Google Scholar] [CrossRef]
- Myasnikova, R.M.; Titova, E.F.; Obolonkova, E.S. Study of 2:1 Poly(Ethylene Oxide)—Resorcinol Molecular Complex. Polymer 1980, 21, 403–407. [Google Scholar] [CrossRef]
- Paternostre, L.; Damman, P.; Dosière, M. Crystalline Structure of PEO-hydroquinone and PEO-2-methylresorcinol Molecular Complexes. Macromol. Symp. 1997, 114, 205–210. [Google Scholar] [CrossRef]
- Point, J.J.; Damman, P. Structure of a New Crystalline Complex: Poly (Ethylene Oxide) with p-Nitrophenol. 1. Macromolecules 1992, 25, 1184–1188. [Google Scholar] [CrossRef]
- Évora, A.O.L.; Castro, R.A.E.; Maria, T.M.R.; Rosado, M.T.S.; Ramos Silva, M.; Matos Beja, A.; Canotilho, J.; Eusébio, M.E.S. Pyrazinamide-Diflunisal: A New Dual-Drug Co-Crystal. Cryst. Growth Des. 2011, 11, 4780–4788. [Google Scholar] [CrossRef]
- Fu, Q.; Xu, X.; Liu, B.; Guo, F. Solid State Transformations of Different Stoichiometric Forms of an Organic Salt Formed from 5-Sulfosalicylic Acid and Hexamethylenetetramine upon Dehydration and Rehydration. CrystEngComm 2018, 20, 1844–1852. [Google Scholar] [CrossRef]
- Mohamed, S.; Tocher, D.A.; Vickers, M.; Karamertzanis, P.G.; Price, S.L. Salt or Cocrystal? A New Series of Crystal Structures Formed from Simple Pyridines and Carboxylic Acids. Cryst. Growth Des. 2009, 9, 2881–2889. [Google Scholar] [CrossRef]
- Adams, C.J.; Haddow, M.F.; Lusi, M.; Orpen, A.G. Crystal Engineering of Lattice Metrics of Perhalometallate Salts and MOFs. Proc. Natl. Acad. Sci. USA 2010, 107, 16033–16038. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Velaga, S.P. Formation of Cocrystals from Stoichiometric Solutions of Incongruently Saturating Systems by Spray Drying. Cryst. Growth Des. 2010, 10, 3302–3305. [Google Scholar] [CrossRef]
- Panzade, P.S.; Shendarkar, G.R.; Kulkarni, D.A. Hot Melt Extrusion: An Emerging Green Technique for the Synthesis of High-Quality Pharmaceutical Cocrystals. J. Pharm. Innov. 2020, 17, 283–293. [Google Scholar] [CrossRef]
- Todaro, V.; Worku, Z.A.; Cabral, L.M.; Healy, A.M. In Situ Cocrystallization of Dapsone and Caffeine during Fluidized Bed Granulation Processing. AAPS PharmSciTech 2019, 20, 28. [Google Scholar] [CrossRef]
- Todaro, V.; Healy, A.M. Development and Characterization of Ibuprofen Co-Crystals Granules Prepared via Fluidized Bed Granulation in a One-Step Process—A Design of Experiment Approach. Drug Dev. Ind. Pharm. 2021, 47, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Gajda, M.; Nartowski, K.P.; Pluta, J.; Karolewicz, B. The Role of the Polymer Matrix in Solvent-Free Hot Melt Extrusion Continuous Process for Mechanochemical Synthesis of Pharmaceutical Cocrystal. Eur. J. Pharm. Biopharm. 2018, 131, 48–59. [Google Scholar] [CrossRef]
- Serrano, D.R.; Walsh, D.; O’Connell, P.; Mugheirbi, N.A.; Worku, Z.A.; Bolas-Fernandez, F.; Galiana, C.; Dea-Ayuela, M.A.; Healy, A.M. Optimising the in Vitro and in Vivo Performance of Oral Cocrystal Formulations via Spray Coating. Eur. J. Pharm. Biopharm. 2018, 124, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Fukui, K.; Otaka, K.; Suzuki, T.; Fukami, T. Monitoring of Cocrystal Dissociation during the Wet Granulation Process in the Presence of Disintegrants by Using Low-Frequency Raman Spectroscopy. Chem. Pharm. Bull. 2021, 69, 877–885. [Google Scholar] [CrossRef]
- De, A.; Dey, S.; Pradhan, P.; Chaudhari, F.U.; Patel, M. Estimation of dapsone in bulk & dosage form by uv spectroscopic method. Indo Am. J. Pharm. Res. 2014, 4, 312–319. [Google Scholar]
- British Pharmacopoeia. Volume III. Dapsone Tablets. Available online: https://www.pharmacopoeia.com/ (accessed on 14 March 2022).
- Craig, D.Q.M. Polyethyelene Glycols and Drug Release. Drug Dev. Ind. Pharm. 1990, 16, 2501–2526. [Google Scholar] [CrossRef]
- Braun, D.E.; Vickers, M.; Griesser, U.J. Dapsone Form V: A Late Appearing Thermodynamic Polymorph of a Pharmaceutical. Mol. Pharm. 2019, 16, 3221–3236. [Google Scholar] [CrossRef]
- Rouiller, M.; Gurny, R.; Doelker, E. Possibilites de Production Avec Un Appareil a Lit Fluidise de Laboratoire. Acta Pharm. Technol. 1975, 21, 129–138. [Google Scholar]
- Gao, J.Z.H.; Jain, A.; Motheram, R.; Gray, D.B.; Hussain, M.A. Fluid Bed Granulation of a Poorly Water Soluble, Low Density, Micronized Drug: Comparison with High Shear Granulation. Int. J. Pharm. 2002, 237, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rohera, B.D.; Zahir, A. Granulations in a Fluidized-Bed: Effect of Binders and Their Concentrations on Granule Growth and Modeling the Relationship Between Granule Size and Binder Concentration. Drug Dev. Ind. Pharm. 1993, 19, 773–792. [Google Scholar] [CrossRef]
- Davies, W.L.; Gloor, W.T. Batch Production of Pharmaceutical Granulations in a Fluidized Bed I: Effects of Process Variables on Physical Properties of Final Granulation. J. Pharm. Sci. 1971, 60, 1869–1874. [Google Scholar] [CrossRef]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of Indomethacin from the Amorphous State below and above Its Glass Transition Temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef]
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; Van den Mooter, G.; Rades, T. Refining Stability and Dissolution Rate of Amorphous Drug Formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef]
- Wouters, J.; Quéré, L. Pharmaceutical Salts and Co-Crystals; Wouters, J., Quéré, L., Eds.; Drug Discovery; The Royal Society of Chemistry: London, UK, 2012; ISBN 978-1-84973-158-4. [Google Scholar]
- Aakeröy, C.B.; Sinha, A.S. Co-Crystals: Preparation, Characterization and Applications; Royal Society of Chemistry: London, UK, 2018; Volume 24, ISBN 1788011155. [Google Scholar]
- Ervasti, T.; Aaltonen, J.; Ketolainen, J. Theophylline–Nicotinamide Cocrystal Formation in Physical Mixture during Storage. Int. J. Pharm. 2015, 486, 121–130. [Google Scholar] [CrossRef]
- Lin, H.-L.; Zhang, G.-C.; Hsu, P.-C.; Lin, S.-Y. A Portable Fiber-Optic Raman Analyzer for Fast Real-Time Screening and Identifying Cocrystal Formation of Drug-Coformer via Grinding Process. Microchem. J. 2013, 110, 15–20. [Google Scholar] [CrossRef]
- Lukin, S.; Užarević, K.; Halasz, I. Raman Spectroscopy for Real-Time and in Situ Monitoring of Mechanochemical Milling Reactions. Nat. Protoc. 2021, 16, 3492–3521. [Google Scholar] [CrossRef]
- Carneiro, R.L.; de Melo, C.C.; de Alvarenga, B.R.; Dayo Owoyemi, B.C.; Ellena, J.; da Silva, C.C.P. Mechanochemical Synthesis and Characterization of a Novel AAs–Flucytosine Drug–Drug Cocrystal: A Versatile Model System for Green Approaches. J. Mol. Struct. 2022, 1251, 132052. [Google Scholar] [CrossRef]
- Yamini, D.; Devanand Venkatasubbu, G.; Kumar, J.; Ramakrishnan, V. Raman Scattering Studies on PEG Functionalized Hydroxyapatite Nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 117, 299–303. [Google Scholar] [CrossRef] [PubMed]
- D’Cunha, R.; Kartha, V.B.; Gurnani, S. Raman and i.r. Studies of the Antileprotic Drug Dapsone. Spectrochim. Acta Part A Mol. Spectrosc. 1983, 39, 331–336. [Google Scholar] [CrossRef]
- Khan, E.; Shukla, A.; Jadav, N.; Telford, R.; Ayala, A.P.; Tandon, P.; Vangala, V.R. Study of Molecular Structure, Chemical Reactivity and H-Bonding Interactions in the Cocrystal of Nitrofurantoin with Urea. New J. Chem. 2017, 41, 11069–11078. [Google Scholar] [CrossRef]
- Zhao, C.; Li, W.; Li, Z.; Hu, W.; Zhang, S.; Wu, S. Preparation and Solid-State Characterization of Dapsone Pharmaceutical Cocrystals through the Supramolecular Synthon Strategy. CrystEngComm 2021, 23, 6690–6702. [Google Scholar] [CrossRef]
- Hemati, M.; Cherif, R.; Saleh, K.; Pont, V. Fluidized Bed Coating and Granulation: Influence of Process-Related Variables and Physicochemical Properties on the Growth Kinetics. Powder Technol. 2003, 130, 18–34. [Google Scholar] [CrossRef]
- Shah, R.B.; Tawakkul, M.A.; Khan, M.A. Comparative Evaluation of Flow for Pharmaceutical Powders and Granules. AAPS PharmSciTech 2008, 9, 250–258. [Google Scholar] [CrossRef]
- Sharma, R.; Setia, G. Mechanical Dry Particle Coating on Cohesive Pharmaceutical Powders for Improving Flowability—A Review. Powder Technol. 2019, 356, 458–479. [Google Scholar] [CrossRef]
- Deng, T.; Paul, K.A.; Bradley, M.S.A.; Immins, L.; Preston, C.; Scott, J.F.; Welfare, E.H. Investigations on Air Induced Segregation of Pharmaceutical Powders and Effect of Material Flow Functions. Powder Technol. 2010, 203, 354–358. [Google Scholar] [CrossRef]
- Otsuka, T.; Iwao, Y.; Miyagishima, A.; Itai, S. Application of Principal Component Analysis Enables to Effectively Find Important Physical Variables for Optimization of Fluid Bed Granulator Conditions. Int. J. Pharm. 2011, 409, 81–88. [Google Scholar] [CrossRef]
- Powder Flow; United States Pharmacopeia: North Bethesda, MD, USA, 2013. [CrossRef]
- Kristensen, J.; Hansen, V.W. Wet Granulation in Rotary Processor and Fluid Bed: Comparison of Granule and Tablet Properties. AAPS PharmSciTech 2006, 7, E153–E162. [Google Scholar] [CrossRef] [PubMed]
- British Pharmacopoeia. Appendix XII. Dissolution. Available online: https://www.pharmacopoeia.com/ (accessed on 14 March 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | PEG MW Used | DAP:PEG Ratio (w/w) |
|---|---|---|
| SECC400A | 400 | 55.0:45.0 |
| SECC400B | 400 | 57.5:42.5 |
| SECC600A | 600 | 55.0:45.0 |
| SECC600B | 600 | 57.5:42.5 |
| SECC1000A | 1000 | 55.0:45.0 |
| SECC1000B | 1000 | 57.5:42.5 |
| SECC1500A | 1500 | 55.0:45.0 |
| SECC1500B | 1500 | 57.5:42.5 |
| SECC4000A | 4000 | 55.0:45.0 |
| SECC4000B | 4000 | 57.5:42.5 |
| SECC6000A | 6000 | 55.0:45.0 |
| SECC6000B | 6000 | 57.5:42.5 |
| FBG Process Variables | Airflow Rate (m3/h) | Temperature (°C) | Atomization (bar) | Liquid Feed Rate (mL/min) | Blowback Cycle |
|---|---|---|---|---|---|
| P1 | 20 | 30 | 0.5 | 7.5 | 10 |
| P2 | 20 | 40 | 0.5 | 3.0 | 10 |
| Formulation Code | PEG MW | [Filler (MCC) + Binder (HPMC)]: (DAP+PEG) (w/w) | Solvent Volume (mL) | FBG Process Variables | Granulation Outcome |
|---|---|---|---|---|---|
| F1 | PEG 400 | [70 + 0]:(30) | 30 | P1 | Y |
| F2 | PEG 600 | [70 + 0]:(30) | 200 | P1 | N |
| F3 | PEG 400 | [70 + 0]:(30) | 30 | P2 | N |
| F4 | PEG 400 | [65 + 5]:(30) | 100 | P2 | N |
| F5 | PEG 600 | [65 + 5]:(30) | 200 | P2 | Y |
| F6 | PEG 600 | [65 + 5]:(30) | 500 | P2 | N |
| F7 | PEG 1000 | [65 + 5]:(30) | 200 | P2 | N |
| F8 | PEG 1500 | [65 + 5]:(30) | 200 | P2 | N |
| F9 | PEG 4000 | [65 + 5]:(30) | 500 | P2 | Y |
| F10 | PEG 6000 | [65 + 5]:(30) | 1000 | P2 | Y |
| Formulation Code | PEG MW | [Filler (MCC) + Binder (HPMC)]: (DAP) (w/w) | Solvent Volume (mL) | FBG Process Variables | Granulation Outcome |
| F11 | N/A * | [65 + 5]:(30) | 100 | P2 | N |
| F12 | N/A * | [65 + 5]:(30) | 200 | P2 | N |
| F13 | N/A * | [65 + 5]:(30) | 500 | P2 | N |
| Co-Crystal Endothermic Melting | PEG Endothermic Melting | 3rd Endothermic Event | |
|---|---|---|---|
| SECC400A | 77.5 ± 2.9 °C (72.8 ± 5.4 J/g) | −9.0 ± 0.4 °C (2.1 ± 1.2 J/g) | / |
| SECC400B | 74.6 ± 9.3 °C (45.8 ± 8.3 J/g) | / | 151.4 ± 2.3 °C (9.4 ± 1.7 J/g) |
| SECC600A | 99.5 ± 0.6 °C (86.4 ± 8.2 J/g) | 10.6 ± 0.7 °C (6.9 ± 1.1 J/g) | 136.6 °C * (3.1 J/g) * |
| SECC600B | 99.6 ± 0.1 °C (69.2 ± 11.2 J/g) | 10.2 ± 0.3 °C (9.7 ± 1.2 J/g) | 153.4 ± 2.6 °C (12.0 ± 4.6 J/g) |
| SE Samples | Co-Crystal Endothermic Melting | PEG Endothermic Melting | 3rd Endothermic Event |
|---|---|---|---|
| SECC1000A | 114.7 ± 0.2 °C (114.3 ± 1.1 J/g) | / | / |
| SECC1000B | 115.0 ± 0.7 °C (114.0 ± 1.6 J/g) | / | / |
| SECC1500A | 122.8 ± 0.4 °C (125.5 ± 2.9 J/g) | 41.5 ± 0.7 °C (0.8 ± 0.4 J/g) | / |
| SECC1500B | 123.1 ± 0.6 °C (32.7 ± 3.6 J/g) | / | / |
| SECC4000A | 133.0 ± 0.5 °C (137.7 ± 5.9 J/g) | 53.9 ± 0.3 °C (1.2 ± 0.1 J/g) | / |
| SECC4000B | 132.8 ± 0.5 °C (89.2 ± 9.1 J/g) | / | 145.8 ± 0.3 °C (1.5 ± 0.3 J/g) |
| SECC6000A | 134.8 ± 0.7 °C (133.1 ± 1.4 J/g) | / | / |
| SECC6000B | 134.7 ± 0.3 °C (63.7 ± 2.5 J/g) | / | 147.7 ± 1.0 °C (1.0 ± 0.3 J/g) |
| Granules | Co-Crystal Endothermic Melting | PEG Endothermic Melting | 3rd Endothermic Event |
|---|---|---|---|
| F1 | 81.5 ± 1.2 °C (46.1 ± 5.0 J/g) | / | / |
| F5 | 98.3 ± 0.8 °C (48.6 ± 4.1 J/g) | / | / |
| F9 | 130.2 ± 0.1 °C (72.4 ± 0.8 J/g) | / | / |
| F10 | 133.7 ± 0.1 °C (71.8 ± 0.9 J/g) | / | / |
| Granules | RSC (%) |
|---|---|
| F1 | 1.7 ± 0.1 |
| F5 | 1.0 ± 0.1 |
| F9 | 1.1 ± 0.1 |
| F10 | 1.2 ± 0.1 |
| Raman Shift (1/cm) | Assignment |
|---|---|
| 1500 (DAP) → 1507 (co-crystals) | C-C stretch of DAP |
| 1487 (PEG) → None | CH2–CH2 symmetric bending vibration |
| 1008 (DAP) → 1005 (co-crystals) | CCC in-plane bend |
| 966 and 952 (DAP) → None | γCH |
| FTIR Bond Stretching Frequency (1/cm) | Assignment |
|---|---|
| 3455 (DAP) → 3471 (co-crystals) | NH2 asymmetric stretching |
| 3396, 3366, 3344 (DAP) → 3437, 3375, 3347 (co-crystals) | N-H stretching |
| 3234 (DAP) → 3241 (co-crystals) | N-H symmetric and asymmetric stretching |
| 1626 (DAP) → 1630 (co-crystals) | NH2 bending |
| Granules | DAP Loading (%) | Co-Crystal Loading (%) |
|---|---|---|
| F1 | 23.1 ± 0.5 | 42.0 ± 0.9 |
| F5 | 28.1 ± 1.4 | 51.1 ± 2.6 |
| F9 | 29.7 ± 0.9 | 53.9 ± 1.7 |
| F10 | 32.5 ± 0.8 | 59.1 ± 1.4 |
| d10 (μm) | d50 (μm) | d90 (μm) | Span | |
|---|---|---|---|---|
| DAP | 10.3 ± 0.3 | 60.6 ± 2.3 | 926.3 ± 215.1 | 12.1 |
| DAP * | 7.6 ± 0.2 | 41.6 ± 0.7 | 150.4 ± 2.7 | 3.4 |
| MCC | 30.4 ± 0.5 | 108.0 ± 4.2 | 218.3 ± 27.7 | 1.9 |
| F1 | 84.1 ± 2.3 | 190.0 ± 3.8 | 350.5 ± 11.9 | 1.5 |
| F5 | 166.7 ± 2.3 | 315.2 ± 7.4 | 568.0 ± 30.9 | 1.3 |
| F9 | 238.7 ± 6.9 | 418.9 ± 20.2 | 701.9 ± 44.9 | 1.1 |
| F10 | 262.2 ± 14.7 | 449.7 ± 19.4 | 736.7 ± 38.6 | 1.1 |
| Granules | Hausner Ratio | Flow Description |
|---|---|---|
| DAP | 1.37 ± 0.05 | “Poor” |
| F1 | 1.15 ± 0.01 | “Good” |
| F5 | 1.18 ± 0.01 | “Good” |
| F9 | 1.08 ± 0.02 | “Excellent” |
| F10 | 1.16 ± 0.03 | “Good” |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, S.; Bonner, D.; Twamley, B.; Singh, A.; Healy, A.M. One Step In Situ Co-Crystallization of Dapsone and Polyethylene Glycols during Fluidized Bed Granulation. Pharmaceutics 2023, 15, 2330. https://doi.org/10.3390/pharmaceutics15092330
Shao S, Bonner D, Twamley B, Singh A, Healy AM. One Step In Situ Co-Crystallization of Dapsone and Polyethylene Glycols during Fluidized Bed Granulation. Pharmaceutics. 2023; 15(9):2330. https://doi.org/10.3390/pharmaceutics15092330
Chicago/Turabian StyleShao, Shizhe, David Bonner, Brendan Twamley, Abhishek Singh, and Anne Marie Healy. 2023. "One Step In Situ Co-Crystallization of Dapsone and Polyethylene Glycols during Fluidized Bed Granulation" Pharmaceutics 15, no. 9: 2330. https://doi.org/10.3390/pharmaceutics15092330
APA StyleShao, S., Bonner, D., Twamley, B., Singh, A., & Healy, A. M. (2023). One Step In Situ Co-Crystallization of Dapsone and Polyethylene Glycols during Fluidized Bed Granulation. Pharmaceutics, 15(9), 2330. https://doi.org/10.3390/pharmaceutics15092330