
Advanced In Vivo Prediction by Introducing Biphasic Dissolution Data into PBPK Models



Abstract
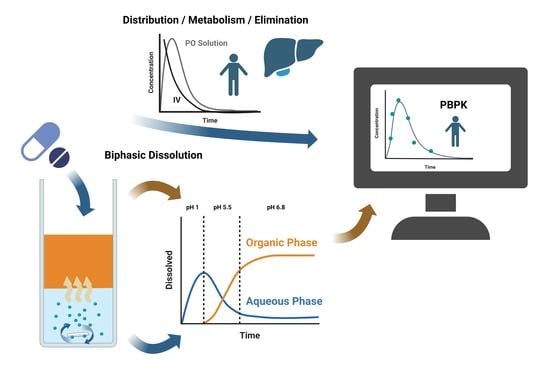
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physiochemical Characterisation
2.3. In Vitro Data
2.4. Model Development and Evaluation

2.4.1. Aprepitant (Nanocrystal)
2.4.2. Celecoxib (Microcrystal)
2.4.3. Fenofibrate (Microcrystal)
2.4.4. Itraconazole (ASD)
2.4.5. Nimodipine (ASD)
2.4.6. Ritonavir (ASD)
3. Results
3.1. Model Development
3.2. IVIVE Using Organic Biphasic Partitioning Profiles
3.2.1. Aprepitant (Nanocrystal)
3.2.2. Celecoxib (Microcrystal)
3.2.3. Fenofibrate (Microcrystal)
3.2.4. Itraconazole (ASD)
3.2.5. Nimodipine (ASD)
3.2.6. Ritonavir (ASD)
3.3. Predictive Performance of PBPK Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grady, H.; Elder, D.; Webster, G.K.; Mao, Y.; Lin, Y.; Flanagan, T.; Mann, J.; Blanchard, A.; Cohen, M.J.; Lin, J.; et al. Industry’s View on Using Quality Control, Biorelevant, and Clinically Relevant Dissolution Tests for Pharmaceutical Development, Registration, and Commercialization. J. Pharm. Sci. 2018, 107, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, P.A.; Lee, W.W.; Stott, P.W.; Townsend, A.I.; Smart, J.P.; Ghahramani, P.; Hammett, T.; Billett, L.; Behn, S.; Gibb, R.C.; et al. Clinical Relevance of Dissolution Testing in Quality by Design. AAPS J. 2008, 10, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.J.; Pygall, S.R.; Cooper, V.B.; Mann, J.C. Overcoming Sink Limitations in Dissolution Testing: A Review of Traditional Methods and the Potential Utility of Biphasic Systems: Dissolution Testing: Biphasic Systems. J. Pharm. Pharmacol. 2012, 64, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Polster, C.S.; Atassi, F.; Wu, S.-J.; Sperry, D.C. Use of Artificial Stomach−Duodenum Model for Investigation of Dosing Fluid Effect on Clinical Trial Variability. Mol. Pharm. 2010, 7, 1533–1538. [Google Scholar] [CrossRef]
- Gao, Y.; Carr, R.A.; Spence, J.K.; Wang, W.W.; Turner, T.M.; Lipari, J.M.; Miller, J.M. A PH-Dilution Method for Estimation of Biorelevant Drug Solubility along the Gastrointestinal Tract: Application to Physiologically Based Pharmacokinetic Modeling. Mol. Pharm. 2010, 7, 1516–1526. [Google Scholar] [CrossRef]
- Pestieau, A.; Evrard, B. In Vitro Biphasic Dissolution Tests and Their Suitability for Establishing in Vitro-in Vivo Correlations: A Historical Review. Eur. J. Pharm. Sci. 2017, 102, 203–219. [Google Scholar] [CrossRef]
- Sironi, D.; Rosenberg, J.; Bauer-Brandl, A.; Brandl, M. PermeaLoopTM, a Novel in Vitro Tool for Small-Scale Drug-Dissolution/Permeation Studies. J. Pharm. Biomed. Anal. 2018, 156, 247–251. [Google Scholar] [CrossRef]
- Denninger, A.; Westedt, U.; Wagner, K.G. Shared IVIVR for Five Commercial Enabling Formulations Using the BiPHa+ Biphasic Dissolution Assay. Pharmaceutics 2021, 13, 285. [Google Scholar] [CrossRef] [PubMed]
- Heigoldt, U.; Sommer, F.; Daniels, R.; Wagner, K.-G. Predicting in Vivo Absorption Behavior of Oral Modified Release Dosage Forms Containing PH-Dependent Poorly Soluble Drugs Using a Novel PH-Adjusted Biphasic in Vitro Dissolution Test. Eur. J. Pharm. Biopharm. 2010, 76, 105–111. [Google Scholar] [CrossRef]
- Locher, K.; Borghardt, J.M.; Frank, K.J.; Kloft, C.; Wagner, K.G. Evolution of a Mini-Scale Biphasic Dissolution Model: Impact of Model Parameters on Partitioning of Dissolved API and Modelling of in Vivo-Relevant Kinetics. Eur. J. Pharm. Biopharm. 2016, 105, 166–175. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK Models for the Prediction of in Vivo Performance of Oral Dosage Forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Narang, A.; Bansal, A.K. Use of Biorelevant Dissolution and PBPK Modeling to Predict Oral Drug Absorption. Eur. J. Pharm. Biopharm. 2018, 129, 222–246. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.M.; Schaefer, K.J.; Jamei, M.; Turner, D.B. Biopharmaceutic IVIVE—Mechanistic Modeling of Single- and Two-Phase In Vitro Experiments to Obtain Drug-Specific Parameters for Incorporation Into PBPK Models. J. Pharm. Sci. 2018, 108, 1604–1618. [Google Scholar] [CrossRef]
- Xu, H.; Vela, S.; Shi, Y.; Marroum, P.; Gao, P. In Vitro Characterization of Ritonavir Drug Products and Correlation to Human in Vivo Performance. Mol. Pharm. 2017, 14, 3801–3814. [Google Scholar] [CrossRef]
- Denninger, A.; Westedt, U.; Rosenberg, J.; Wagner, K.G. A Rational Design of a Biphasic DissolutionSetup—Modelling of Biorelevant Kinetics for a Ritonavir Hot-Melt Extruded Amorphous Solid Dispersion. Pharmaceutics 2020, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, D.; Wollatz, U.; Haznar-Garbacz, D.; Hanke, U.; Box, K.J.; Taylor, R.; Ruiz, R.; Beato, S.; Becker, D.; Weitschies, W. Characterisation of Selected Active Agents Regarding PKa Values, Solubility Concentrations and PH Profiles by SiriusT3. Eur. J. Pharm. Biopharm. 2015, 92, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.F.; Pescatore, M.C. Method for Measuring the Logarithm of the Octanol–Water Partition Coefficient by Using Short Octadecyl–Poly(Vinyl Alcohol) High-Performance Liquid Chromatography Columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a Detailed Physiological Model to Simulate the Gastrointestinal Transit and Absorption Process in Humans, Part 1: Oral Solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Lou, X.; Zhang, G.G.Z.; Taylor, L.S. Insights into the Dissolution Mechanism of Ritonavir–Copovidone Amorphous Solid Dispersions: Importance of Congruent Release for Enhanced Performance. Mol. Pharm. 2019, 16, 1327–1339. [Google Scholar] [CrossRef]
- Saboo, S.; Mugheirbi, N.A.; Zemlyanov, D.Y.; Kestur, U.S.; Taylor, L.S. Congruent Release of Drug and Polymer: A “Sweet Spot” in the Dissolution of Amorphous Solid Dispersions. J. Control Release 2019, 298, 68–82. [Google Scholar] [CrossRef]
- Deac, A.; Qi, Q.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Dissolution Mechanisms of Amorphous Solid Dispersions: A Close Look at the Dissolution Interface. Mol. Pharm. 2023, 20, 2217–2234. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, E.S.; Steidel, A.; Rosenblatt, K.M.; Gessner, D.; Liepold, B. Assessment of the Amorphous Solid Dispersion Erosion Behavior Following a Novel Small-Scale Predictive Approach. Eur. J. Pharm. Sci. 2021, 158, 105682. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C. Comparison of Methods for Predicting Dissolution and the Theoretical Implications of Particle-Size-Dependent Solubility. J. Pharm. Sci. 2012, 101, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Hirlak, O.; Dieluweit, S.; Merkel, R.; Wagner, K.G. Polymer-Mediated Drug Supersaturation—A Spotlight on the Interplay between Phase-Separated Amorphous Drug Colloids and Dissolved Molecules. J. Colloid Interface Sci. 2021, 603, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Rathi, S.; Chavan, R.B.; Shastri, N.R. Classification of the Crystallization Tendency of Active Pharmaceutical Ingredients (APIs) and Nutraceuticals Based on Their Nucleation and Crystal Growth Behaviour in Solution State. Drug Deliv. Transl. Res. 2020, 10, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.G. Perorale Retardarzneiformen. Pharmakon 2016, 4, 107–116. [Google Scholar] [CrossRef]
- Emend: EPAR—Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/emend-epar-product-information_en.pdf (accessed on 23 May 2023).
- Majumdar, A.K.; Howard, L.; Goldberg, M.R.; Hickey, L.; Constanzer, M.; Rothenberg, P.L.; Crumley, T.M.; Panebianco, D.; Bradstreet, T.E.; Bergman, A.J.; et al. Pharmacokinetics of Aprepitant After Single and Multiple Oral Doses in Healthy Volunteers. J. Clin. Pharmacol. 2006, 46, 291–300. [Google Scholar] [CrossRef]
- Fachinfo CELEBREX® 100 mg/200 mg Hartkapseln. Available online: https://fachinformation.srz.de/pdf/pfizerpharma/celebrexhartkapseln.pdf (accessed on 23 May 2023).
- Paulson, S.K.; Vaughn, M.B.; Jessen, S.M.; Lawal, Y.; Gresk, C.J.; Yan, B.; Maziasz, T.J.; Cook, C.S.; Karim, A. Pharmacokinetics of Celecoxib after Oral Administration in Dogs and Humans: Effect of Food and Site of Absorption. J. Pharmacol. Exp. Ther. 2001, 297, 638–645. [Google Scholar]
- Pal, A.; Shenoy, S.; Gautam, A.; Munjal, S.; Niu, J.; Gopalakrishnan, M.; Gobburru, J. Pharmacokinetics of DFN-15, a Novel Oral Solution of Celecoxib, Versus Celecoxib 400-Mg Capsules: A Randomized Crossover Study in Fasting Healthy Volunteers. Clin. Drug Investig. 2017, 37, 937–946. [Google Scholar] [CrossRef]
- Miller, D.B.; Spence, J.D. Clinical Pharmacokinetics of Fibric Acid Derivatives (Fibrates). Clin. Pharmacokinet. 1998, 34, 155–162. [Google Scholar] [CrossRef]
- Chapman, M.J. Pharmacology of Fenofibrate. Am. J. Med. 1987, 83, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shi, Y.; Vela, S.; Marroum, P.; Gao, P. Developing Quantitative In Vitro—In Vivo Correlation for Fenofibrate Immediate-Release Formulations With the Biphasic Dissolution-Partition Test Method. J. Pharm. Sci. 2018, 107, 476–487. [Google Scholar] [CrossRef]
- Zhu, T.; Ansquer, J.-C.; Kelly, M.T.; Sleep, D.J.; Pradhan, R.S. Comparison of the Gastrointestinal Absorption and Bioavailability of Fenofibrate and Fenofibric Acid in Humans. J. Clin. Pharmacol. 2010, 50, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Sauron, R.; Wilkins, M.; Jessent, V.; Dubois, A.; Maillot, C.; Weil, A. Absence of a Food Effect with a 145 Mg Nanoparticle Fenofibrate Tablet Formulation. Int. J. Clin. Pharmacol. Ther. 2006, 44, 64–70. [Google Scholar] [CrossRef]
- Fei, Y.; Kostewicz, E.S.; Sheu, M.-T.; Dressman, J.B. Analysis of the Enhanced Oral Bioavailability of Fenofibrate Lipid Formulations in Fasted Humans Using an in Vitro–in Silico–in Vivo Approach. Eur. J. Pharm. Biopharm. 2013, 85, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Barone, J.A.; Koh, J.G.; Bierman, R.H.; Colaizzi, J.L.; Swanson, K.A.; Gaffar, M.C.; Moskovitz, B.L.; Mechlinski, W.; de Velde, V.V. Food Interaction and Steady-State Pharmacokinetics of Itraconazole Capsules in Healthy Male Volunteers. Antimicrob. Agents Chemother. 1993, 37, 778–784. [Google Scholar] [CrossRef]
- Grabowski, T.; Świerczewska, A.; Borucka, B.; Sawicka, R.; Sasinowska-Motyl, M.; Gumulka, S.W. Chromatographic/Mass Spectrometric Method for the Estimation of Itraconazole and its Metabolite in Human Plasma. Arzneimittelforschung 2009, 59, 422–428. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.M.; Jacobs, R.F.; Massarella, J.; Kauffman, R.E.; Bradley, J.S.; Kimko, H.C.; Kearns, G.L.; Shalayda, K.; Curtin, C.; Maldonado, S.D.; et al. Single-Dose Pharmacokinetics of Intravenous Itraconazole and Hydroxypropyl- -Cyclodextrin in Infants, Children, and Adolescents. Antimicrob. Agents Chemother. 2007, 51, 2668–2673. [Google Scholar] [CrossRef]
- SPORANOX® (Itraconazole) Oral Solution. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020657s027lbl.pdf (accessed on 23 May 2023).
- SPORANOX® (Itraconazole) Capsules. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020083s048s049s050lbl.pdf (accessed on 6 August 2018).
- Bayer Vital GmbH Geschäftsbereich Pharma Fachinformation: Nimotop®, 30 Mg, Filmtabletten 2014. Available online: https://www.fachinfo.de/pdf/001480 (accessed on 12 July 2023).
- Agilent 5989-7668EN. 2007. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.615.4436&rep=rep1&type=pdf (accessed on 12 July 2023).
- Blardi, P. Nimodipine: Drug Pharmacokinetics and Plasma Adenosine Levels in Patients Affected by Cerebral Ischemia. Clin. Pharmacol. Ther. 2002, 72, 556–561. [Google Scholar] [CrossRef]
- Norvir: EPAR -Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/norvir-epar-product-information_en.pdf (accessed on 23 May 2023).
- Salem, A.H.; Chiu, Y.-L.; Valdes, J.M.; Nilius, A.M.; Klein, C.E. A Novel Ritonavir Paediatric Powder Formulation Is Bioequivalent to Ritonavir Oral Solution with a Similar Food Effect. Antivir. Ther. 2015, 20, 425–432. [Google Scholar] [CrossRef]
- Klein, C.; Ng, J.; Kim, D.; Chui, Y.; Awni, W.; Morris, J.; Podsadecki, T.; Cui, Y.; Bernstein, B. The Effect of Food on Ritonavir Bioavailability Following Administration of Ritonavir 100 Mg Film-Coated Tablet in Healthy Adult Subjects. J. Int. AIDS Soc. 2008, 11, P247. [Google Scholar] [CrossRef]
- Open Systems Pharmacology Suite Community. Open Systems Pharmacology Suite Manual; Version 7.0.; Open Systems Pharmacology. 2017. Available online: https://docs.open-systems-pharmacology.org/copyright (accessed on 12 July 2023).
- Schlender, J.-F.; Teutonico, D.; Coboeken, K.; Schnizler, K.; Eissing, T.; Willmann, S.; Jaehde, U.; Stass, H. A Physiologically-Based Pharmacokinetic Model to Describe Ciprofloxacin Pharmacokinetics Over the Entire Span of Life. Clin. Pharmacokinet. 2018, 57, 1613–1634. [Google Scholar] [CrossRef]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on Success Criteria for Predictability of Pharmacokinetic Parameters from In Vitro Studies: An Analysis Based on In Vivo Observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Abrahamsson, B.; Brown, J.; Bevernage, J.; Bolger, M.B.; Heimbach, T.; Karlsson, E.; Kotzagiorgis, E.; Lindahl, A.; McAllister, M.; et al. Current Status and Future Opportunities for Incorporation of Dissolution Data in PBPK Modeling for Pharmaceutical Development and Regulatory Applications: OrBiTo Consortium Commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Zhu, W.; Kesisoglou, F. Physiologically Based Absorption Modeling for Amorphous Solid Dispersion Formulations. Mol. Pharm. 2016, 13, 3206–3215. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Nakagawa, H.; Mikkaichi, T.; Miyano, T.; Matsumoto, Y.; Ando, S. Establishment of a Clinically Relevant Specification for Dissolution Testing Using Physiologically Based Pharmacokinetic (PBPK) Modeling Approaches. Eur. J. Pharm. Biopharm. 2020, 151, 45–52. [Google Scholar] [CrossRef]
- Butler, J.; Hens, B.; Vertzoni, M.; Brouwers, J.; Berben, P.; Dressman, J.; Andreas, C.J.; Schaefer, K.J.; Mann, J.; McAllister, M.; et al. In Vitro Models for the Prediction of in Vivo Performance of Oral Dosage Forms: Recent Progress from Partnership through the IMI OrBiTo Collaboration. Eur. J. Pharm. Biopharm. 2019, 136, 70–83. [Google Scholar] [CrossRef]
- Emami Riedmaier, A.; Lindley, D.J.; Hall, J.A.; Castleberry, S.; Slade, R.T.; Stuart, P.; Carr, R.A.; Borchardt, T.B.; Bow, D.A.J.; Nijsen, M. Mechanistic Physiologically Based Pharmacokinetic Modeling of the Dissolution and Food Effect of a Biopharmaceutics Classification System IV Compound—The Venetoclax Story. J. Pharm. Sci. 2018, 107, 495–502. [Google Scholar] [CrossRef]
- Hens, B.; Pathak, S.M.; Mitra, A.; Patel, N.; Liu, B.; Patel, S.; Jamei, M.; Brouwers, J.; Augustijns, P.; Turner, D.B. In Silico Modeling Approach for the Evaluation of Gastrointestinal Dissolution, Supersaturation, and Precipitation of Posaconazole. Mol. Pharm. 2017, 14, 4321–4333. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Trade Name | Dose [mg] | Formulation Type |
|---|---|---|---|
| Aprepitant | Emend ® (MSD, Munich, Germany) | 125 | Nanocrystal |
| Celecoxib | Celebrex ® (Pfizer, Vienna, Austria) | 200 | Microcrystal |
| Fenofibrate | Lipidil ® (Viatris, Bad Homburg, Germany) | 200 | Microcrystal |
| Itraconazole | Sempera 7 ® (JANSSEN-CILAG GmbH, Neuss, Germany) | 100 | Amorphous solid dispersion |
| Nimodipine | Nimotop ® (Bayer AG, Leverkusen, Germany) | 30 | Amorphous solid dispersion |
| Ritonavir | Norvir ® (AbbVie, Wiesbaden, Germany) | 100 | Amorphous solid dispersion |
| Parameter | Aprepitant | Celecoxib | Fenofibrate | Fenofibric acid | Itraconazole | Nimodipine | Ritonavir |
|---|---|---|---|---|---|---|---|
| pKa | 2.8 | 10.7 (A*) | N/A | 4.0 (A*) | 3.8 | 2.6 | 1.9 2.5 |
| S (0.1N HCl) [µg/mL] | 62.0 | 2.67 | 0.62 | N/A | 6.1 | 3.21 | 382.8 |
| S (6.8N Buffer) [µg/mL] | 1.39 | 1.76 | 1.08 | 1.04 × 103 | 0.88 | 2.90 | 0.96 |
| S (FaSSIF-V2) [µg/mL] | 14.0 | 4.52 | 1.03 | N/A | 0.60 | 5.18 | 4.3 |
| Log P | 4.8 | 3.7 | 5.4 | 3.0 | 5.4 | 3.5 | 4.4 |
| Fa | 0.59 | 0.39 | 0.46 | N/A | 0.16 | 0.035 | 0.80 |
| Peff [cm/min] | 1.67 × 10−3 | 3.71 × 10−4 | 1.64 × 10−3 | 8.78 × 10−5 | 8.13 × 10−4 | 6.00 × 10−4 | 3.30 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denninger, A.; Becker, T.; Westedt, U.; Wagner, K.G. Advanced In Vivo Prediction by Introducing Biphasic Dissolution Data into PBPK Models. Pharmaceutics 2023, 15, 1978. https://doi.org/10.3390/pharmaceutics15071978
Denninger A, Becker T, Westedt U, Wagner KG. Advanced In Vivo Prediction by Introducing Biphasic Dissolution Data into PBPK Models. Pharmaceutics. 2023; 15(7):1978. https://doi.org/10.3390/pharmaceutics15071978
Chicago/Turabian StyleDenninger, Alexander, Tim Becker, Ulrich Westedt, and Karl G. Wagner. 2023. "Advanced In Vivo Prediction by Introducing Biphasic Dissolution Data into PBPK Models" Pharmaceutics 15, no. 7: 1978. https://doi.org/10.3390/pharmaceutics15071978
APA StyleDenninger, A., Becker, T., Westedt, U., & Wagner, K. G. (2023). Advanced In Vivo Prediction by Introducing Biphasic Dissolution Data into PBPK Models. Pharmaceutics, 15(7), 1978. https://doi.org/10.3390/pharmaceutics15071978