

Protein Corona Attenuates the Targeting of Antitumor Sialyl Lewis X-Decorated Liposomes to Vascular Endothelial Cells under Flow Conditions

 , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Preparation of Liposomes
2.3. Liposome Hydrodynamic Diameter and Zeta Potential Measurements
2.4. Determination of the Zeta Potential of Liposome–Protein Complexes
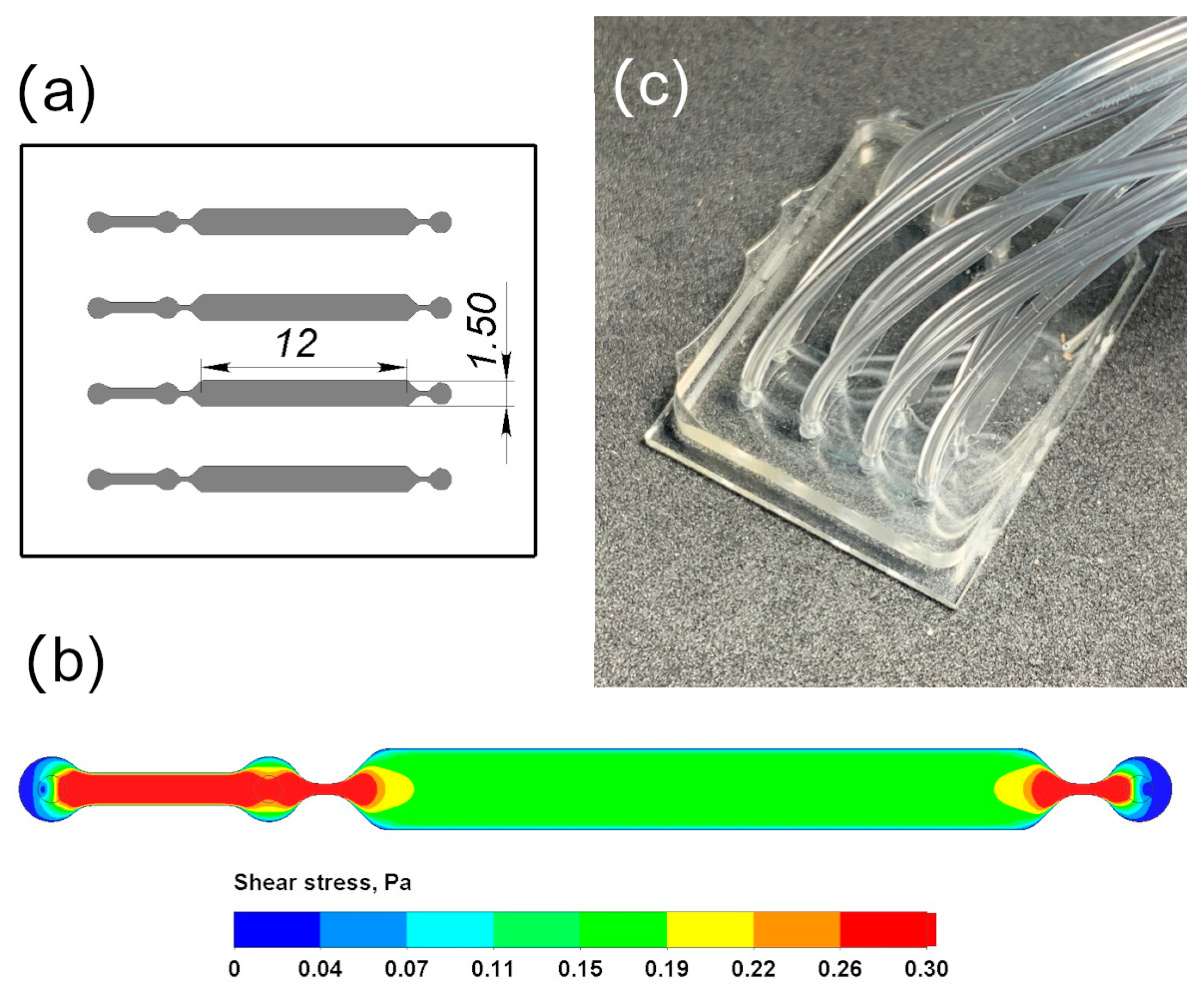
2.5. Hydrodynamic Modeling: Fabrication of Microfluidic Chips
2.6. Primary Culture of Human Umbilical Vein Endothelial Cells
2.7. Cell Culturing in Microfluidic Chips
2.8. Liposome Uptake by HUVECs in Microfluidic Chips under Flow
2.9. Liposome Uptake by HUVECs under Static Conditions
2.10. Isolation of Liposome–Protein Complexes from Plasma
2.11. HPLC-MS/MS Sample Preparation
2.12. HPLC-MS/MS Data Acquisition and Analysis
2.13. Delipidization of Liposome–Protein Complexes, SDS-PAGE, and Immunoblotting
2.14. Statistics
3. Results and Discussion
3.1. Characteristics of Liposomes

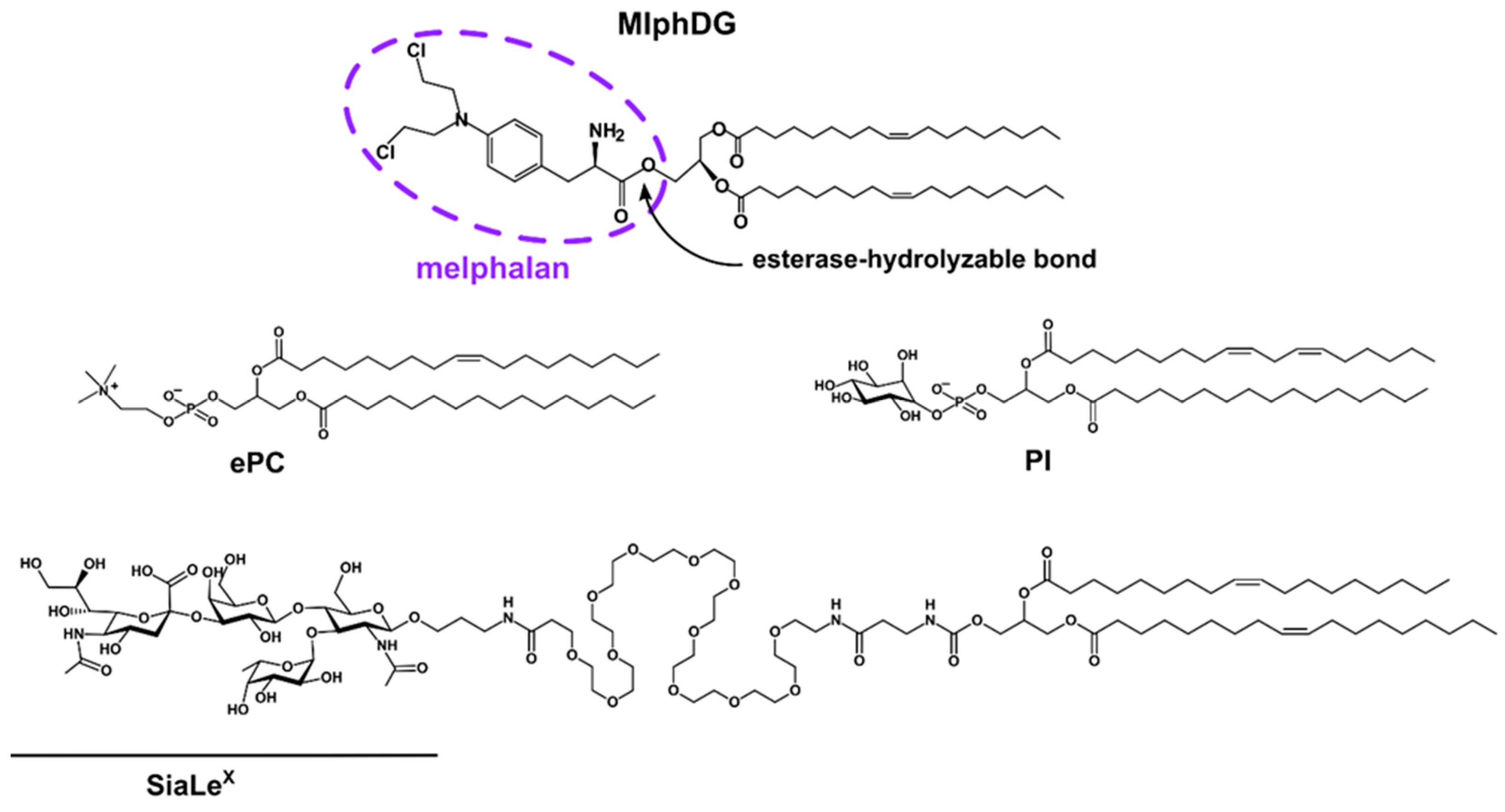{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Liposome Composition, Mol Ratio | D, nm 1 Average ± SD | PdI 1, Average ± SD | Zeta-Potential, mV 2 Average ± SD | |
|---|---|---|---|---|---|
| Liposomes | Liposome–Protein Complex | ||||
| ePC | ePC | 120.0 ± 1.1 | 0.028 ± 0.020 | −3.3 ± 0.4 | N/A |
| MLPH | ePC–PI–MlphDG, 8:1:1 | 130.2 ± 1.4 | 0.087 ± 0.044 | −8.8 ± 0.8 | −11.7 ± 0.5 |
| 5SX | ePC–PI–MlphDG–SiaLeX, 7.5:1:1:0.5 | 122.7 ± 0.8 | 0.069 ± 0.025 | −25.0 ± 0.9 | −22.1 ± 0.5 |
| 10SX | ePC–PI–MlphDG–SiaLeX, 7:1:1:1 | 102.7 ± 1.0 | 0.079 ± 0.042 | −31.7 ± 1.5 | −28.1 ± 0.6 |
3.2. Hydrodynamic Conditions in Microfluidic Chips
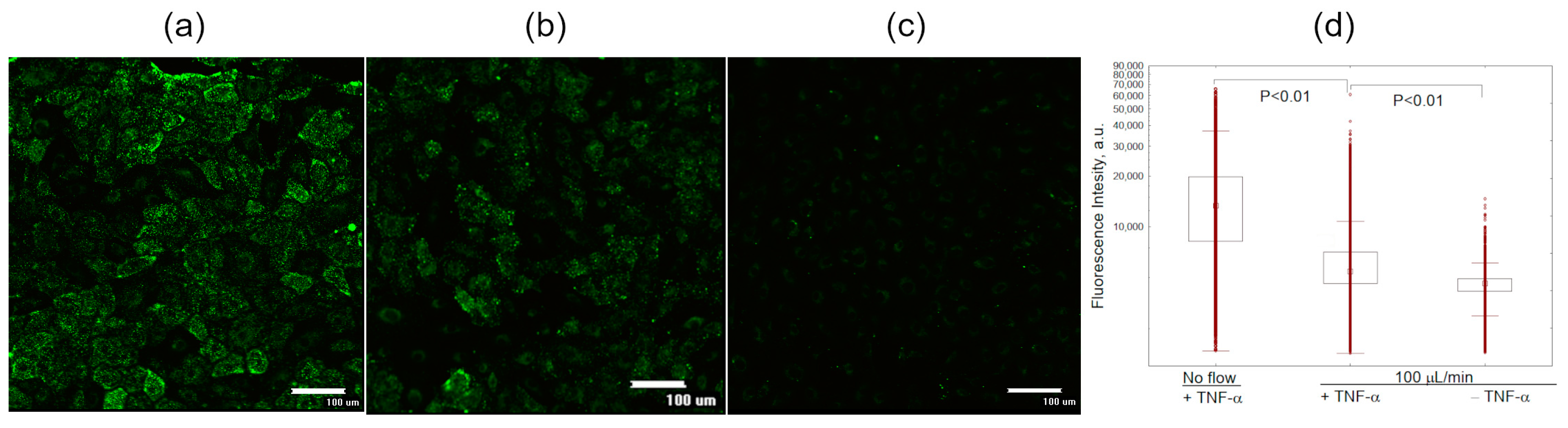
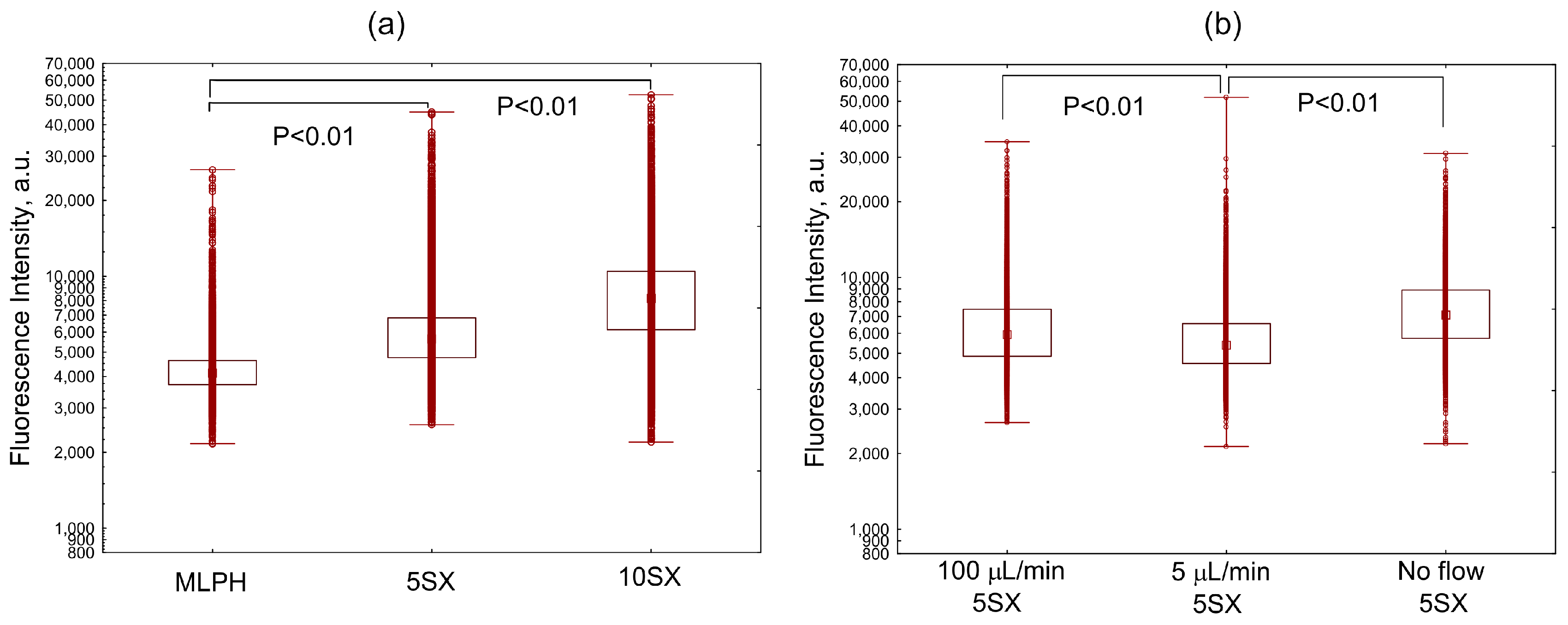
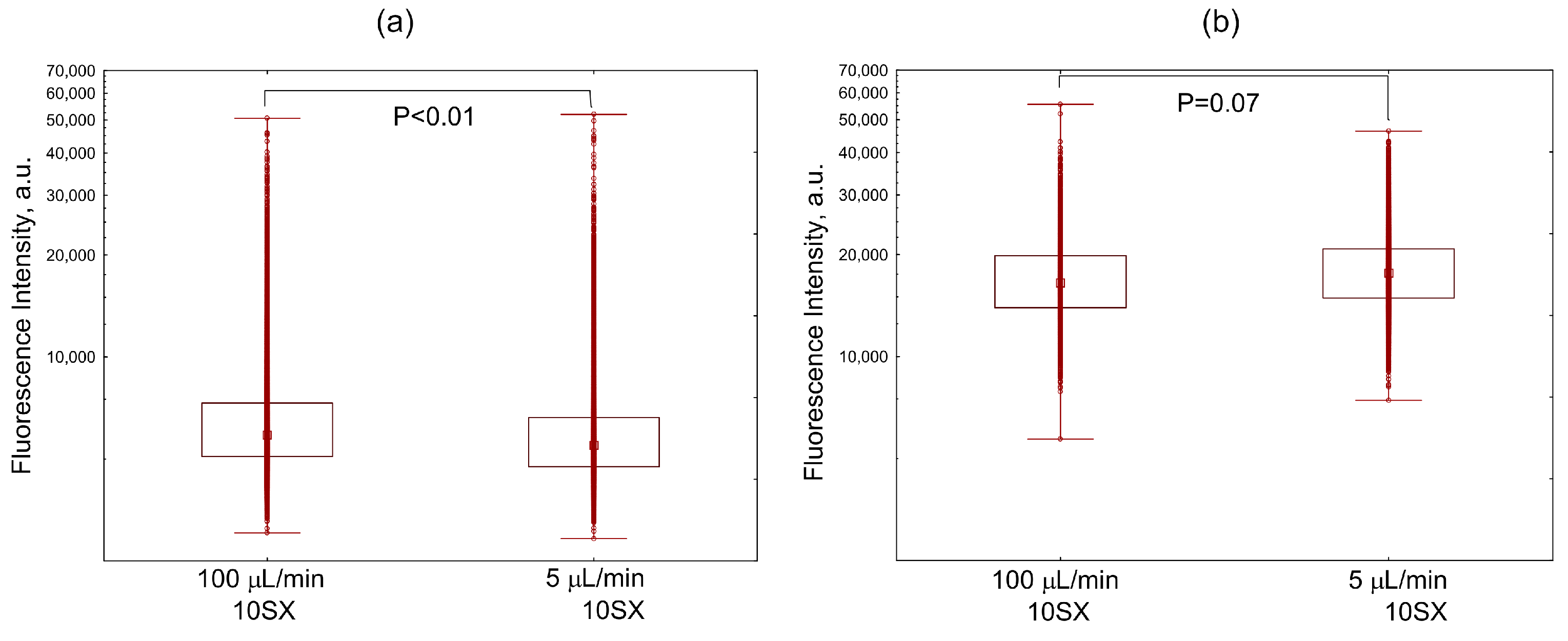
3.3. Liposome Uptake by HUVEC Cells in Microfluidic Chips
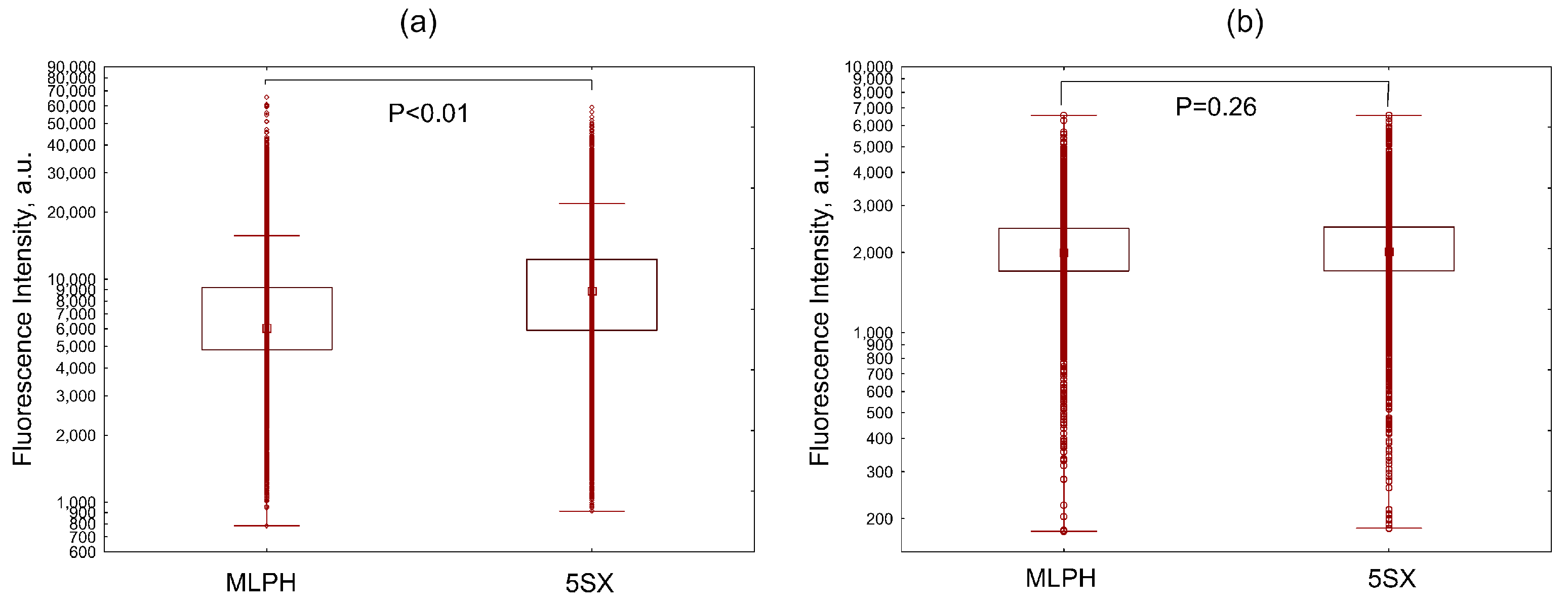
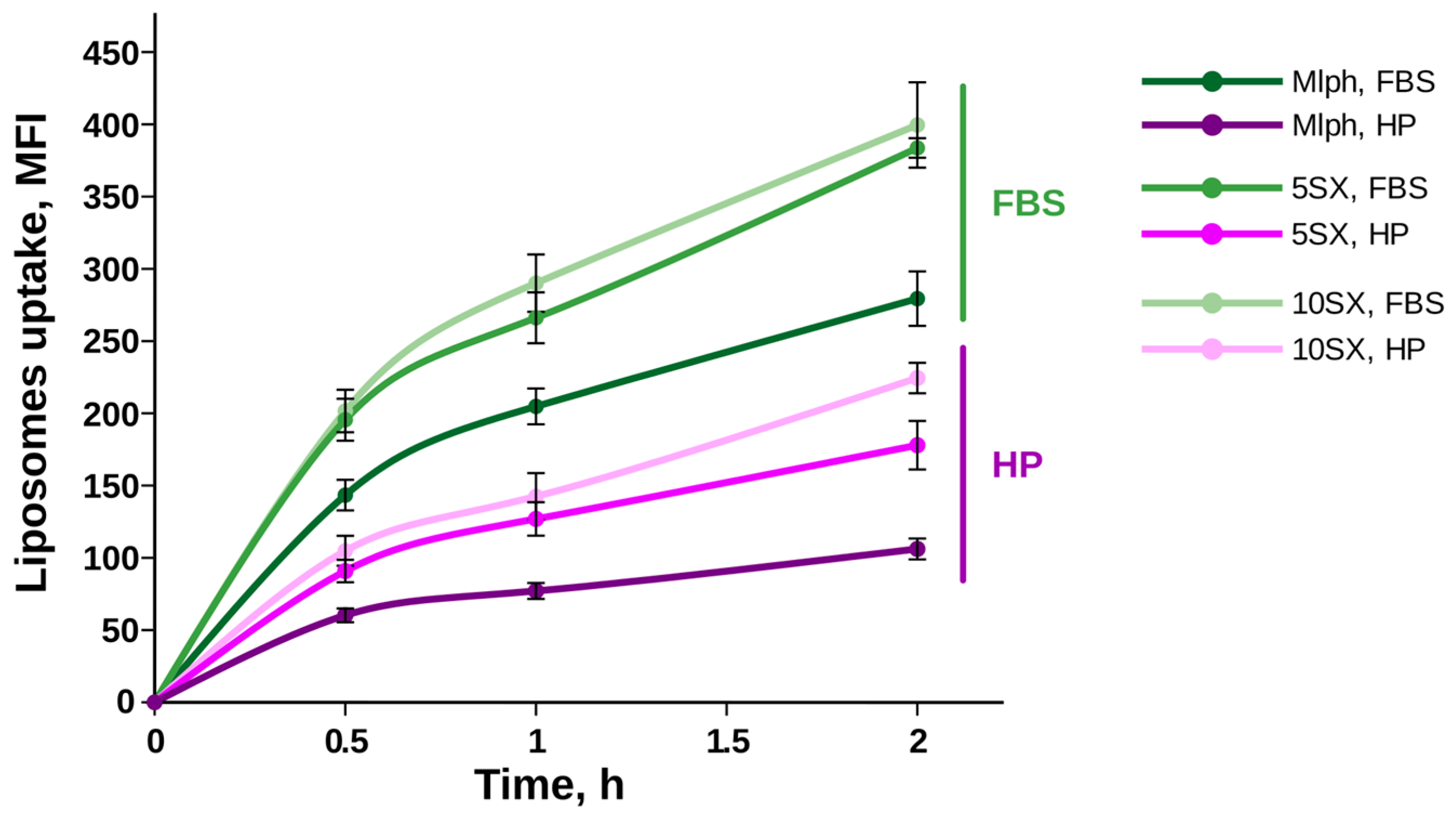
3.4. Liposome Uptake by Activated HUVECs under Static Conditions
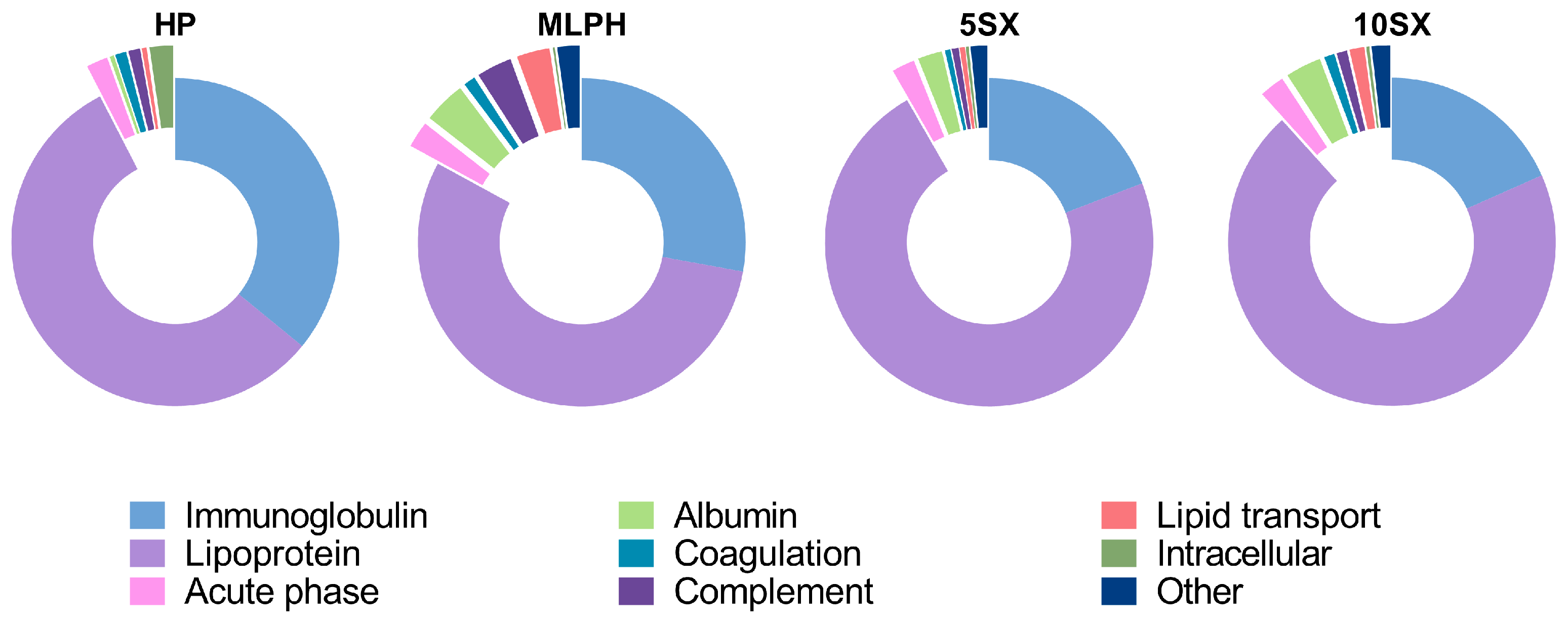
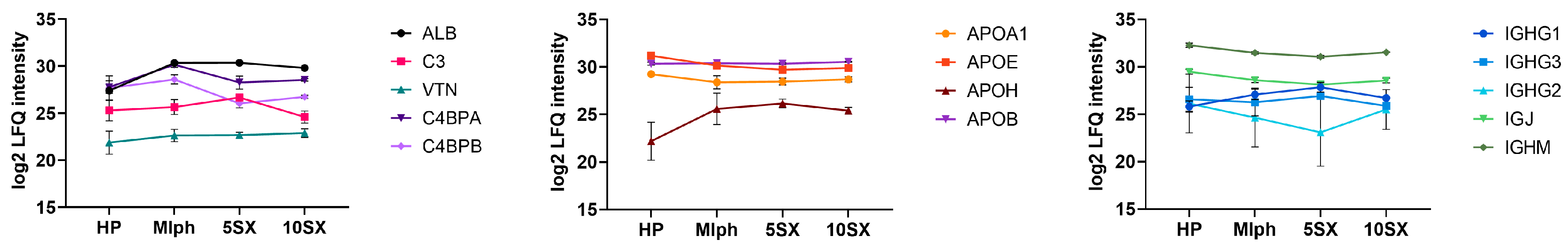
3.5. Plasma Protein Binding by Liposomes: General Results of the Proteome Analysis
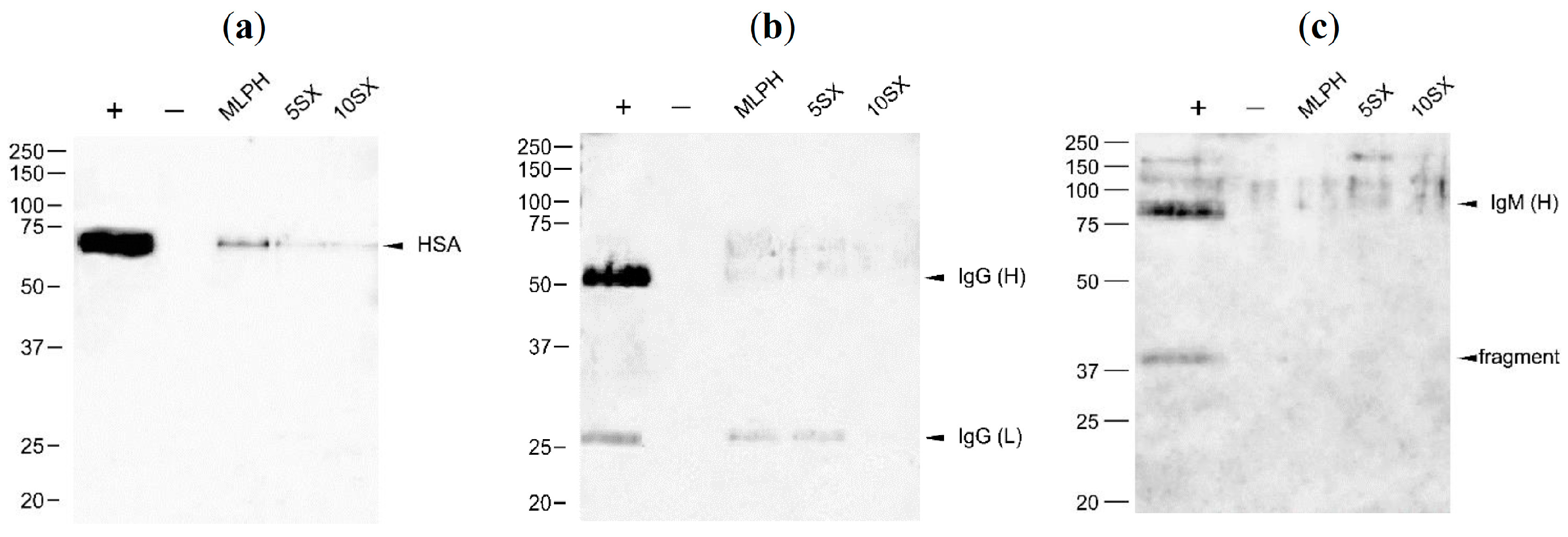
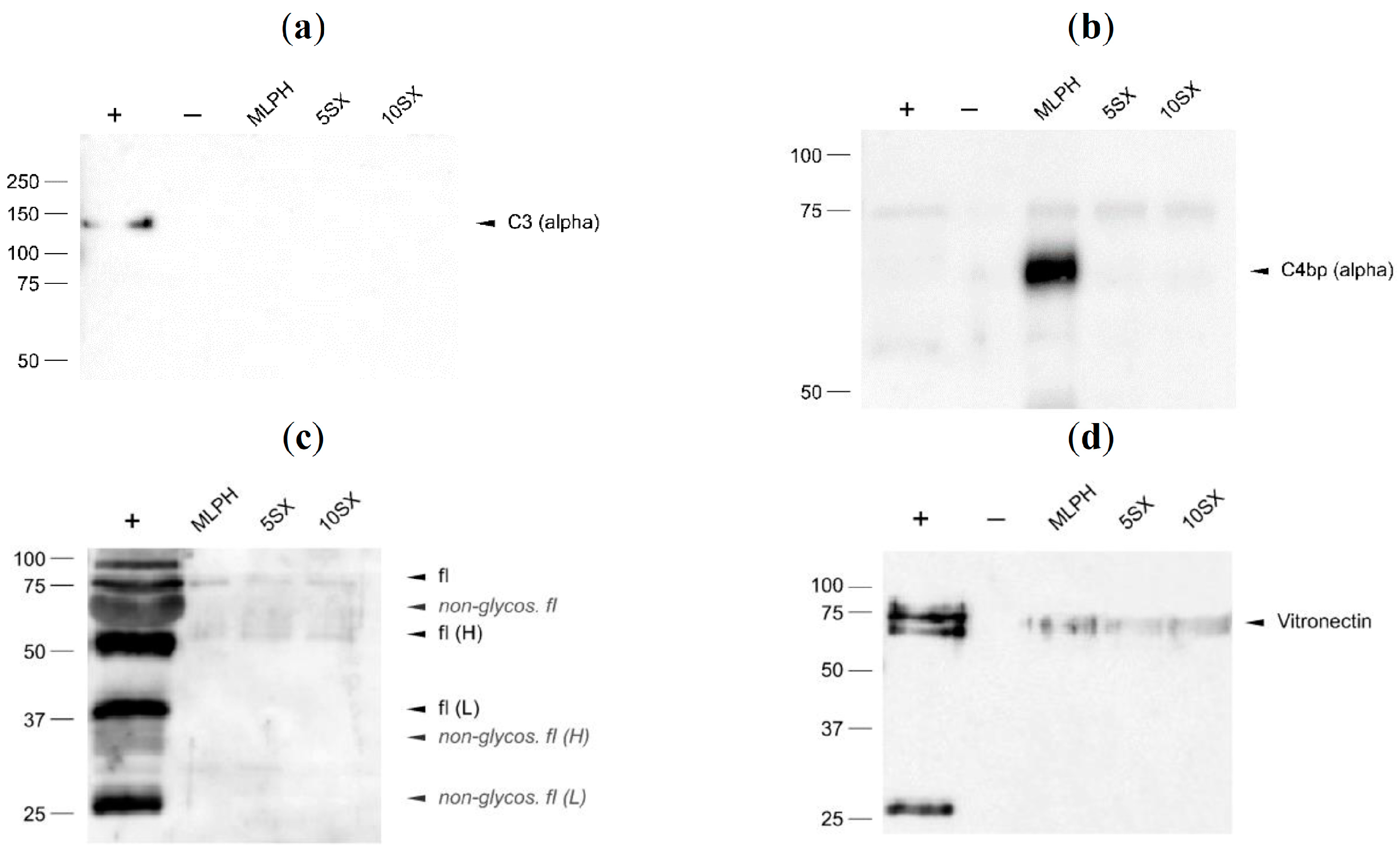
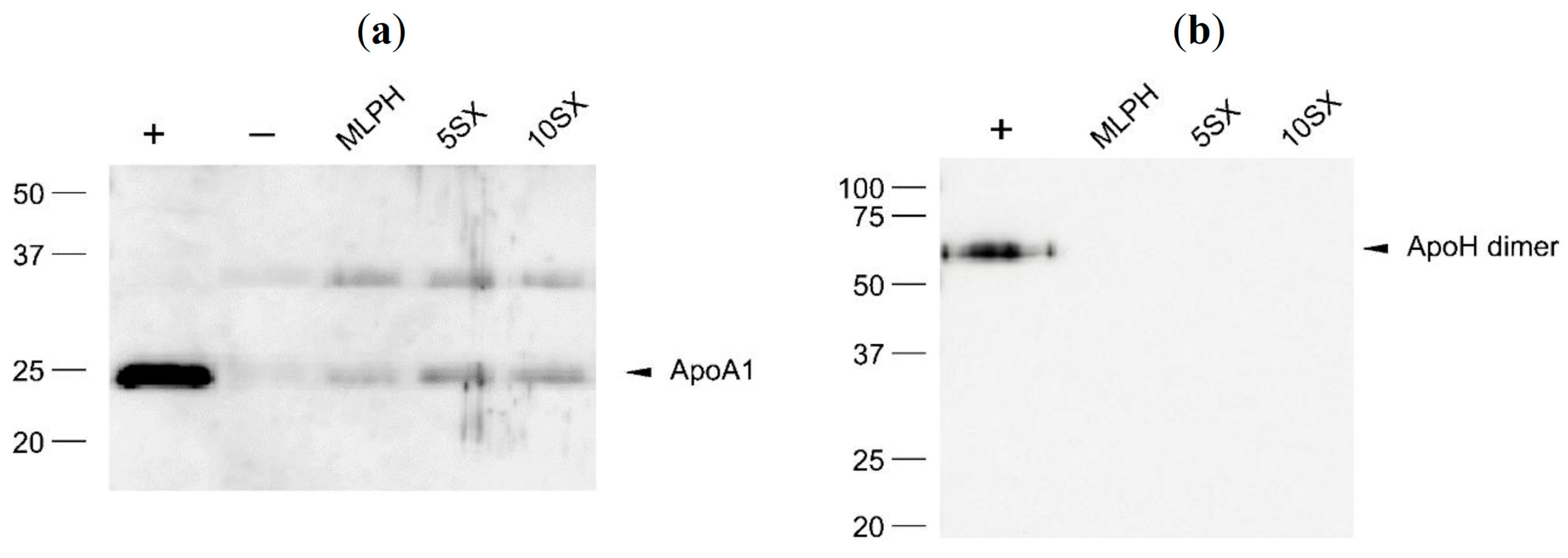
3.6. Plasma Protein Binding by Liposomes: Selected proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Allen, C. Why I’m holding onto hope for nano in oncology. Mol. Pharm. 2016, 13, 2603–2604. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Andra, V.V.S.N.; Pammi, S.V.N.; Bhatraju, L.V.K.P.; Ruddaraju, L.K. A comprehensive review on novel liposomal methodologies, commercial formulations, clinical trials and patents. Bionanoscience 2022, 12, 274–291. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Ojha, T.; Pathak, V.; Shi, Y.; Hennink, W.E.; Moonen, C.T.W.; Storm, G.; Kiessling, F.; Lammers, T. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv. Drug Deliv. Rev. 2017, 119, 44–60. [Google Scholar] [CrossRef]
- Anchordoquy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.A.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and barriers in cancer nanomedicine: Addressing challenges, looking for solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef]
- Yan, W.; Leung, S.S.; To, K.K. Updates on the use of liposomes for active tumor targeting in cancer therapy. Nanomedicine 2020, 15, 303–318. [Google Scholar] [CrossRef]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef]
- Khodabakhsh, F.; Muyldermans, S.; Behdani, M.; Kazemi-Lomedasht, F. Liposomal delivery of vascular endothelial growth factor/receptors and their inhibitors. J. Drug Target. 2020, 28, 379–385. [Google Scholar] [CrossRef]
- Wicki, A.; Rochlitz, C.; Orleth, A.; Ritschard, R.; Albrecht, I.; Herrmann, R.; Christofori, G.; Mamot, C. Targeting tumor-associated endothelial cells: Anti-VEGFR2 immunoliposomes mediate tumor vessel disruption and inhibit tumor growth. Clin. Cancer Res. 2012, 18, 454–464. [Google Scholar] [CrossRef]
- Shein, S.A.; Kuznetsov, I.I.; Abakumova, T.O.; Chelushkin, P.S.; Melnikov, P.A.; Korchagina, A.A.; Bychkov, D.A.; Seregina, I.F.; Bolshov, M.A.; Kabanov, A.V.; et al. VEGF- and VEGFR2-targeted liposomes for cisplatin delivery to glioma cells. Mol. Pharm. 2016, 13, 3712–3723. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; De Santis, A.; Portioli, E.; Russo Krauss, I.; Battistini, L.; Curti, C.; Peppicelli, S.; Calorini, L.; D’Errico, G.; Zanardi, F.; et al. Integrin-targeted AmpRGD sunitinib liposomes as integrated antiangiogenic tools. Nanomedicine 2019, 18, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ma, G.; Yuan, Z.; Qian, H.; Xu, L.; Sidransky, E.; Chen, S. Development of zwitterionic polypeptide nanoformulation with high doxorubicin loading content for targeted drug delivery. Langmuir 2019, 35, 1273–1283. [Google Scholar] [CrossRef]
- Alzamely, K.O.; Hajizadeh, F.; Heydari, M.; Ghaderi Sede, M.J.; Asl, S.H.; Peydaveisi, M.; Masjedi, A.; Izadi, S.; Nikkhoo, A.; Atyabi, F.; et al. Combined inhibition of CD73 and ZEB1 by Arg-Gly-Asp (RGD)-targeted nanoparticles inhibits tumor growth. Colloids Surf. B Biointerfaces 2021, 197, 111421. [Google Scholar] [CrossRef]
- Ailuno, G.; Baldassari, S.; Zuccari, G.; Schlich, M.; Caviglioli, G. Peptide-based nanosystems for vascular cell adhesion molecule-1 targeting: A real opportunity for therapeutic and diagnostic agents in inflammation associated disorders. J. Drug Deliv. Sci. Techn. 2020, 55, 101461. [Google Scholar] [CrossRef]
- Pastorino, S.; Baldassari, S.; Ailuno, G.; Zuccari, G.; Drava, G.; Petretto, A.; Cossu, V.; Marini, C.; Alfei, S.; Florio, T.; et al. Two novel PET radiopharmaceuticals for endothelial vascular cell adhesion molecule-1 (VCAM-1) targeting. Pharmaceutics 2021, 13, 1025. [Google Scholar] [CrossRef]
- Distasio, N.; Salmon, H.; Dierick, F.; Ebrahimian, T.; Tabrizian, M.; Lehoux, S. VCAM-1-targeted gene delivery nanoparticles localize to inflamed endothelial cells and atherosclerotic plaques. Adv. Ther. 2020, 4, 2000196. [Google Scholar] [CrossRef]
- Fuior, E.V.; Deleanu, M.; Constantinescu, C.A.; Rebleanu, D.; Voicu, G.; Simionescu, M.; Calin, M. Functional role of VCAM-1 targeted flavonoid-loaded lipid nanoemulsions in reducing endothelium inflammation. Pharmaceutics 2019, 11, 391. [Google Scholar] [CrossRef]
- Manthe, R.L.; Muro, S. ICAM-1-targeted nanocarriers attenuate endothelial release of soluble ICAM-1, an inflammatory regulator. Bioeng. Transl. Med. 2017, 2, 109–119. [Google Scholar] [CrossRef]
- Garnacho, C.; Muro, S. ICAM-1 targeting, intracellular trafficking, and functional activity of polymer nanocarriers coated with a fibrinogen-derived peptide for lysosomal enzyme replacement. J. Drug. Target. 2017, 25, 786–795. [Google Scholar] [CrossRef]
- Jubeli, E.; Moine, L.; Vergnaud-Gauduchon, J.; Barratt, G. E-selectin as a target for drug delivery and molecular imaging. J. Control. Release 2012, 158, 194–206. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Kneuer, C.; Bakowsky, U. Selectins—An emerging target for drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 527–549. [Google Scholar] [CrossRef]
- Barthel, S.R.; Gavino, J.D.; Descheny, L.; Dimitroff, C.J. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin. Ther. Targets 2007, 11, 1473–1491. [Google Scholar] [CrossRef]
- St Hill, C.A. Interactions between endothelial selectins and cancer cells regulate metastasis. Front. Biosci. 2011, 16, 3233–3251. [Google Scholar] [CrossRef] [PubMed]
- Foxall, C.; Watson, S.R.; Dowbenko, D.; Fennie, C.; Lasky, L.A.; Kiso, M.; Hasegawa, A.; Asa, D.; Brandley, B.K. The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J. Cell Biol. 1992, 117, 895–902. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Visweswaran, G.R.R.; Storm, G.; Hennink, W.E.; Kamps, J.A.A.M.; Kok, R.J. E-selectin targeted immunoliposomes for rapamycin delivery to activated endothelial cells. Int. J. Pharm. 2018, 548, 759–770. [Google Scholar] [CrossRef]
- Jin, F.; Wang, F. The physiological and pathological roles and applications of sialyl Lewis x, a common carbohydrate ligand of the three selectins. Glycoconj. J. 2020, 37, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Chantarasrivong, C.; Ueki, A.; Ohyama, R.; Unga, J.; Nakamura, S.; Nakanishi, I.; Higuchi, Y.; Kawakami, S.; Ando, H.; Imamura, A.; et al. Synthesis and functional characterization of novel Sialyl LewisX mimic-decorated liposomes for E-selectin-mediated targeting to inflamed endothelial cells. Mol. Pharm. 2017, 14, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Chantarasrivong, C.; Higuchi, Y.; Tsuda, M.; Yamane, Y.; Hashida, M.; Konishi, M.; Komura, N.; Ando, H.; Yamashita, F. Sialyl LewisX mimic-decorated liposomes for anti-angiogenic everolimus delivery to E-selectin expressing endothelial cells. RSC Adv. 2019, 9, 20518–20527. [Google Scholar] [CrossRef]
- Kuznetsova, N.R.; Stepanova, E.V.; Peretolchina, N.M.; Khochenkov, D.A.; Boldyrev, I.A.; Bovin, N.V.; Vodovozova, E.L. Targeting liposomes loaded with melphalan prodrug to tumour vasculature via the sialyl Lewis X selectin ligand. J. Drug Target. 2014, 22, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, A.; Kapkaeva, M.; Shcheglovitova, O.; Boldyrev, I.; Pazynina, G.; Bovin, N.; Vodovozova, E. Interactions of antitumour Sialyl Lewis X liposomes with vascular endothelial cells. Biochim. Biophys. Acta 2015, 1848, 1099–1110. [Google Scholar] [CrossRef]
- Gabizon, A.; Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef]
- Muller, M.; Zschornig, O.; Ohki, S.; Arnold, K. Fusion, leakage and surface hydrophobicity of vesicles containing phosphoinositides: Influence of steric and electrostatic effects. J. Membr. Biol. 2003, 192, 33–43. [Google Scholar] [CrossRef]
- Kozma, G.T.; Shimizu, T.; Ishida, T.; Szebeni, J. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nanobiopharmaceuticals. Adv. Drug Deliv. Rev. 2020, 154–155, 163–175. [Google Scholar] [CrossRef]
- Zalba, S.; Ten Hagen, T.L.M.; Burgui, C.; Garrido, M.J. Stealth nanoparticles in oncology: Facing the PEG dilemma. J. Control. Release 2022, 351, 22–36. [Google Scholar] [CrossRef]
- Tretiakova, D.; Le-Deigen, I.; Onishchenko, N.; Kuntsche, J.; Kudryashova, E.; Vodovozova, E. Phosphatidylinositol stabilizes fluid-phase liposomes loaded with a melphalan lipophilic prodrug. Pharmaceutics 2021, 13, 473. [Google Scholar] [CrossRef]
- Tretiakova, D.; Onishchenko, N.; Boldyrev, I.; Mikhalyov, I.; Tuzikov, A.; Bovin, N.; Evtushenko, E.; Vodovozova, E. Influence of stabilizing components on the integrity of antitumor liposomes loaded with lipophilic prodrug in the bilayer. Colloids Surf. B Biointerfaces 2018, 166, 45–53. [Google Scholar] [CrossRef]
- Kuznetsova, N.R.; Sevrin, C.; Lespineux, D.; Bovin, N.V.; Vodovozova, E.L.; Mészáros, T.; Szebeni, J.; Grandfils, C. Hemocompatibility of liposomes loaded with lipophilic prodrugs of methotrexate and melphalan in the lipid bilayer. J. Control. Release 2012, 160, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechn. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Onishchenko, N.; Tretiakova, D.; Vodovozova, E. Spotlight on the protein corona of liposomes. Acta Biomater. 2021, 134, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Caracciolo, G.; Digiacomo, L.; Colapicchioni, V.; Palchetti, S.; Capriotti, A.L.; Cavaliere, C.; Zenezini Chiozzi, R.; Puglisi, A.; Laganà, A. The biomolecular corona of nanoparticles in circulating biological media. Nanoscale 2015, 7, 13958–13966. [Google Scholar] [CrossRef] [PubMed]
- Palchetti, S.; Pozzi, D.; Capriotti, A.L.; Barbera, G.; Chiozzi, R.Z.; Digiacomo, L.; Peruzzi, G.; Caracciolo, G.; Laganà, A. Influence of dynamic flow environment on nanoparticle-protein corona: From protein patterns to uptake in cancer cells. Colloids Surf. B Biointerfaces 2017, 153, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.C.G.; Kempe, K.; Förster, S.; Caruso, F. Microfluidic examination of the “hard” biomolecular corona formed on engineered particles in different biological milieu. Biomacromolecules 2018, 19, 2580–2594. [Google Scholar] [CrossRef]
- Digiacomo, L.; Palchetti, S.; Giulimondi, F.; Pozzi, D.; Zenezini Chiozzi, R.; Capriotti, A.L.; Laganà, A.; Caracciolo, G. The biomolecular corona of gold nanoparticles in a controlled microfluidic environment. Lab Chip 2019, 19, 2557–2567. [Google Scholar] [CrossRef]
- Meghani, N.M.; Amin, H.; Park, C.; Cui, J.H.; Cao, Q.R.; Choi, K.H.; Lee, B.J. Combinatory interpretation of protein corona and shear stress for active cancer targeting of bioorthogonally clickable gelatin-oleic nanoparticles. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 111, 110760. [Google Scholar] [CrossRef]
- Huang, K.; Boerhan, R.; Liu, C.; Jiang, G. Nanoparticles penetrate into the multicellular spheroid-on-chip: Effect of surface charge, protein corona, and exterior flow. Mol. Pharm. 2017, 14, 4618–4627. [Google Scholar] [CrossRef]
- Tretiakova, D.; Svirshchevskaya, E.; Onishchenko, N.; Alekseeva, A.; Boldyrev, I.; Kamyshinsky, R.; Natykan, A.; Lokhmotov, A.; Arantseva, D.; Shobolov, D.; et al. Liposomal formulation of a melphalan lipophilic prodrug: Studies of acute toxicity, tolerability, and antitumor efficacy. Curr. Drug Deliv. 2020, 17, 312–323. [Google Scholar] [CrossRef]
- Tuzikov, A.B.; Ryabukhina, E.V.; Paramonov, A.S.; Chizhov, A.O.; Bovin, N.V.; Vodovozova, E.L. A convenient route to conjugates of 1,2-diglycerides with functionalized oligoethylene glycol spacer arms. Mendeleev Commun. 2021, 31, 538–541. [Google Scholar] [CrossRef]
- Boldyrev, I.A.; Zhai, X.; Momsen, M.M.; Brockman, H.L.; Brown, R.E.; Molotkovsky, J.G. New BODIPY lipid probes for fluorescence studies of membranes. J. Lipid Res. 2007, 48, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Kobanenko, M.K.; Tretiakova, D.S.; Shchegravina, E.S.; Antipova, N.V.; Boldyrev, I.A.; Fedorov, A.Y.; Vodovozova, E.L.; Onishchenko, N.R. Liposomal formulation of a PLA2-sensitive phospholipid–allocolchicinoid conjugate: Stability and activity studies in vitro. Int. J. Mol. Sci. 2022, 23, 1034. [Google Scholar] [CrossRef] [PubMed]
- Doane, T.L.; Chuang, C.H.; Hill, R.J.; Burda, C. Nanoparticle ζ-potentials. Acc. Chem. Res. 2012, 45, 317–326. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.C.; Duffy, D.C.; Anderson, J.R.; Chiu, D.T.; Wu, H.; Schueller, O.J.; Whitesides, G.M. Fabrication of microfluidic systems in poly(dimethylsiloxane). Electrophoresis 2000, 21, 27–40. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Scheglovitova, O.N.; Romanov, Y.A.; Maksianina, E.V.; Svintsitskaya, V.A.; Pronin, A.G. Herpes simplex type I virus infected human vascular endothelial cells induce the production of anti-viral and proinflammatory factors by peripheral blood leukocytes in vitro. Russ. J. Immunol. 2002, 7, 115–122. [Google Scholar]
- Rutkowski, J.M.; Swartz, M.A. A driving force for change: Interstitial flow as a morphoregulator. Trends Cell Biol. 2007, 17, 44–50. [Google Scholar] [CrossRef]
- Zougman, A.; Banks, R.E. C-STrap sample preparation method—In-situ cysteinyl peptide capture for bottom-up proteomics analysis in the STrap format. PLoS ONE 2015, 10, e0138775. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Dos Santos, N.; Allen, C.; Doppen, A.M.; Anantha, M.; Cox, K.A.K.; Gallagher, R.C.; Karlsson, G.; Edwards, K.; Kenner, G.; Samuels, L.; et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim. Biophys. Acta 2007, 1768, 1367–1377. [Google Scholar] [CrossRef]
- Walkey, C.D.; Chan, W.C. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem. Soc. Rev. 2012, 41, 2780–2799. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Palchetti, S.; Digiacomo, L.; Pozzi, D.; Capriotti, A.L.; Frati, L.; Oliva, M.A.; Tsaouli, G.; Rota, R.; Screpanti, I.; et al. Brain targeting by liposome-biomolecular corona boosts anticancer efficacy of temozolomide in glioblastoma cells. ACS Chem. Neurosci. 2018, 9, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Mancera-Andrade, E.I.; Parsaeimehr, A.; Arevalo-Gallegos, A.; Ascencio-Favela, G.; Parra-Saldivar, R. Microfluidics technology for drug delivery: A review. Front. Biosci. 2018, 10, 74–91. [Google Scholar] [CrossRef]
- Godoy-Gallardo, M.; Ek, P.K.; Jansman, M.M.T.; Wohl, B.M.; Hosta-Rigau, L. Interaction between drug delivery vehicles and cells under the effect of shear stress. Biomicrofluidics 2015, 9, 052605. [Google Scholar] [CrossRef] [PubMed]
- Paszkowiak, J.J.; Dardik, A. Arterial wall shear stress: Observations from the bench to the bedside. Vasc. Endovasc. Surg. 2003, 37, 47–57. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial Fluid Shear Stress Sensing in Vascular Health and Disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hellenic J. Cardiol. 2005, 46, 9–15. [Google Scholar]
- Ballermann, B.J.; Dardik, A.; Eng, E.; Liu, A. Shear stress and the endothelium. Kidney Int. Suppl. 1998, 54, 100–108. [Google Scholar] [CrossRef]
- Lipowsky, H.H.; Kovalcheck, S.; Zweifach, B.W. The distribution of blood rheological parameters in the microvasculature of cat mesentery. Circ. Res. 1978, 43, 738–749. [Google Scholar] [CrossRef]
- Simone, E.; Ding, B.-S.; Muzykantov, V. Targeted delivery of therapeutics to endothelium. Cell Tissue Res. 2009, 335, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Caputo, K.E.; Lee, D.; King, M.R.; Hammer, D.A. Adhesive dynamics simulations of the shear threshold effect for leukocytes. Biophys. J. 2007, 92, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Wu, J.; Wey, C.D.; Klopocki, A.G.; Zhu, C.; McEver, R.P. Catch bonds govern adhesion through L-selectin at threshold shear. J. Cell Biol. 2004, 166, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Reker-Smit, C.; Stuart, M.C.A.; Salvati, A. Effects of protein source on liposome uptake by cells: Corona composition and impact of the excess free proteins. Adv. Healthc. Mater. 2021, 10, e2100370. [Google Scholar] [CrossRef]
- Kristensen, K.; Münter, R.; Kempen, P.J.; Thomsen, M.E.; Stensballe, A.; Andresen, T.L. Isolation methods commonly used to study the liposomal protein corona suffer from contamination issues. Acta Biomater. 2021, 130, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Dumont, L.; Gautier, T.; Pais De Barros, J.P.; Laplanche, H.; Blache, D.; Ducoroy, P.; Fruchart, J.; Fruchart, J.C.; Gambert, P.; Masson, D.; et al. Molecular mechanism of the blockade of plasma cholesteryl ester transfer protein by its physiological inhibitor apolipoprotein CI. J. Biol. Chem. 2005, 280, 38108–38116. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.A.; Deguchi, H.; Elias, D.J.; Griffin, J.H. Serum amyloid A4 is a procoagulant apolipoprotein that it is elevated in venous thrombosis patients. Res. Pract. Thromb. Haemost. 2019, 4, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, G.; Palchetti, S.; Colapicchioni, V.; Digiacomo, L.; Pozzi, D.; Capriotti, A.L.; La Barbera, G.; Laganà, A. Stealth effect of biomolecular corona on nanoparticle uptake by immune cells. Langmuir 2015, 31, 10764–10773. [Google Scholar] [CrossRef]
- Chernecky, C.; Berger, B. Laboratory Tests and Diagnostic Procedures, 6th ed.; Elsevier/Saunders: Amsterdam, The Netherlands, 2012; pp. 84–180. ISBN 9781455745029. [Google Scholar]
- Taguchi, K.; Okamoto, Y.; Matsumoto, K.; Otagiri, M.; Chuang, V.T.G. When albumin meets liposomes: A feasible drug carrier for biomedical applications. Pharmaceuticals 2021, 14, 296. [Google Scholar] [CrossRef]
- Savay, S.; Szebeni, J.; Baranyi, L.; Alving, C.R. Potentiation of liposome-induced complement activation by surface-bound albumin. Biochim. Biophys. Acta 2002, 1559, 79–86. [Google Scholar] [CrossRef]
- Vuarchey, C.; Kumar, S.; Schwendener, R.A. Albumin coated liposomes: A novel platform for macrophage specific drug delivery. Nanotechnol. Dev. 2011, 1, e2. [Google Scholar] [CrossRef]
- Fleischer, C.C.; Payne, C.K. Secondary structure of corona proteins determines the cell surface receptors used by nanoparticles. J. Phys. Chem. B 2014, 118, 14017–14026. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Lichtman, A.; Pillai, S. Basic Immunology, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/humanproteome/blood+protein (accessed on 11 June 2023).
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Tsiftsoglou, S.A.; Arnold, J.N.; Roversi, P.; Crispin, M.D.; Radcliffe, C.; Lea, S.M.; Dwek, R.A.; Rudd, P.M.; Sim, R.B. Human complement factor I glycosylation: Structural and functional characterisation of the N-linked oligosaccharides. Biochim. Biophys. Acta 2006, 764, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Ermert, D.; Blom, A.M. C4b-binding protein: The good, the bad and the deadly. Novel functions of an old friend. Immunol. Lett. 2016, 169, 82–92. [Google Scholar] [CrossRef]
- Baroni, M.; Pavani, G.; Marescotti, D.; Kaabache, T.; Borgel, D.; Gandrille, S.; Marchetti, G.; Legnani, C.; D’Angelo, A.; Pinotti, M.; et al. Membrane binding and anticoagulant properties of protein S natural variants. Thromb. Res. 2010, 125, E33–E39. [Google Scholar] [CrossRef]
- Nilsson, S.C.; Sim, R.B.; Lea, S.M.; Fremeaux-Bacchi, V.; Blom, A.M. Complement factor I in health and disease. Mol. Immunol. 2011, 48, 1611–1620. [Google Scholar] [CrossRef]
- Du Clos, T.W.; Mold, C. Complement and complement deficiencies. In Clinical Immunology, 3rd ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Mosby: Maryland Heights, MO, USA, 2008; Chapter 20; pp. 305–325. [Google Scholar] [CrossRef]
- Palchetti, S.; Digiacomo, L.; Pozzi, D.; Peruzzi, G.; Micarelli, E.; Mahmoudi, M.; Caracciolo, G. Nanoparticles-cell association predicted by protein corona fingerprints. Nanoscale 2016, 8, 12755–12763. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Caracciolo, G.; Cardarelli, F.; Pozzi, D.; Salomone, F.; Maccari, G.; Bardi, G.; Capriotti, A.L.; Cavaliere, C.; Papi, M.; Laganà, A. Selective targeting capability acquired with a protein corona adsorbed on the surface of 1,2-dioleoyl-3-trimethylammonium propane/dna nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 13171–13179. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, G.; Lorkowski, S.W.; Gulshan, K.; Hazen, S.L.; Gogonea, V.; Smith, J.D. First eight residues of apolipoprotein A-I mediate the C-terminus control of helical bundle unfolding and its lipidation. PLoS ONE 2020, 15, e0221915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guan, J.; Jiang, Z.; Yang, Y.; Liu, J.; Hua, W.; Mao, Y.; Li, C.; Lu, W.; Qian, J.; et al. Brain-targeted drug delivery by manipulating protein corona functions. Nat. Commun. 2019, 10, 3561. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, J.B.; Münter, R. Pay attention to biological nanoparticles when studying the protein corona on nanomedicines. Angew. Chem. Int. Ed. Engl. 2020, 59, 12584–12588. [Google Scholar] [CrossRef] [PubMed]
- de Groot, P.G.; Meijers, J.C. β(2)-Glycoprotein I: Evolution, structure and function. J. Thromb. Haemost. 2011, 9, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Erkan, D.; Salmon, J.E.; Lockshin, M.D. Anti-phospholipid Syndrome. In Kelley and Firestein’s Textbook of Rheumatology, 10th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 82; pp. 1389–1399. ISBN 9780323316965. [Google Scholar] [CrossRef]












| Plasma Control | MLPH | 5SX | 10SX | |||||
|---|---|---|---|---|---|---|---|---|
| Gene Name * | RPA % | Gene Name | RPA % | Gene Name | RPA % | Gene Name | RPA % | |
| 1 | APOC4-APOC2 ** | 27.6 ± 5.4 | APOC4-APOC2 | 20.0 ± 9.8 | APOC1 | 27.3 ± 4.1 | APOC1 | 30.4 ± 1.1 |
| 2 | IGHM | 13.3 ± 2.9 | APOC1 | 14.0 ± 0.8 | APOC4-APOC2 | 24.2 ± 3.0 | APOC4-APOC2 | 25.8 ± 1.8 |
| 3 | APOE | 9.3 ± 0.6 | IGHM | 11.0 ± 2.4 | APOC3 | 8.4 ± 2.3 | IGHM | 8.1 ± 0.6 |
| 4 | APOC1 | 7.1 ± 2.4 | APOC3 | 8.0 ± 2.2 | IGHM | 7.7 ± 1.7 | APOC3 | 6.8 ± 0.7 |
| 5 | IGKC | 7.0 ± 3.0 | IGKC | 6.3 ± 0.5 | IGKC | 4.2 ± 0.8 | IGKC | 4.1 ± 1.3 |
| 6 | APOC3 | 6.6 ± 1.9 | APOE | 5.5 ± 0.5 | APOE | 4.1 ± 0.4 | APOE | 4.1 ± 0.5 |
| 7 | IGLL5; IGLC1 | 3.8 ± 1.3 | ALB | 4.1 ± 0.5 | ALB | 3.5 ± 0.5 | ALB | 2.4 ± 0.2 |
| 8 | IGJ | 3.3 ± 1.0 | APOD | 3.2 ± 0.5 | SAA2-SAA4 | 1.8 ± 0.3 | SAA2-SAA4 | 1.7 ± 0.2 |
| 9 | CD5L | 1.7 ± 0.5 | IGJ | 2.4 ± 0.7 | APOA1 | 1.4 ± 0.2 | APOA1 | 1.4 ± 0.3 |
| 10 | APOA2 | 1.5 ± 0.9 | IGLL5; IGLC1 | 2.4 ± 0.5 | APOM | 1.4 ± 0.3 | IGJ | 1.4 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onishchenko, N.R.; Moskovtsev, A.A.; Kobanenko, M.K.; Tretiakova, D.S.; Alekseeva, A.S.; Kolesov, D.V.; Mikryukova, A.A.; Boldyrev, I.A.; Kapkaeva, M.R.; Shcheglovitova, O.N.; et al. Protein Corona Attenuates the Targeting of Antitumor Sialyl Lewis X-Decorated Liposomes to Vascular Endothelial Cells under Flow Conditions. Pharmaceutics 2023, 15, 1754. https://doi.org/10.3390/pharmaceutics15061754
Onishchenko NR, Moskovtsev AA, Kobanenko MK, Tretiakova DS, Alekseeva AS, Kolesov DV, Mikryukova AA, Boldyrev IA, Kapkaeva MR, Shcheglovitova ON, et al. Protein Corona Attenuates the Targeting of Antitumor Sialyl Lewis X-Decorated Liposomes to Vascular Endothelial Cells under Flow Conditions. Pharmaceutics. 2023; 15(6):1754. https://doi.org/10.3390/pharmaceutics15061754
Chicago/Turabian StyleOnishchenko, Natalia R., Alexey A. Moskovtsev, Maria K. Kobanenko, Daria S. Tretiakova, Anna S. Alekseeva, Dmitry V. Kolesov, Anna A. Mikryukova, Ivan A. Boldyrev, Marina R. Kapkaeva, Olga N. Shcheglovitova, and et al. 2023. "Protein Corona Attenuates the Targeting of Antitumor Sialyl Lewis X-Decorated Liposomes to Vascular Endothelial Cells under Flow Conditions" Pharmaceutics 15, no. 6: 1754. https://doi.org/10.3390/pharmaceutics15061754
APA StyleOnishchenko, N. R., Moskovtsev, A. A., Kobanenko, M. K., Tretiakova, D. S., Alekseeva, A. S., Kolesov, D. V., Mikryukova, A. A., Boldyrev, I. A., Kapkaeva, M. R., Shcheglovitova, O. N., Bovin, N. V., Kubatiev, A. A., Tikhonova, O. V., & Vodovozova, E. L. (2023). Protein Corona Attenuates the Targeting of Antitumor Sialyl Lewis X-Decorated Liposomes to Vascular Endothelial Cells under Flow Conditions. Pharmaceutics, 15(6), 1754. https://doi.org/10.3390/pharmaceutics15061754