
Next-Generation Adjuvants: Applying Engineering Methods to Create and Evaluate Novel Immunological Responses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction to Adjuvants
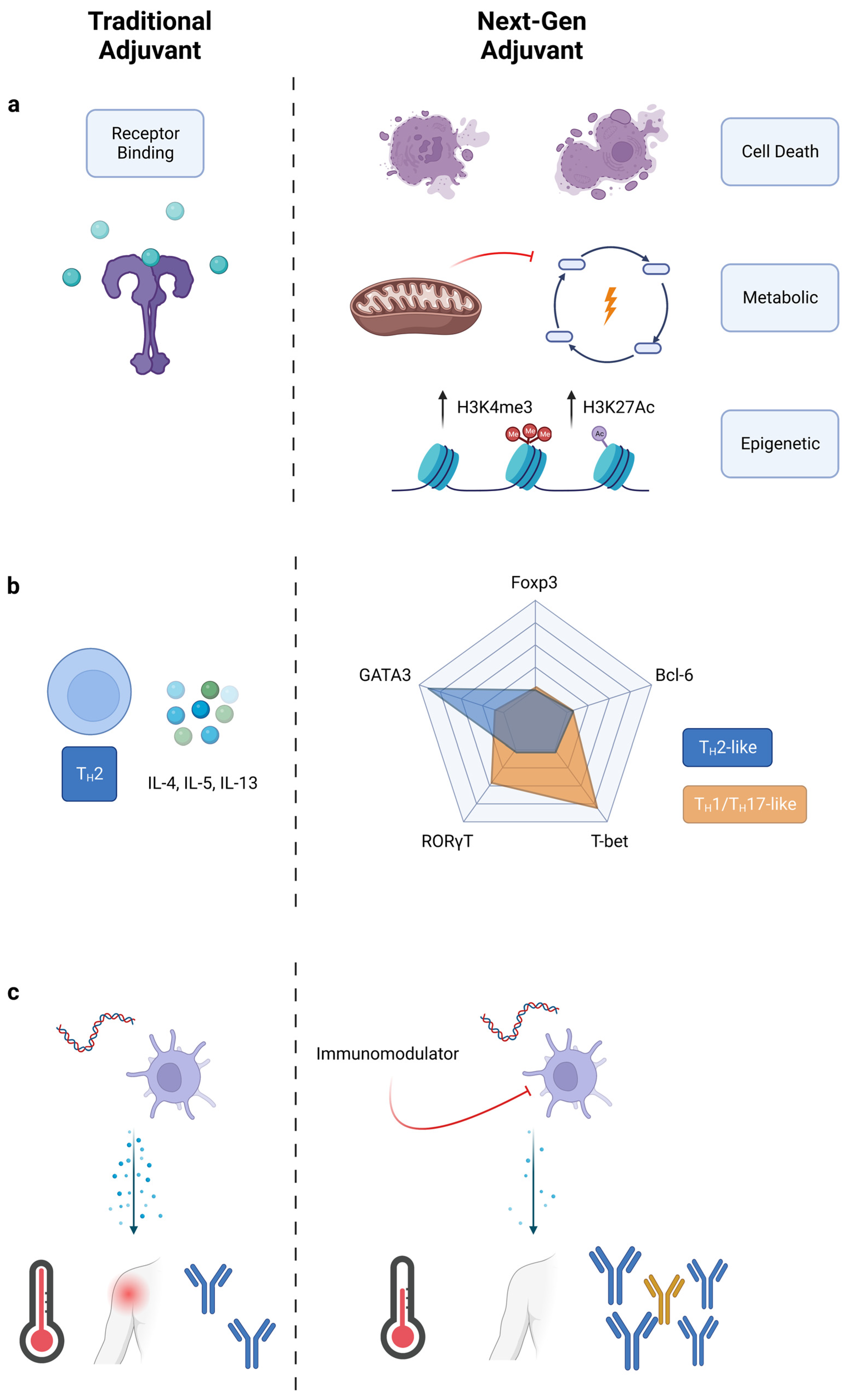
2. Traditional Adjuvant Development
3. Engineering New Outcomes
3.1. Target Identification and Signal Engineering
3.1.1. Alternative Pathways for Adjuvanticity
3.1.2. TH Polarization: Going beyond TH1, TH2, and TH17
3.1.3. Reducing Reactogenicity of Adjuvants
3.2. Formulation and Targeted Delivery
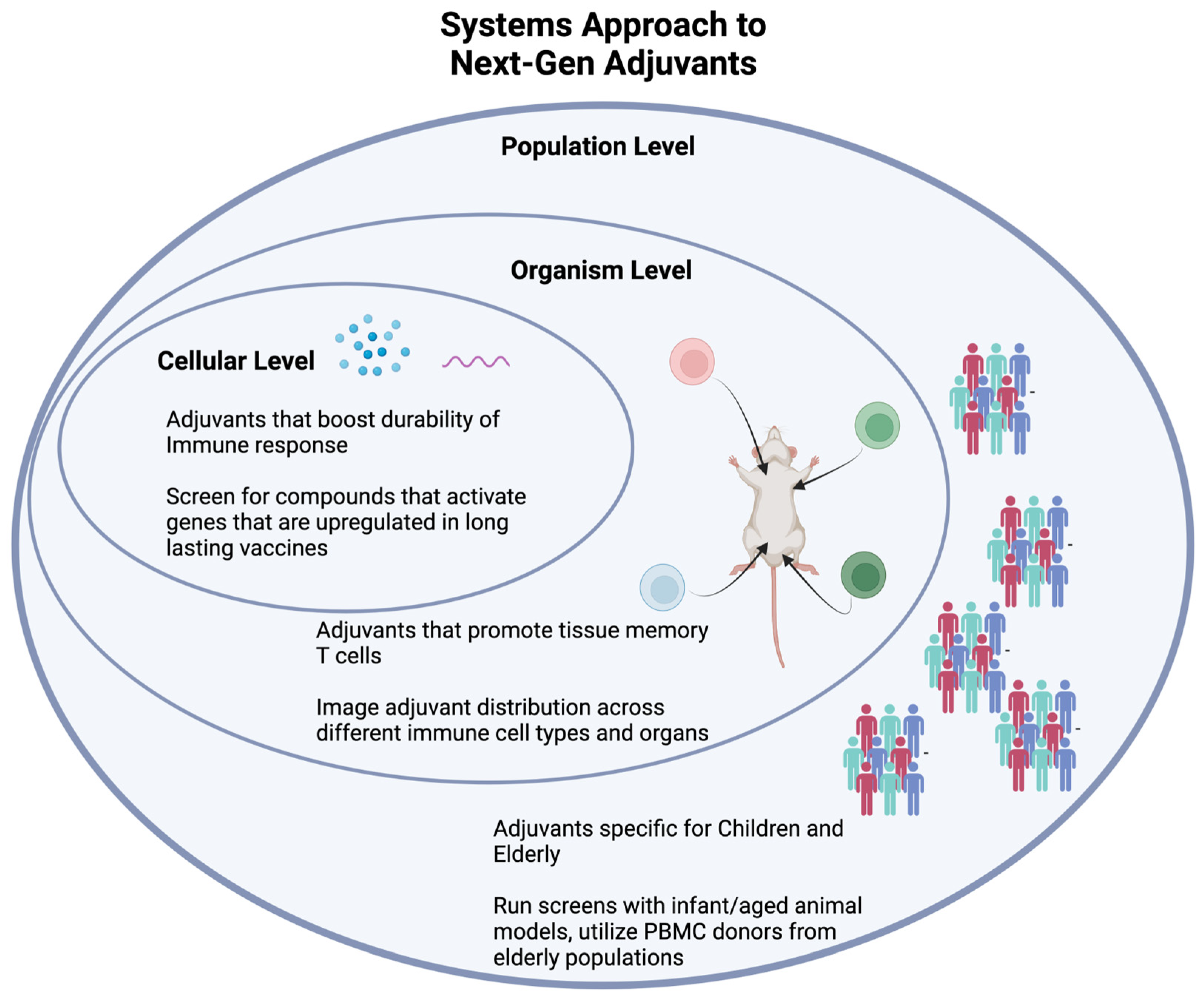
4. Engineering New Evaluation Methods
4.1. Computational and “Big Data” Approaches
4.1.1. Systems Vaccinology
4.1.2. In Silico and Machine Learning
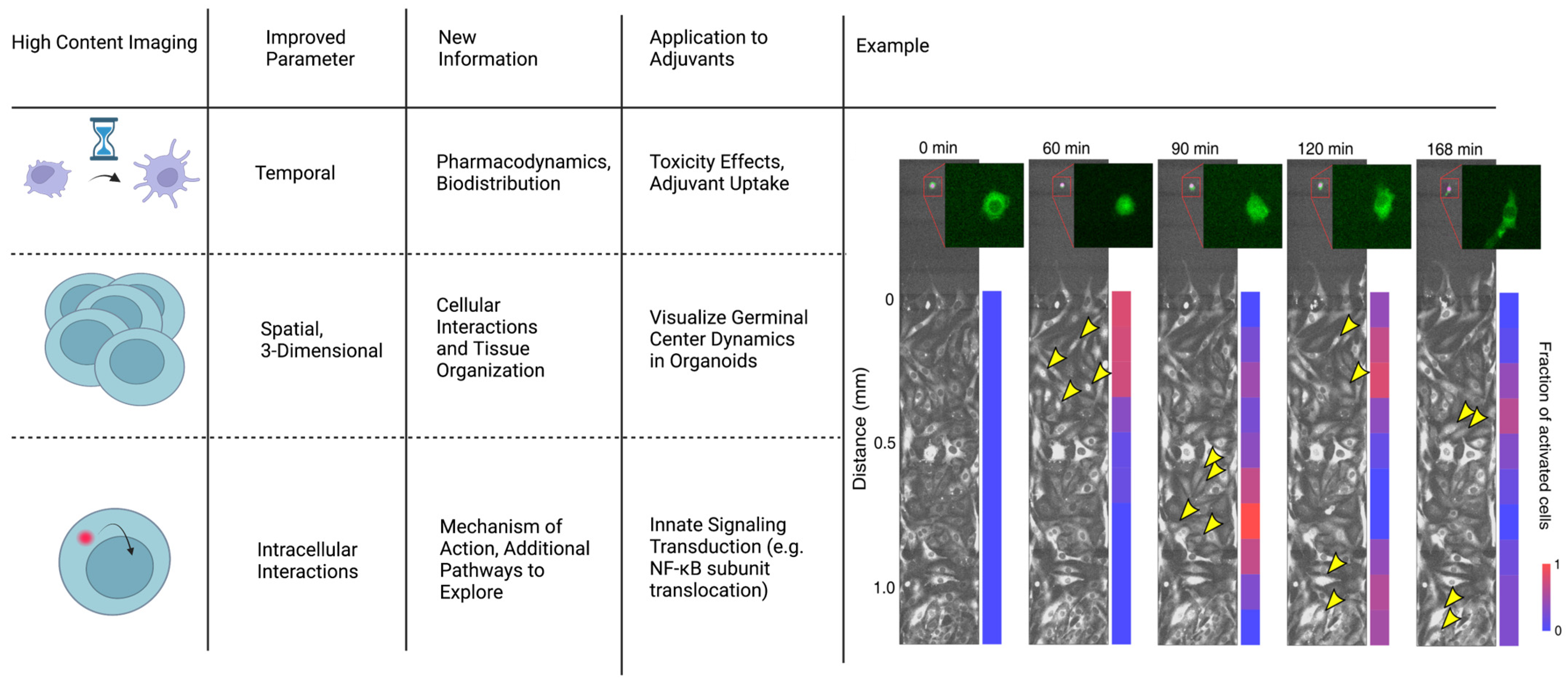
4.1.3. High-Content Imaging
4.2. Rethinking Animal Models
5. Engineering New Applications
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Apostólico, J.d.S.; Lunardelli, V.A.S.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1459394. [Google Scholar] [CrossRef] [PubMed]
- Gerberding, J.L.; Haynes, B.F. Vaccine Innovations—Past and Future. N. Engl. J. Med. 2021, 384, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Bergmann-Leitner, E.; Leitner, W. Adjuvants in the Driver’s Seat: How Magnitude, Type, Fine Specificity and Longevity of Immune Responses Are Driven by Distinct Classes of Immune Potentiators. Vaccines 2014, 2, 252–296. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H. Mechanism of Immunopotentiation and Safety of Aluminum Adjuvants. Front. Immun. 2013, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, A.; Preiss, S.; Tavares Da Silva, F.; Garçon, N. Vaccine Adjuvants: From 1920 to 2015 and Beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef]
- Pulendran, B.; Arunachalam, P.S.; O’Hagan, D.T. Emerging Concepts in the Science of Vaccine Adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- De Gregorio, E.; Tritto, E.; Rappuoli, R. Alum Adjuvanticity: Unraveling a Century Old Mystery. Eur. J. Immunol. 2008, 38, 2068–2071. [Google Scholar] [CrossRef]
- Dowling, D.J. Recent Advances in the Discovery and Delivery of TLR7/8 Agonists as Vaccine Adjuvants. ImmunoHorizons 2018, 2, 185–197. [Google Scholar] [CrossRef]
- Smith, A.J.; Li, Y.; Bazin, H.G.; St-Jean, J.R.; Larocque, D.; Evans, J.T.; Baldridge, J.R. Evaluation of Novel Synthetic TLR7/8 Agonists as Vaccine Adjuvants. Vaccine 2016, 34, 4304–4312. [Google Scholar] [CrossRef]
- Petrovsky, N. Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf. 2015, 38, 1059–1074. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic Cells Acquire Antigen from Apoptotic Cells and Induce Class I-Restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-ΚB Signaling in Dying Cells Determines Cross-Priming of CD8+ T Cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Ballas, Z.K.; Krieg, A.M.; Warren, T.; Rasmussen, W.; Davis, H.L.; Waldschmidt, M.; Weiner, G.J. Divergent Therapeutic and Immunologic Effects of Oligodeoxynucleotides with Distinct CpG Motifs. J. Immunol. 2001, 167, 4878–4886. [Google Scholar] [CrossRef]
- Bungener, L.; Geeraedts, F.; Ter Veer, W.; Medema, J.; Wilschut, J.; Huckriede, A. Alum Boosts TH2-Type Antibody Responses to Whole-Inactivated Virus Influenza Vaccine in Mice but Does Not Confer Superior Protection. Vaccine 2008, 26, 2350–2359. [Google Scholar] [CrossRef]
- Miyauchi, K.; Sugimoto-Ishige, A.; Harada, Y.; Adachi, Y.; Usami, Y.; Kaji, T.; Inoue, K.; Hasegawa, H.; Watanabe, T.; Hijikata, A.; et al. Protective Neutralizing Influenza Antibody Response in the Absence of T Follicular Helper Cells. Nat. Immunol. 2016, 17, 1447–1458. [Google Scholar] [CrossRef]
- Zambrano-Zaragoza, J.F.; Romo-Martínez, E.J.; Durán-Avelar, M.d.J.; García-Magallanes, N.; Vibanco-Pérez, N. Th17 Cells in Autoimmune and Infectious Diseases. Int. J. Inflamm. 2014, 2014, 651503. [Google Scholar] [CrossRef]
- Shen, H.; Chen, Z.W. The Crucial Roles of Th17-Related Cytokines/Signal Pathways in M. Tuberculosis Infection. Cell. Mol. Immunol. 2018, 15, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Lyadova, I.V.; Panteleev, A.V. Th1 and Th17 Cells in Tuberculosis: Protection, Pathology, and Biomarkers. Mediat. Inflamm. 2015, 2015, 854507. [Google Scholar] [CrossRef]
- Tuzlak, S.; Dejean, A.S.; Iannacone, M.; Quintana, F.J.; Waisman, A.; Ginhoux, F.; Korn, T.; Becher, B. Repositioning TH Cell Polarization from Single Cytokines to Complex Help. Nat. Immunol. 2021, 22, 1210–1217. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 Cells: Different Patterns of Lymphokine Secretion Lead to Different Functional Properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An Effector CD4 T Cell Lineage with Regulatory T Cell Ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef]
- Corthay, A. How Do Regulatory T Cells Work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Kaplan, M.H. Th9 Cells: Differentiation and Disease. Immunol. Rev. 2013, 252, 104–115. [Google Scholar] [CrossRef]
- Jia, L.; Wu, C. The Biology and Functions of Th22 Cells. Adv. Exp. Med. Biol. 2014, 841, 209–230. [Google Scholar] [CrossRef]
- Zhang, J.; Roberts, A.I.; Liu, C.; Ren, G.; Xu, G.; Zhang, L.; Devadas, S.; Shi, Y. A Novel Subset of Helper T Cells Promotes Immune Responses by Secreting GM-CSF. Cell Death Differ. 2013, 20, 1731–1741. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Weinmann, A.S. Master Regulators or Lineage-Specifying? Changing Views on CD4+ T Cell Transcription Factors. Nat. Rev. Immunol. 2012, 12, 799–804. [Google Scholar] [CrossRef]
- Mirlekar, B. Co-Expression of Master Transcription Factors Determines CD4+ T Cell Plasticity and Functions in Auto-Inflammatory Diseases. Immunol. Lett. 2020, 222, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Christian, L.M.; Porter, K.; Karlsson, E.; Schultz-Cherry, S. Proinflammatory Cytokine Responses Correspond with Subjective Side Effects after Influenza Virus Vaccination. Vaccine 2015, 33, 3360–3366. [Google Scholar] [CrossRef] [PubMed]
- Eigler, A.; Sinha, B.; Hartmann, G.; Endres, S. Taming TNF: Strategies to Restrain This Proinflammatory Cytokine. Immunol. Today 1997, 18, 487–492. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF Biology, Pathogenic Mechanisms and Emerging Therapeutic Strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.-S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. The IL-1β, IL-6, and TNF Cytokine Triad Is Associated with Post-Acute Sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three Decades of Messenger RNA Vaccine Development. Nano Today 2019, 28, 100766. [Google Scholar] [CrossRef]
- Tahtinen, S.; Tong, A.-J.; Himmels, P.; Oh, J.; Paler-Martinez, A.; Kim, L.; Wichner, S.; Oei, Y.; McCarron, M.J.; Freund, E.C.; et al. IL-1 and IL-1ra Are Key Regulators of the Inflammatory Response to RNA Vaccines. Nat. Immunol. 2022, 23, 532–542. [Google Scholar] [CrossRef]
- Matias, J.; Kurokawa, C.; Sajid, A.; Narasimhan, S.; Arora, G.; Diktas, H.; Lynn, G.E.; DePonte, K.; Pardi, N.; Valenzuela, J.G.; et al. Tick Immunity Using MRNA, DNA and Protein-Based Salp14 Delivery Strategies. Vaccine 2021, 39, 7661–7668. [Google Scholar] [CrossRef]
- Sajid, A.; Matias, J.; Arora, G.; Kurokawa, C.; DePonte, K.; Tang, X.; Lynn, G.; Wu, M.-J.; Pal, U.; Strank, N.O.; et al. MRNA Vaccination Induces Tick Resistance and Prevents Transmission of the Lyme Disease Agent. Sci. Transl. Med. 2021, 13, eabj9827. [Google Scholar] [CrossRef]
- Zhu, D.; Tuo, W. QS-21: A Potent Vaccine Adjuvant. Nat. Prod. Chem. Res. 2016, 3, e113. [Google Scholar] [CrossRef] [PubMed]
- Wang, P. Natural and Synthetic Saponins as Vaccine Adjuvants. Vaccines 2021, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-X.; Xie, Y.; Ye, Y.-P. Advances in Saponin-Based Adjuvants. Vaccine 2009, 27, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.; Orme, A.; El-Demerdash, A.; Owen, C.; Martin, L.B.B.; Misra, R.C.; Kikuchi, S.; Rejzek, M.; Martin, A.C.; Harkess, A.; et al. Elucidation of the Pathway for Biosynthesis of Saponin Adjuvants from the Soapbark Tree. Science 2023, 379, 1252–1264. [Google Scholar] [CrossRef]
- Moser, B.A.; Steinhardt, R.C.; Escalante-Buendia, Y.; Boltz, D.A.; Barker, K.M.; Cassaidy, B.J.; Rosenberger, M.G.; Yoo, S.; McGonnigal, B.G.; Esser-Kahn, A.P. Increased Vaccine Tolerability and Protection via NF-ΚB Modulation. Sci. Adv. 2020, 6, eaaz8700. [Google Scholar] [CrossRef]
- Moser, B.A.; Escalante-Buendia, Y.; Steinhardt, R.C.; Rosenberger, M.G.; Cassaidy, B.J.; Naorem, N.; Chon, A.C.; Nguyen, M.H.; Tran, N.T.; Esser-Kahn, A.P. Small Molecule NF-ΚB Inhibitors as Immune Potentiators for Enhancement of Vaccine Adjuvants. Front. Immunol. 2020, 11, 511513. [Google Scholar] [CrossRef]
- Garçon, N.; Di Pasquale, A. From Discovery to Licensure, the Adjuvant System Story. Hum. Vaccin Immunother. 2016, 13, 19–33. [Google Scholar] [CrossRef]
- Lövgren, K.; Morein, B. The Requirement of Lipids for the Formation of Immunostimulating Complexes (Iscoms). Biotechnol. Appl. Biochem. 1988, 10, 161–172. [Google Scholar] [CrossRef]
- Reimer, J.M.; Karlsson, K.H.; Lövgren-Bengtsson, K.; Magnusson, S.E.; Fuentes, A.; Stertman, L. Matrix-MTM Adjuvant Induces Local Recruitment, Activation and Maturation of Central Immune Cells in Absence of Antigen. PLoS ONE 2012, 7, e41451. [Google Scholar] [CrossRef] [PubMed]
- Koshy, S.T.; Cheung, A.S.; Gu, L.; Graveline, A.R.; Mooney, D.J. Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv. Biosyst. 2017, 1, 1600013. [Google Scholar] [CrossRef] [PubMed]
- Schudel, A.; Francis, D.M.; Thomas, S.N. Material Design for Lymph Node Drug Delivery. Nat. Rev. Mater. 2019, 4, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.N.; Vokali, E.; Lund, A.W.; Hubbell, J.A.; Swartz, M.A. Targeting the Tumor-Draining Lymph Node with Adjuvanted Nanoparticles Reshapes the Anti-Tumor Immune Response. Biomaterials 2014, 35, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Wolf, N.; Ward, R.W.; Hearnden, C.H.; Sharp, F.A.; Geoghegan, J.; O’Grady, K.; McEntee, C.P.; Shanahan, K.A.; Guy, C.; Bowie, A.G.; et al. Non-Canonical Inflammasome Activation Mediates the Adjuvanticity of Nanoparticles. Cell Rep. Med. 2023, 4, 4024552. [Google Scholar] [CrossRef]
- Oyewumi, M.O.; Kumar, A.; Cui, Z. Nano-Microparticles as Immune Adjuvants: Correlating Particle Sizes and the Resultant Immune Responses. Expert Rev. Vaccines 2010, 9, 1095–1107. [Google Scholar] [CrossRef]
- Yang, X.; Coriolan, D.; Murthy, V.; Schultz, K.; Golenbock, D.T.; Beasley, D. Proinflammatory Phenotype of Vascular Smooth Muscle Cells: Role of Efficient Toll-like Receptor 4 Signaling. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1069–H1076. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of Antigen-Specific Immunity with a Vaccine Targeting NY-ESO-1 to the Dendritic Cell Receptor DEC-205. Sci. Transl. Med. 2014, 6, ra51–ra232. [Google Scholar] [CrossRef]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The Dendritic Cell Receptor for Endocytosis, DEC-205, Can Recycle and Enhance Antigen Presentation via Major Histocompatibility Complex Class II-Positive Lysosomal Compartments. J. Cell Biol. 2000, 151, 673–684. [Google Scholar] [CrossRef]
- van Kooyk, Y.; Unger, W.W.J.; Fehres, C.M.; Kalay, H.; García-Vallejo, J.J. Glycan-Based DC-SIGN Targeting Vaccines to Enhance Antigen Cross-Presentation. Mol. Immunol. 2013, 55, 143–145. [Google Scholar] [CrossRef]
- Stack, T.; Vincent, M.; Vahabikashi, A.; Li, G.; Perkumas, K.M.; Stamer, W.D.; Johnson, M.; Scott, E. Targeted Delivery of Cell Softening Micelles to Schlemm’s Canal Endothelial Cells for Treatment of Glaucoma. Small 2020, 16, 2004205. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, B.A.; Burchill, M.A.; Kedl, R.M. Antigen Capture and Archiving by Lymphatic Endothelial Cells Following Vaccination or Viral Infection. Nat. Commun. 2014, 5, 3989. [Google Scholar] [CrossRef]
- Xue, Q.; Lu, Y.; Eisele, M.R.; Sulistijo, E.S.; Khan, N.; Fan, R.; Miller-Jensen, K. Analysis of Single-Cell Cytokine Secretion Reveals a Role for Paracrine Signaling in Coordinating Macrophage Responses to TLR4 Stimulation. Sci. Signal. 2015, 8, ra59. [Google Scholar] [CrossRef] [PubMed]
- Deak, P.; Studnitzer, B.; Ung, T.; Steinhardt, R.; Swartz, M.; Esser-Kahn, A. Isolating and Targeting a Highly Active, Stochastic Dendritic Cell Subpopulation for Improved Immune Responses. Cell Rep. 2022, 41, 111563. [Google Scholar] [CrossRef] [PubMed]
- Tomalka, J.A.; Suthar, M.S.; Deeks, S.G.; Sekaly, R.P. Fighting the SARS-CoV-2 Pandemic Requires a Global Approach to Understanding the Heterogeneity of Vaccine Responses. Nat. Immunol. 2022, 23, 360–370. [Google Scholar] [CrossRef]
- Davies, M.N.; Bayry, J.; Tchilian, E.Z.; Vani, J.; Shaila, M.S.; Forbes, E.K.; Draper, S.J.; Beverley, P.C.L.; Tough, D.F.; Flower, D.R. Toward the Discovery of Vaccine Adjuvants: Coupling In Silico Screening and In Vitro Analysis of Antagonist Binding to Human and Mouse CCR4 Receptors. PLoS ONE 2009, 4, e8084. [Google Scholar] [CrossRef] [PubMed]
- Tomalka, J.A.; Pelletier, A.N.; Fourati, S.; Latif, M.B.; Sharma, A.; Furr, K.; Carlson, K.; Lifton, M.; Gonzalez, A.; Wilkinson, P.; et al. The Transcription Factor CREB1 Is a Mechanistic Driver of Immunogenicity and Reduced HIV-1 Acquisition Following ALVAC Vaccination. Nat. Immunol. 2021, 22, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Kadoki, M.; Patil, A.; Thaiss, C.C.; Brooks, D.J.; Pandey, S.; Deep, D.; Alvarez, D.; von Andrian, U.H.; Wagers, A.J.; Nakai, K.; et al. Organism-Level Analysis of Vaccination Reveals Networks of Protection across Tissues. Cell 2017, 171, 398–413.e21. [Google Scholar] [CrossRef]
- Nanishi, E.; Borriello, F.; Seo, H.-S.; O’Meara, T.R.; McGrath, M.E.; Saito, Y.; Chen, J.; Diray-Arce, J.; Song, K.; Xu, A.Z.; et al. Carbohydrate Fatty Acid Monosulphate: Oil-in-Water Adjuvant Enhances SARS-CoV-2 RBD Nanoparticle-Induced Immunogenicity and Protection in Mice. NPJ Vaccines 2023, 8, 18. [Google Scholar] [CrossRef]
- Hagan, T.; Gerritsen, B.; Tomalin, L.E.; Fourati, S.; Mulè, M.P.; Chawla, D.G.; Rychkov, D.; Henrich, E.; Miller, H.E.R.; Diray-Arce, J.; et al. Transcriptional Atlas of the Human Immune Response to 13 Vaccines Reveals a Common Predictor of Vaccine-Induced Antibody Responses. Nat. Immunol. 2022, 23, 1788–1798. [Google Scholar] [CrossRef]
- Dave, A.; Mitchell, J.; Kandasamy, K.; Wang, H.; Burke, S.; Paria, B.; Póczos, B.; Whitacre, J.; Viswanathan, V. Autonomous Discovery of Battery Electrolytes with Robotic Experimentation and Machine Learning. CR-PHYS-SC 2020, 1, 100264. [Google Scholar] [CrossRef]
- Hie, B.; Bryson, B.D.; Berger, B. Leveraging Uncertainty in Machine Learning Accelerates Biological Discovery and Design. Cell Syst. 2020, 11, 461–477.e9. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-Scale Comparison of Machine Learning Methods for Drug Target Prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [PubMed]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M. Molecular Docking as a Popular Tool in Drug Design, an in Silico Travel. AABC 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Terayama, K.; Sumita, M.; Tamura, R.; Tsuda, K. Black-Box Optimization for Automated Discovery. Acc. Chem. Res. 2021, 54, 1334–1346. [Google Scholar] [CrossRef]
- Harding, J.D. Nonhuman Primates and Translational Research: Progress, Opportunities, and Challenges. ILAR J. 2017, 58, 141–150. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of Clinical Drug Development Fails and How to Improve It? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Lin, S.; Schorpp, K.; Rothenaigner, I.; Hadian, K. Image-Based High-Content Screening in Drug Discovery. Drug Discov. Today 2020, 25, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Løye, A.F.; Mow, T.; Hornberg, J.J. A High Content Screening Assay to Predict Human Drug-Induced Liver Injury during Drug Discovery. J. Pharmacol. Toxicol. Methods 2013, 68, 302–313. [Google Scholar] [CrossRef]
- Reid, B.G.; Jerjian, T.; Patel, P.; Zhou, Q.; Yoo, B.H.; Kabos, P.; Sartorius, C.A.; LaBarbera, D.V. Live Multicellular Tumor Spheroid Models For High-Content Imaging and Screening In Cancer Drug Discovery. CCGTM 2014, 8, 27–35. [Google Scholar] [CrossRef]
- Yu, B.; Li, B.; Chen, T.; Yang, J.; Wang, X.; Peng, B.; Hu, Q. A NF-ΚB-Based High-Throughput Screening for Immune Adjuvants and Inhibitors. Inflammation 2022, 46, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.P.; Carstens, M.R.; Lewis, J.S.; Dolgova, N.; Xia, C.Q.; Clare-Salzler, M.J.; Keselowsky, B.G. A Cell-Based Microarray to Investigate Combinatorial Effects of Microparticle-Encapsulated Adjuvants on Dendritic Cell Activation. J. Mater. Chem. B 2016, 4, 1672–1685. [Google Scholar] [CrossRef]
- Lv, X.; Chen, D.; Yang, L.; Zhu, N.; Li, J.; Zhao, J.; Hu, Z.; Wang, F.-J.; Zhang, L.W. Comparative Studies on the Immunoregulatory Effects of Three Polysaccharides Using High Content Imaging System. Int. J. Biol. Macromol. 2016, 86, 28–42. [Google Scholar] [CrossRef]
- Wilson, D.S.; Hirosue, S.; Raczy, M.M.; Bonilla-Ramirez, L.; Jeanbart, L.; Wang, R.; Kwissa, M.; Franetich, J.-F.; Broggi, M.A.S.; Diaceri, G.; et al. Antigens Reversibly Conjugated to a Polymeric Glyco-Adjuvant Induce Protective Humoral and Cellular Immunity. Nat. Mater. 2019, 18, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jin, R.; Jiang, D.; Chen, H.-Y. Electrochemiluminescence-Based Capacitance Microscopy for Label-Free Imaging of Antigens on the Cellular Plasma Membrane. J. Am. Chem. Soc. 2019, 141, 10294–10299. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Frank, T.; Holst-Hansen, T.; Wang, A.G.; Junkin, M.; Kashaf, S.S.; Trusina, A.; Tay, S. Spatiotemporal NF-κB dynamics encodes the position, amplitude, and duration of local immune inputs. Sci. Adv. 2022, 8, eabn6240. [Google Scholar] [CrossRef]
- Herati, R.S.; Wherry, E.J. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? Cold Spring Harb. Perspect. Biol. 2018, 10, a031583. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.E.; Monie, T.P. Mice, Men and the Relatives: Cross-Species Studies Underpin Innate Immunity. Open Biol. 2012, 2, 120015. [Google Scholar] [CrossRef]
- Cervantes, J.L.; Weinerman, B.; Basole, C.; Salazar, J.C. TLR8: The Forgotten Relative Revindicated. Cell Mol. Immunol. 2012, 9, 434–438. [Google Scholar] [CrossRef]
- Guiducci, C.; Gong, M.; Cepika, A.-M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA Recognition by Human TLR8 Can Lead to Autoimmune Inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef]
- Hu, Z.; Tanji, H.; Jiang, S.; Zhang, S.; Koo, K.; Chan, J.; Sakaniwa, K.; Ohto, U.; Candia, A.; Shimizu, T.; et al. Small-Molecule TLR8 Antagonists via Structure-Based Rational Design. Cell Chem. Biol. 2018, 25, 1286–1291.e3. [Google Scholar] [CrossRef]
- Jiang, S.; Li, X.; Hess, N.J.; Guan, Y.; Tapping, R.I. TLR10 Is a Negative Regulator of Both MyD88-Dependent and -Independent TLR Signaling. J. Immunol. 2016, 196, 3834–3841. [Google Scholar] [CrossRef]
- Rehli, M. Of Mice and Men: Species Variations of Toll-like Receptor Expression. Trends Immunol. 2002, 23, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized Immune System Mouse Models: Progress, Challenges and Opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of Functional Human Blood and Immune Systems in NOD/SCID/IL2 Receptor γ Chainnull Mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Blancher, A.; Inoko, H.; Kulski, J.K. Comparative Genomics of the Human, Macaque and Mouse Major Histocompatibility Complex. Immunology 2017, 150, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of Functional Human T-Cell Subsets with HLA-Restricted Immune Responses in HLA Class I Expressing NOD/SCID/IL2rγnull Humanized Mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef]
- Roßmann, L.; Bagola, K.; Stephen, T.; Gerards, A.-L.; Walber, B.; Ullrich, A.; Schülke, S.; Kamp, C.; Spreitzer, I.; Hasan, M.; et al. Distinct Single-Component Adjuvants Steer Human DC-Mediated T-Cell Polarization via Toll-like Receptor Signaling toward a Potent Antiviral Immune Response. Proc. Natl. Acad. Sci. USA 2021, 118, e2103651118. [Google Scholar] [CrossRef]
- Chew, K.; Lee, B.; van Haren, S.D.; Nanishi, E.; O’Meara, T.; Splaine, J.B.; DeLeon, M.; Soni, D.; Seo, H.-S.; Dhe-Paganon, S.; et al. Adjuvant Discovery via a High Throughput Screen Using Human Primary Mononuclear Cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Fan, Y.; Naglich, J.G.; Koenitzer, J.D.; Ribeiro, H.; Lippy, J.; Blum, J.; Li, X.; Milburn, C.; Barnhart, B.; Zhang, L.; et al. Miniaturized High-Throughput Multiparameter Flow Cytometry Assays Measuring In Vitro Human Dendritic Cell Maturation and T-Cell Activation in Mixed Lymphocyte Reactions. SLAS Discov. Adv. Sci. Drug Discov. 2018, 23, 742–750. [Google Scholar] [CrossRef]
- Allen, C.D.; Okada, T.; Cyster, J.G. Germinal Center Organization and Cellular Dynamics. Immunity 2007, 27, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Lisk, C.; Yuen, R.; Kuniholm, J.; Antos, D.; Reiser, M.L.; Wetzler, L.M. Toll-Like Receptor Ligand Based Adjuvant, PorB, Increases Antigen Deposition on Germinal Center Follicular Dendritic Cells While Enhancing the Follicular Dendritic Cells Network. Front. Immunol. 2020, 11, 1254. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Roh, K.-H.; Song, H.W.; Pradhan, P.; Bai, K.; Bohannon, C.D.; Dale, G.; Leleux, J.; Jacob, J.; Roy, K. A Synthetic Stroma-Free Germinal Center Niche for Efficient Generation of Humoral Immunity Ex Vivo. Biomaterials 2018, 164, 106–120. [Google Scholar] [CrossRef]
- Wagar, L.E.; Salahudeen, A.; Constantz, C.M.; Wendel, B.S.; Lyons, M.M.; Mallajosyula, V.; Jatt, L.P.; Adamska, J.Z.; Blum, L.K.; Gupta, N.; et al. Modeling Human Adaptive Immune Responses with Tonsil Organoids. Nat. Med. 2021, 27, 125–135. [Google Scholar] [CrossRef]
- Purwada, A.; Jaiswal, M.K.; Ahn, H.; Nojima, T.; Kitamura, D.; Gaharwar, A.K.; Cerchietti, L.; Singh, A. Ex Vivo Engineered Immune Organoids for Controlled Germinal Center Reactions. Biomaterials 2015, 63, 24–34. [Google Scholar] [CrossRef]
- Paston, S.J.; Brentville, V.A.; Symonds, P.; Durrant, L.G. Cancer Vaccines, Adjuvants, and Delivery Systems. Front. Immunol. 2021, 12, 627932. [Google Scholar] [CrossRef]
- Kruit, W.H.J.; Suciu, S.; Dreno, B.; Mortier, L.; Robert, C.; Chiarion-Sileni, V.; Maio, M.; Testori, A.; Dorval, T.; Grob, J.-J.; et al. Selection of Immunostimulant AS15 for Active Immunization with MAGE-A3 Protein: Results of a Randomized Phase II Study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 2413–2420. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and Senostatics as Adjuvant Tumour Therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and Senotherapeutics: A New Field in Cancer Therapy. Pharm. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Fletcher-Sananikone, E.; Kanji, S.; Tomimatsu, N.; Di Cristofaro, L.F.M.; Kollipara, R.K.; Saha, D.; Floyd, J.R.; Sung, P.; Hromas, R.; Burns, T.C.; et al. Elimination of Radiation-Induced Senescence in the Brain Tumor Microenvironment Attenuates Glioblastoma Recurrence. Cancer Res. 2021, 81, 5935–5947. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hong, B.; Li, X.; Deng, K.; Li, H.; Yan Lui, V.W.; Lin, W. JQ1 Synergizes with the Bcl-2 Inhibitor ABT-263 against MYCN-Amplified Small Cell Lung Cancer. Oncotarget 2017, 8, 86312–86324. [Google Scholar] [CrossRef]
- Guccini, I.; Revandkar, A.; D’Ambrosio, M.; Colucci, M.; Pasquini, E.; Mosole, S.; Troiani, M.; Brina, D.; Sheibani-Tezerji, R.; Elia, A.R.; et al. Senescence Reprogramming by TIMP1 Deficiency Promotes Prostate Cancer Metastasis. Cancer Cell 2021, 39, 68–82.e9. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Tan, A.C.; Bagley, S.J.; Wen, P.Y.; Lim, M.; Platten, M.; Colman, H.; Ashley, D.M.; Wick, W.; Chang, S.M.; Galanis, E.; et al. Systematic Review of Combinations of Targeted or Immunotherapy in Advanced Solid Tumors. J. Immunother. Cancer 2021, 9, e002459. [Google Scholar] [CrossRef]
- Maeng, H.; Terabe, M.; Berzofsky, J.A. Cancer Vaccines: Translation from Mice to Human Clinical Trials. Curr. Opin. Immunol. 2018, 51, 111–122. [Google Scholar] [CrossRef]
- Lai, X.; Friedman, A. Combination Therapy of Cancer with Cancer Vaccine and Immune Checkpoint Inhibitors: A Mathematical Model. PLoS ONE 2017, 12, e0178479. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.Y.; Rosenberger, M.G.; Rutledge, N.S.; Esser-Kahn, A.P. Next-Generation Adjuvants: Applying Engineering Methods to Create and Evaluate Novel Immunological Responses. Pharmaceutics 2023, 15, 1687. https://doi.org/10.3390/pharmaceutics15061687
Kim JY, Rosenberger MG, Rutledge NS, Esser-Kahn AP. Next-Generation Adjuvants: Applying Engineering Methods to Create and Evaluate Novel Immunological Responses. Pharmaceutics. 2023; 15(6):1687. https://doi.org/10.3390/pharmaceutics15061687
Chicago/Turabian StyleKim, Jeremiah Y., Matthew G. Rosenberger, Nakisha S. Rutledge, and Aaron P. Esser-Kahn. 2023. "Next-Generation Adjuvants: Applying Engineering Methods to Create and Evaluate Novel Immunological Responses" Pharmaceutics 15, no. 6: 1687. https://doi.org/10.3390/pharmaceutics15061687
APA StyleKim, J. Y., Rosenberger, M. G., Rutledge, N. S., & Esser-Kahn, A. P. (2023). Next-Generation Adjuvants: Applying Engineering Methods to Create and Evaluate Novel Immunological Responses. Pharmaceutics, 15(6), 1687. https://doi.org/10.3390/pharmaceutics15061687