
Non-Invasive, Targeted Nanoparticle-Mediated Drug Delivery across a Novel Human BBB Model


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.1.1. Materials
Cell Culture
Transwell Model
2.1.2. Methods
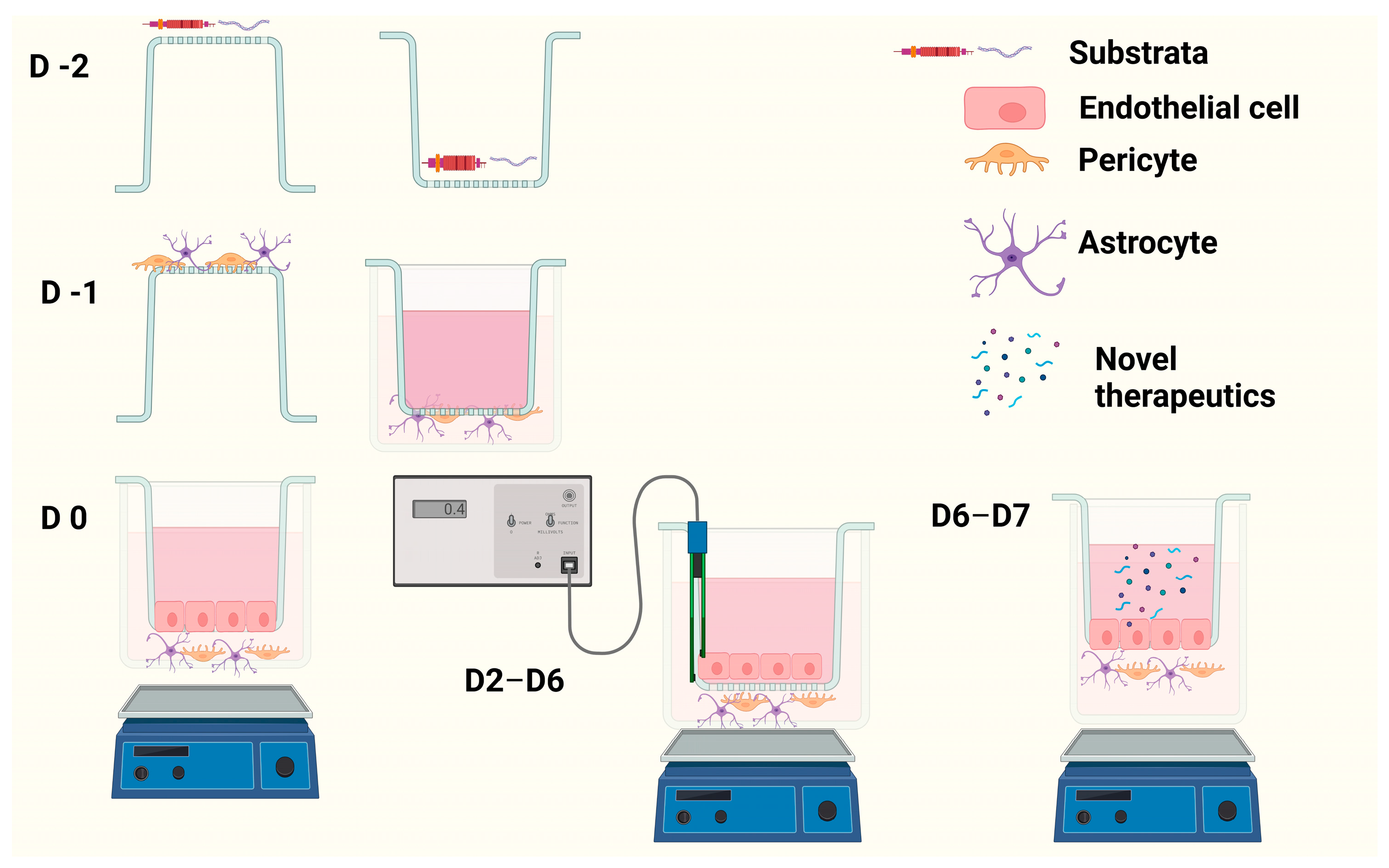
Preparation of Cells for In Vitro EP + A transwell BBB Model
Preparation of In Vitro Tri-Culture Transwell BBB Model
2.2. Evaluation of Barrier Integrity
2.2.1. Materials
2.2.2. Methods: Preparation of DAS-Labelled Fitc-Dextran
2.2.3. Measurement of Barrier Permeability
2.3. Immunocytochemistry
2.3.1. Materials
2.3.2. Methods
2.4. Protein Expression
2.4.1. Materials
2.4.2. Methods
2.4.3. SDS-PAGE and Western Blotting
Materials
Methods
2.5. Nanoparticles (NP)
2.5.1. Nanoparticle Formulation
Materials
Methods
2.5.2. Sizing and PDI
Materials
Methods
2.5.3. Encapsulation Efficiency
Materials
Methods
2.5.4. Release Assay
Materials
Methods
2.5.5. Conjugation Efficiency
Materials
Methods
2.5.6. Delivery across BBB Model
Materials
Methods
3. Results
3.1. Determining Transwell Model Architecture
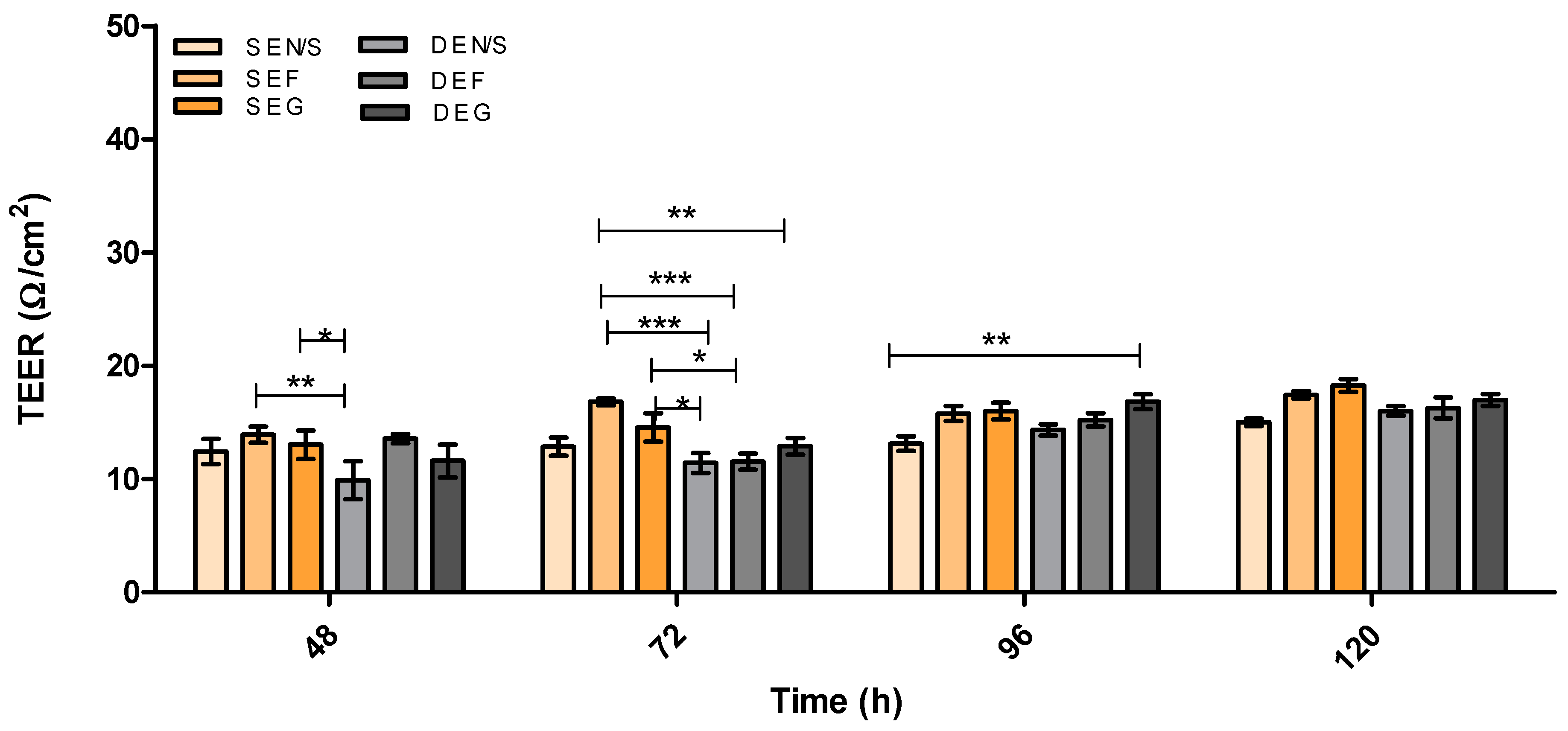
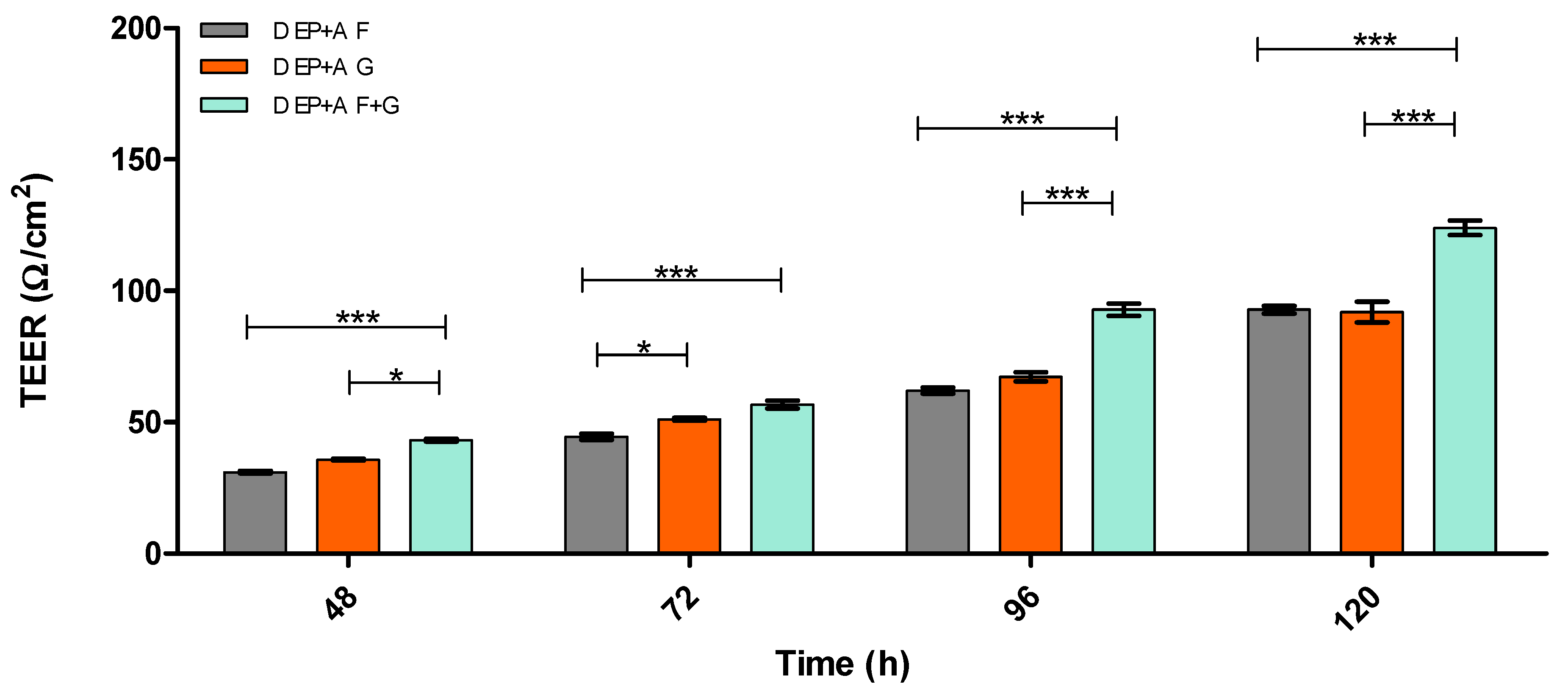
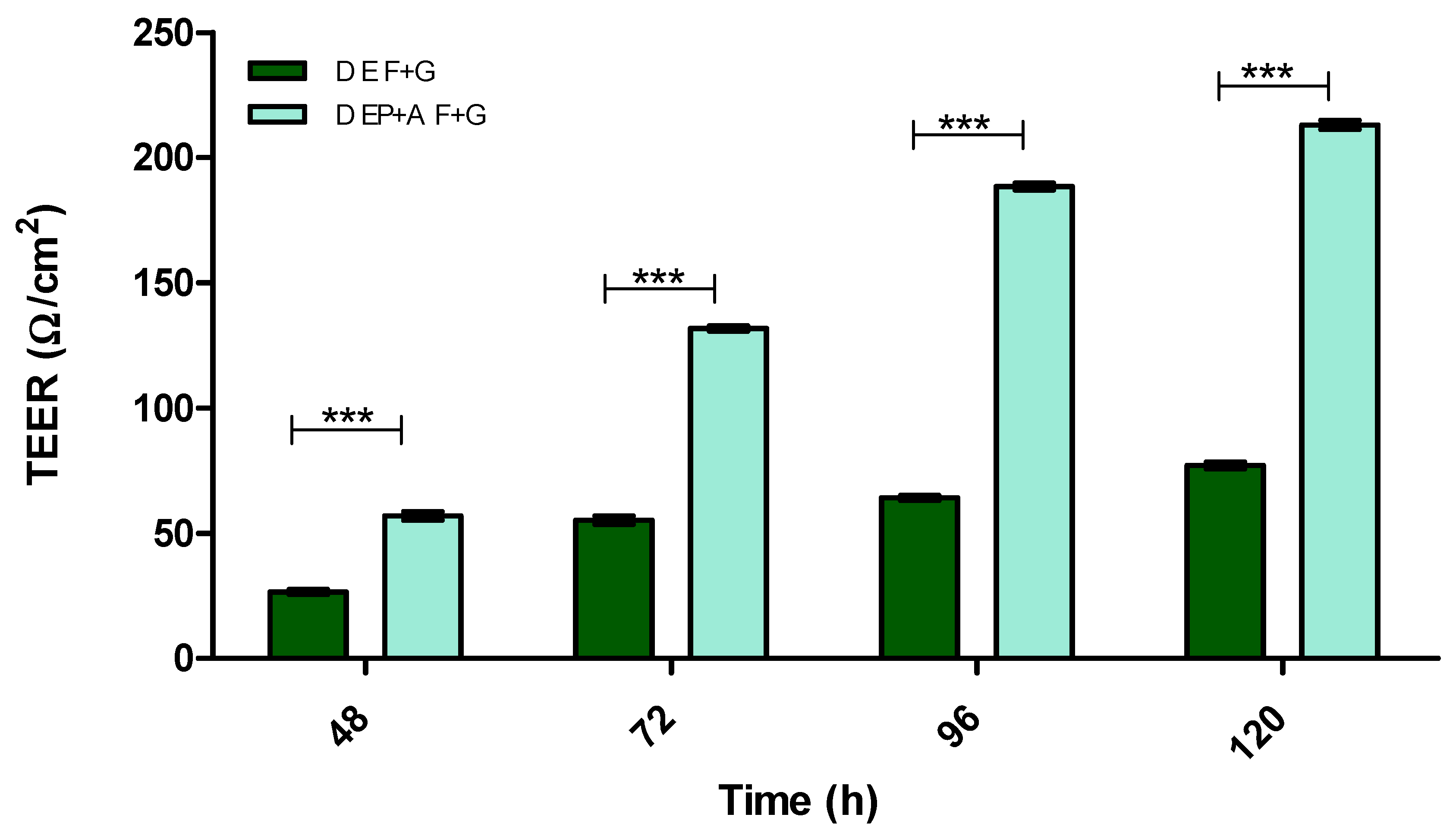
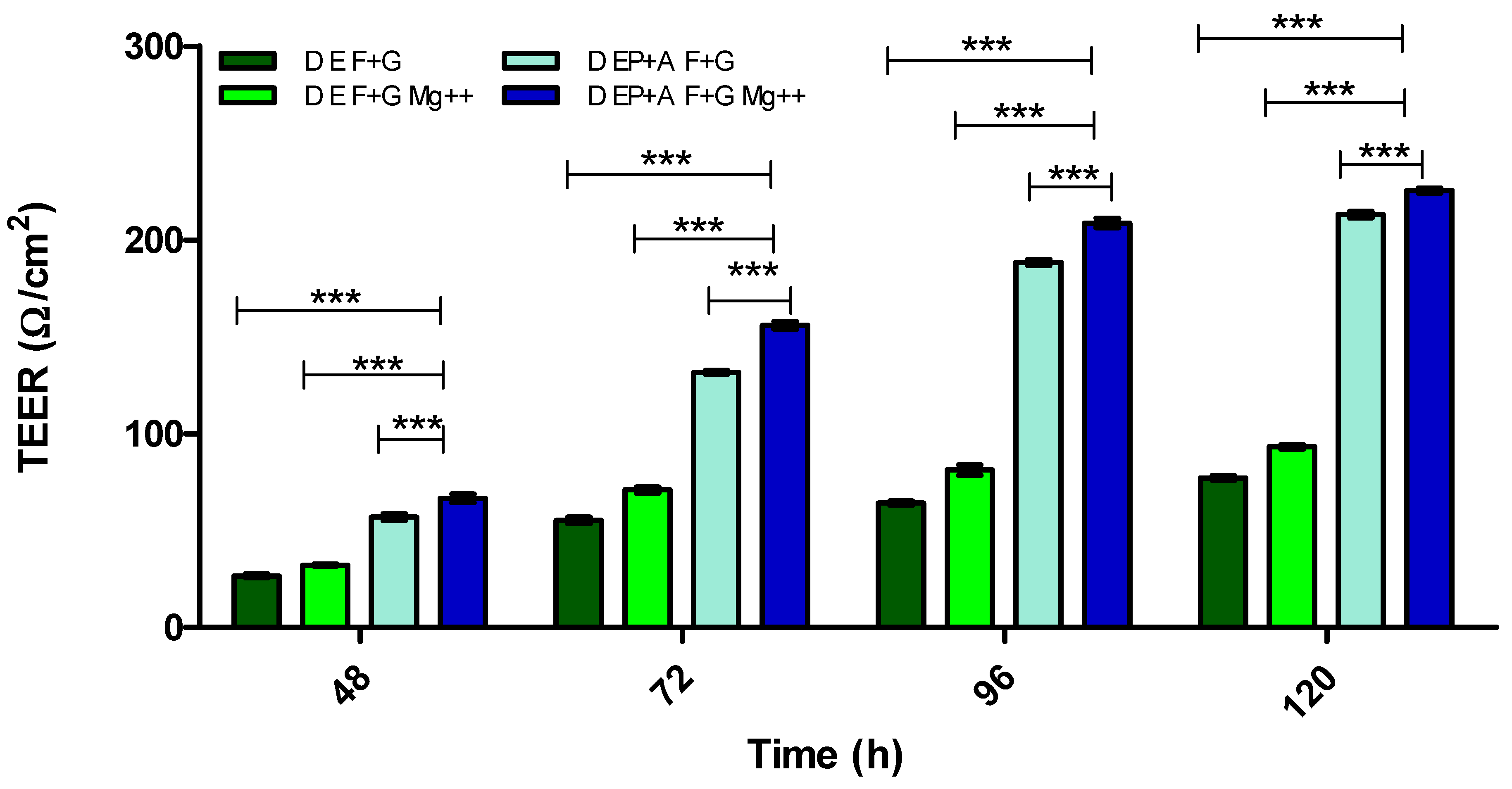
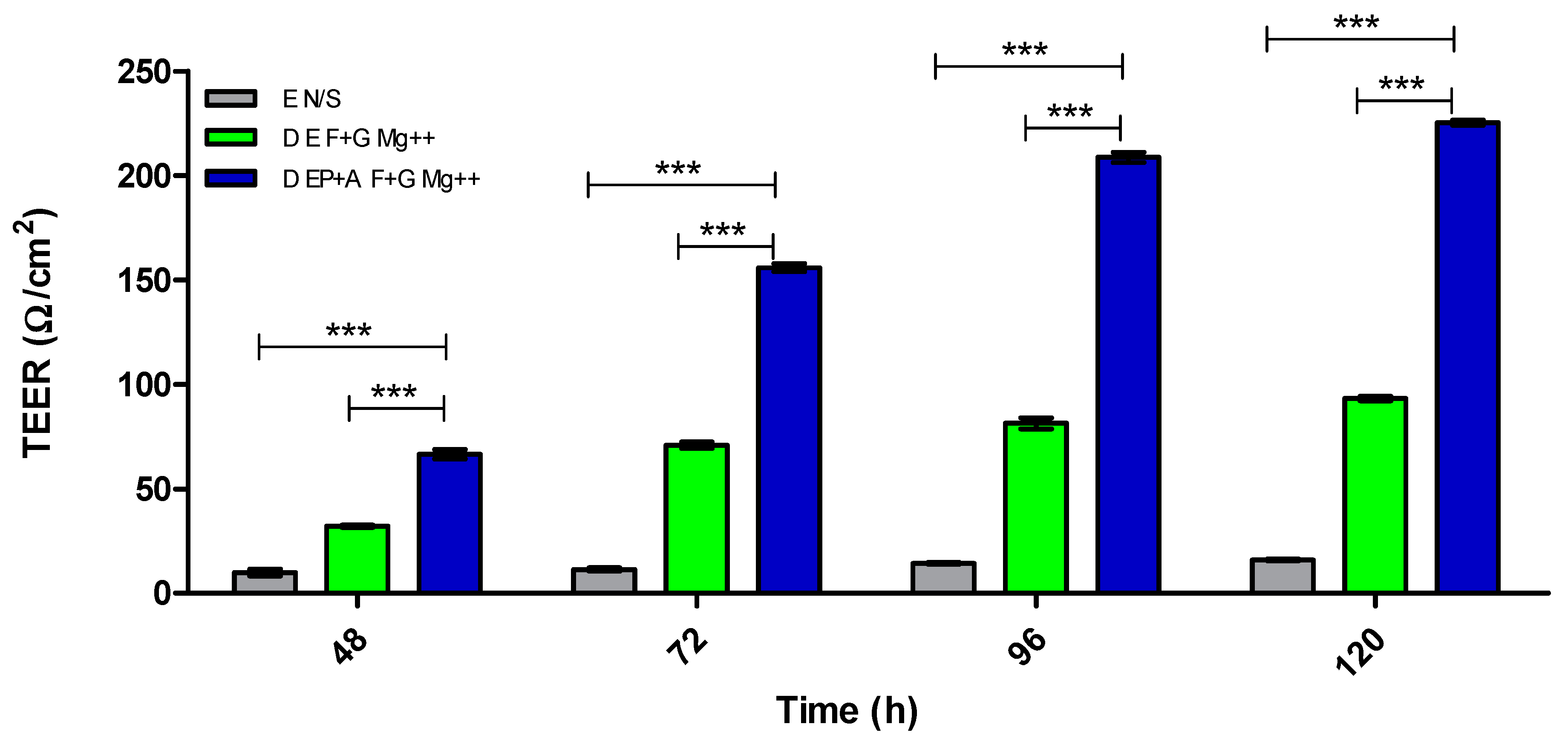
3.2. Evaluation of Barrier Integrity
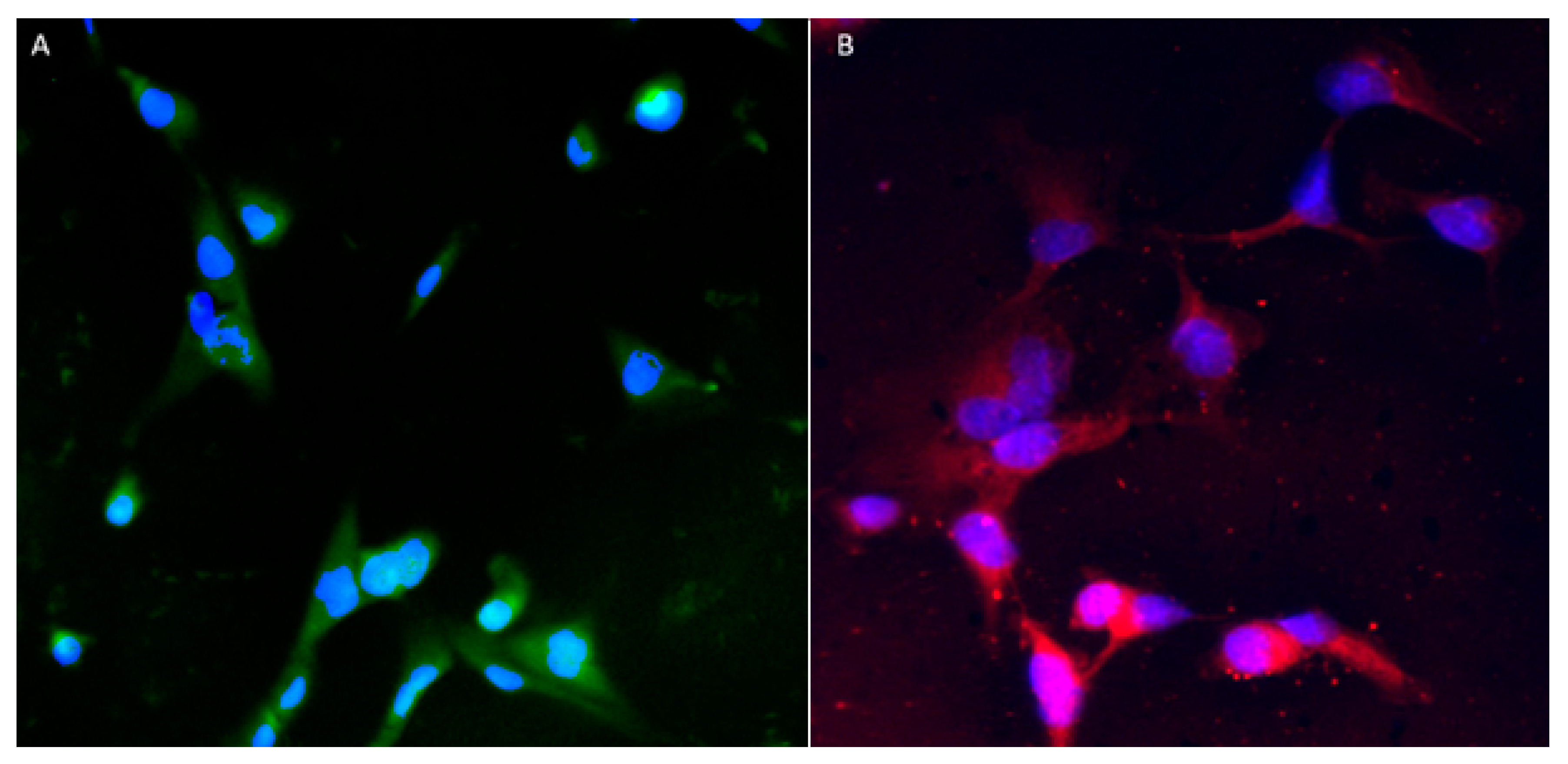
3.3. Immunocytochemistry
3.4. Protein Expression
SDS-PAGE and Western Blotting
3.5. Sizing and PDI
3.6. Encapsulation Efficiency
3.7. Release
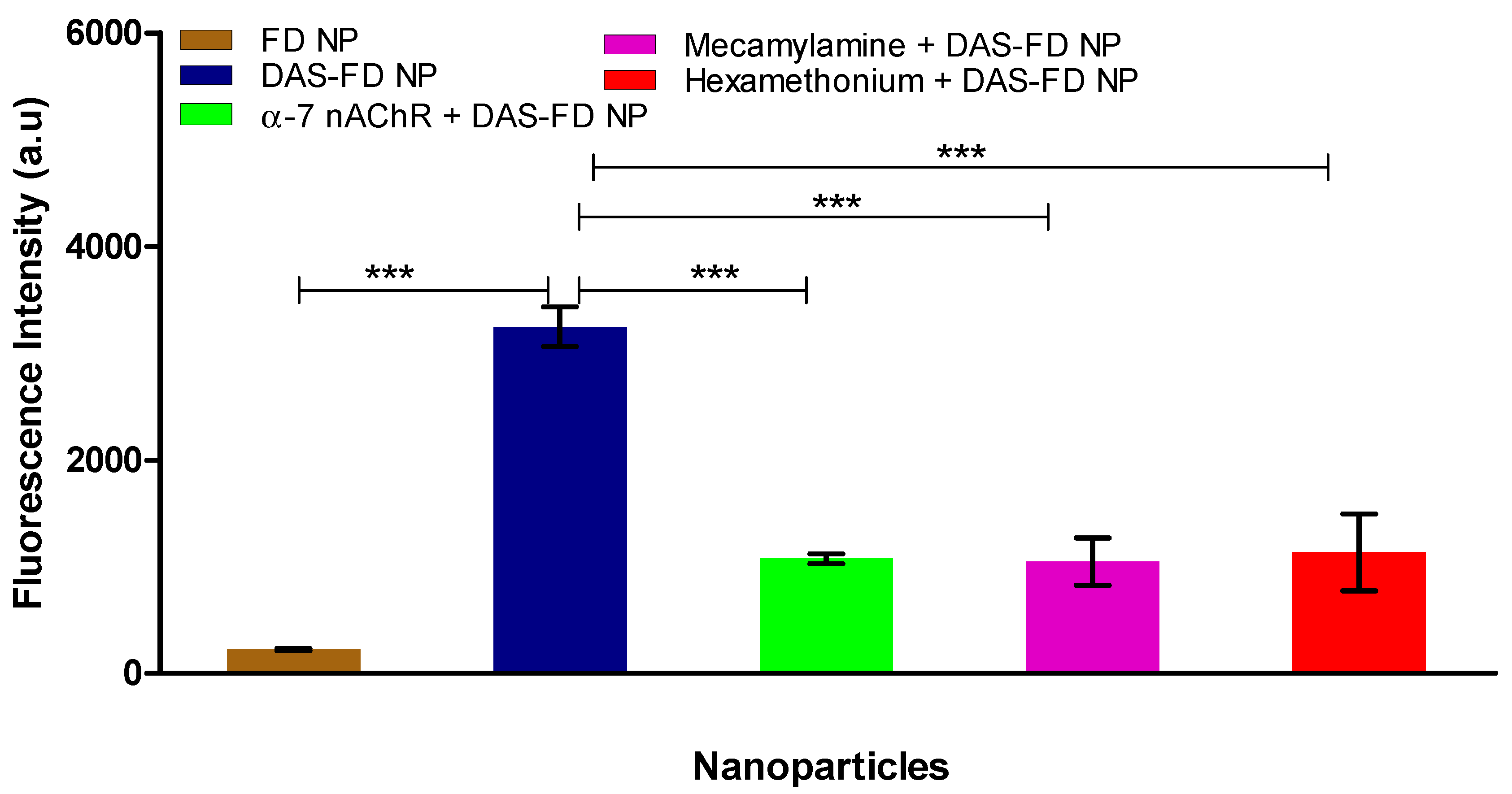
3.8. Delivery across BBB Model
4. Discussion
4.1. In Vitro BBB Construction
4.2. Nanoparticle (NP) Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood–brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 165, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Segarra, M.; Aburto, M.R.; Acker-Palmer, A. Blood-brain barrier dynamics to maintain brain homeostasis. Trends Neurosci. 2021, 44, 393–405. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Engelhardt, B.; Liebner, S. Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res. 2014, 355, 687–699. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 1–24. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Cohen-Pfeffer, J.L.; Gururangan, S.; Lester, T.; Lim, D.A.; Shaywitz, A.J.; Westphal, M.; Slavc, I. Intracerebroventricular delivery as a safe, long-term route of drug administration. Pediatr. Neurol. 2017, 67, 23–35. [Google Scholar] [CrossRef]
- Tobinick, E.L. Perispinal delivery of CNS drugs. CNS Drugs 2016, 30, 469–480. [Google Scholar] [CrossRef]
- Tobinick, E. Perispinal etanercept for neuroinflammatory disorders. Drug Discov. Today 2009, 14, 168–177. [Google Scholar] [CrossRef]
- Tobinick, E. Perispinal etanercept advances as a neurotherapeutic. Expert Rev. Neurother. 2018, 18, 453–455. [Google Scholar] [CrossRef]
- Tobinick, E.; Rodriguez-Romanacce, H.; Levine, A.; Ignatowski, T.A.; Spengler, R.N. Immediate neurological recovery following perispinal etanercept years after brain injury. Clin. Drug Investig. 2014, 34, 361–366. [Google Scholar] [CrossRef]
- Ralph, S.J.; Weissenberger, A.; Bonev, V.; King, L.D.; Bonham, M.D.; Ferguson, S.; Smith, A.D.; Goodman-Jones, A.A.; Espinet, A.J. Phase I/II parallel double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke: Improved mobility and pain alleviation. Expert Opin. Investig. Drugs 2020, 29, 311–326. [Google Scholar] [CrossRef]
- Manrique-Suárez, V.; Macaya, L.; Contreras, M.A.; Parra, N.; Maura, R.; González, A.; Toledo, J.R.; Sánchez, O. Design and characterization of a novel dimeric blood-brain barrier penetrating TNFα inhibitor. Proteins 2021, 89, 1508–1521. [Google Scholar] [CrossRef]
- Kageyama, T.; Nakamura, M.; Matsuo, A.; Yamasaki, Y.; Takakura, Y.; Hashida, M.; Kanai, Y.; Naito, M.; Tsuruo, T.; Minato, N. The 4F2hc/LAT1 complex transports L-DOPA across the blood-brain barrier. Brain Res. 2000, 879, 115–121. [Google Scholar] [CrossRef]
- Cornacchia, C.; Marinelli, L.; Di Rienzo, A.; Dimmito, M.P.; Serra, F.; Di Biase, G.; De Filippis, B.; Turkez, H.; Mardinoglu, A.; Bellezza, I. Development of l-Dopa-containing diketopiperazines as blood-brain barrier shuttle. Eur. J. Med. Chem. 2022, 243, 114746. [Google Scholar] [CrossRef]
- Lane, E.L. L-DOPA for Parkinson’s disease—A bittersweet pill. Eur. J. Neurosci. 2019, 49, 384–398. [Google Scholar] [CrossRef]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; De Deurwaerdère, P. L-DOPA in parkinson’s disease: Looking at the “false” neurotransmitters and their meaning. Int. J. Mol. Sci. 2019, 21, 294. [Google Scholar] [CrossRef]
- Bello, F.D.; Giannella, M.; Giorgioni, G.; Piergentili, A.; Quaglia, W. Receptor ligands as helping hands to L-DOPA in the treatment of Parkinson’s disease. Biomolecules 2019, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Shusta, E.V. Blood–brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Pulgar, V.M. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front. Neurosci. 2019, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T. Smart strategies for therapeutic agent delivery into brain across the blood-brain barrier using receptor-mediated transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.K. Ligand-appended BBB-targeted nanocarriers (LABTNs). Crit. Rev. Ther. Drug Carr. Syst. 2015, 32, 149–180. [Google Scholar] [CrossRef]
- Kou, L.; Hou, Y.; Yao, Q.; Guo, W.; Wang, G.; Wang, M.; Fu, Q.; He, Z.; Ganapathy, V.; Sun, J. L-Carnitine-conjugated nanoparticles to promote permeation across blood-brain barrier and to target glioma cells for drug delivery via the novel organic cation/carnitine transporter OCTN2. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1605–1616. [Google Scholar] [CrossRef]
- Lee, S.W.L.; Campisi, M.; Osaki, T.; Possenti, L.; Mattu, C.; Adriani, G.; Kamm, R.D.; Chiono, V. Modeling nanocarrier transport across a 3D in vitro human blood-brain-barrier microvasculature. Adv. Healthc. Mater. 2020, 9, 1901486. [Google Scholar] [CrossRef]
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car, H. Nanoparticles as drug delivery systems. Pharmacol. Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef]
- Huey, R.; O’Hagan, B.; McCarron, P.; Hawthorne, S. Targeted drug delivery system to neural cells utilizes the nicotinic acetylcholine receptor. Int. J. Pharm. 2017, 525, 12–20. [Google Scholar] [CrossRef]
- Bhalerao, A.; Sivandzade, F.; Archie, S.R.; Chowdhury, E.A.; Noorani, B.; Cucullo, L. In vitro modeling of the neurovascular unit: Advances in the field. Fluids Barriers CNS 2020, 17, 1–20. [Google Scholar] [CrossRef]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying human neurological disorders using induced pluripotent stem cells: From 2D monolayer to 3D organoid and blood brain barrier models. Compr. Physiol. 2019, 9, 565. [Google Scholar]
- Sivandzade, F.; Cucullo, L. In-vitro blood-brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 2018, 38, 1667–1681. [Google Scholar] [CrossRef]
- Cho, C.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Naik, P.; Cucullo, L. In vitro blood-brain barrier models: Current and perspective technologies. J. Pharm. Sci. 2012, 101, 1337–1354. [Google Scholar] [CrossRef]
- Jagtiani, E.; Yeolekar, M.; Naik, S.; Patravale, V. In vitro blood brain barrier models: An overview. J. Control. Release 2022, 343, 13–30. [Google Scholar] [CrossRef]
- Jain, A.K.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A.K. Models and methods for in vitro toxicity. In In Vitro Toxicology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 45–65. [Google Scholar]
- Huey, R.; Rathbone, D.; McCarron, P.; Hawthorne, S. Design, stability and efficacy of a new targeting peptide for nanoparticulate drug delivery to SH-SY5Y neuroblastoma cells. J. Drug Target. 2019, 27, 959–970. [Google Scholar] [CrossRef]
- Kaya, S.; Callan, B.; Hawthorne, S. Human in vitro blood brain barrier models—A review. PLoS ONE 2023. submitted. [Google Scholar]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Appelt-Menzel, A.; Cubukova, A.; Günther, K.; Edenhofer, F.; Piontek, J.; Krause, G.; Stüber, T.; Walles, H.; Neuhaus, W.; Metzger, M. Establishment of a human blood-brain barrier co-culture model mimicking the neurovascular unit using induced pluri-and multipotent stem cells. Stem Cell Rep. 2017, 8, 894–906. [Google Scholar] [CrossRef]
- Gaillard, P.J.; de Boer, A.G. Relationship between permeability status of the blood–brain barrier and in vitro permeability coefficient of a drug. Eur. J. Pharm. Sci. 2000, 12, 95–102. [Google Scholar] [CrossRef]
- Reichel, A.; Begley, D.J.; Abbott, N.J. An overview of in vitro techniques for blood-brain barrier studies. Blood-Brain Barrier Biol. Res. Protoc. 2003, 89, 307–324. [Google Scholar]
- Deli, M.A.; Ábrahám, C.S.; Kataoka, Y.; Niwa, M. Permeability studies on in vitro blood–brain barrier models: Physiology, pathology, and pharmacology. Cell Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Dev. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Elbakary, B.; Badhan, R.K. A dynamic perfusion-based blood-brain barrier model for cytotoxicity testing and drug permeation. Sci. Rep. 2020, 10, 3788. [Google Scholar] [CrossRef]
- Ferrell, N.; Cheng, J.; Miao, S.; Roy, S.; Fissell, W.H. Orbital shear stress regulates differentiation and barrier function of primary renal tubular epithelial cells. ASAIO J. 2018, 64, 766. [Google Scholar] [CrossRef]
- Dardik, A.; Chen, L.; Frattini, J.; Asada, H.; Aziz, F.; Kudo, F.A.; Sumpio, B.E. Differential effects of orbital and laminar shear stress on endothelial cells. J. Vasc. Surg. 2005, 41, 869–880. [Google Scholar] [CrossRef]
- Salvador, E.; Burek, M.; Löhr, M.; Nagai, M.; Hagemann, C.; Förster, C.Y. Senescence and associated blood-brain barrier alterations in vitro. Histochem. Cell Biol. 2021, 156, 283–292. [Google Scholar] [CrossRef]
- Eigenmann, D.E.; Xue, G.; Kim, K.S.; Moses, A.V.; Hamburger, M.; Oufir, M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS 2013, 10, 1–17. [Google Scholar] [CrossRef]
- Xu, L.; Nirwane, A.; Yao, Y. Basement membrane and blood-brain barrier. Stroke Vasc. Neurol. 2019, 4, 78–82. [Google Scholar] [CrossRef]
- Banerjee, J.; Shi, Y.; Azevedo, H.S. In vitro blood-brain barrier models for drug research: State-of-the-art and new perspectives on reconstituting these models on artificial basement membrane platforms. Drug Discov. Today 2016, 21, 1367–1386. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Seltzer, J.L.; Weingarten, H.; Akers, K.T.; Eschbach, M.L.; Grant, G.A.; Eisen, A.Z. Cleavage specificity of type IV collagenase (gelatinase) from human skin. Use of synthetic peptides as model substrates. J. Biol. Chem. 1989, 264, 19583–19586. [Google Scholar] [CrossRef]
- Maherally, Z.; Fillmore, H.L.; Tan, S.L.; Tan, S.F.; Jassam, S.A.; Quack, F.I.; Hatherell, K.E.; Pilkington, G.J. Real-time acquisition of transendothelial electrical resistance in an all-human, in vitro, 3-dimensional, blood-brain barrier model exemplifies tight-junction integrity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 168–182. [Google Scholar] [CrossRef]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, A.; Tanaka, K.; Niwa, M. A new blood–brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef]
- Malina, K.C.; Cooper, I.; Teichberg, V.I. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res. 2009, 1284, 12–21. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Zhu, D.; Su, Y.; Fu, B.; Xu, H. Magnesium reduces blood-brain barrier permeability and regulates amyloid-β transcytosis. Mol. Neurobiol. 2018, 55, 7118–7131. [Google Scholar] [CrossRef]
- León, J.; Acurio, J.; Bergman, L.; López, J.; Karin Wikström, A.; Torres-Vergara, P.; Troncoso, F.; Castro, F.O.; Vatish, M.; Escudero, C. Disruption of the blood-brain barrier by extracellular vesicles from preeclampsia plasma and hypoxic placentae: Attenuation by magnesium sulfate. Hypertension 2021, 78, 1423–1433. [Google Scholar] [CrossRef]
- Alarcon-Martinez, L.; Yilmaz-Ozcan, S.; Yemisci, M.; Schallek, J.; Kılıç, K.; Can, A.; Di Polo, A.; Dalkara, T. Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. eLife 2018, 7, e34861. [Google Scholar] [CrossRef]
- Su, X.; Huang, L.; Qu, Y.; Xiao, D.; Mu, D. Pericytes in Cerebrovascular Diseases: An Emerging Therapeutic Target. Front. Cell Neurosci. 2019, 13, 519. [Google Scholar] [CrossRef]
- Reilly, J.F.; Maher, P.A.; Kumari, V.G. Regulation of astrocyte GFAP expression by TGF-β1 and FGF-2. Glia 1998, 22, 202–210. [Google Scholar] [CrossRef]
- Baba, H.; Nakahira, K.; Morita, N.; Tanaka, F.; Akita, H.; Ikenaka, K. GFAP gene expression during development of astrocyte. Dev. Neurosci. 1997, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hajal, C.; Campisi, M.; Mattu, C.; Chiono, V.; Kamm, R.D. In vitro models of molecular and nano-particle transport across the blood-brain barrier. Biomicrofluidics 2018, 12, 042213. [Google Scholar] [CrossRef] [PubMed]
- Chigumira, W.; Maposa, P.; Gadaga, L.L.; Dube, A.; Tagwireyi, D.; Maponga, C.C. Preparation and evaluation of pralidoxime-loaded PLGA nanoparticles as potential carriers of the drug across the blood brain barrier. J. Nanomater. 2015, 2015, 8. [Google Scholar] [CrossRef]
- Sadat Tabatabaei Mirakabad, F.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y.; et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 517–535. [Google Scholar] [CrossRef]
- Monge, M.; Fornaguera, C.; Quero, C.; Dols-Perez, A.; Calderó, G.; Grijalvo, S.; García-Celma, M.J.; Rodríguez-Abreu, C.; Solans, C. Functionalized PLGA nanoparticles prepared by nano-emulsion templating interact selectively with proteins involved in the transport through the blood-brain barrier. Eur. J. Pharm. Biopharm. 2020, 156, 155–164. [Google Scholar] [CrossRef]
- Azizi, M.; Farahmandghavi, F.; Joghataei, M.; Zandi, M.; Imani, M.; Bakhtiary, M.; Dorkoosh, F.A.; Ghazizadeh, F. Fabrication of protein-loaded PLGA nanoparticles: Effect of selected formulation variables on particle size and release profile. J. Polym. Res. 2013, 20, 1–14. [Google Scholar] [CrossRef]
- Carter, D.C.; Ho, J.X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar]
- Hughes, J.M.; Budd, P.M.; Tiede, K.; Lewis, J. Polymerized high internal phase emulsion monoliths for the chromatographic separation of engineered nanoparticles. J. Appl. Polym. Sci. 2015, 132, 41229. [Google Scholar] [CrossRef]
- Clayton, K.N.; Salameh, J.W.; Wereley, S.T.; Kinzer-Ursem, T.L. Physical characterization of nanoparticle size and surface modification using particle scattering diffusometry. Biomicrofluidics 2016, 10, 054107. [Google Scholar] [CrossRef]
- Fornaguera, C.; Dols-Perez, A.; Calderó, G.; García-Celma, M.J.; Camarasa, J.; Solans, C. PLGA nanoparticles prepared by nano-emulsion templating using low-energy methods as efficient nanocarriers for drug delivery across the blood–brain barrier. J. Control Release 2015, 211, 134–143. [Google Scholar] [CrossRef]
- Patel, J.; Amrutiya, J.; Bhatt, P.; Javia, A.; Jain, M.; Misra, A. Targeted delivery of monoclonal antibody conjugated docetaxel loaded PLGA nanoparticles into EGFR overexpressed lung tumour cells. J. Microencapsul. 2018, 35, 204–217. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA NP | Size (nm) ± SD | PDI ± SD |
|---|---|---|
| Blank PLGA NP | 268.5 ± 36.63 | 0.22 ± 0.052 |
| Fitc-dextran-PLGA NP | 347.7 ± 62.48 | 0.35 ± 0.153 |
| DAS-Fitc-dextran-PLGA-NP | 386.50 ± 11.30 | 0.27 ± 0.08 |
| Fitc-Dextran PLGA NP | EE% | CE% |
|---|---|---|
| Sample 1 | 64.49 | 62.50 |
| Sample 2 | 61.87 | 64.06 |
| Sample 3 | 64.94 | 56.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaya, S.; Callan, B.; Hawthorne, S. Non-Invasive, Targeted Nanoparticle-Mediated Drug Delivery across a Novel Human BBB Model. Pharmaceutics 2023, 15, 1382. https://doi.org/10.3390/pharmaceutics15051382
Kaya S, Callan B, Hawthorne S. Non-Invasive, Targeted Nanoparticle-Mediated Drug Delivery across a Novel Human BBB Model. Pharmaceutics. 2023; 15(5):1382. https://doi.org/10.3390/pharmaceutics15051382
Chicago/Turabian StyleKaya, Shona, Bridgeen Callan, and Susan Hawthorne. 2023. "Non-Invasive, Targeted Nanoparticle-Mediated Drug Delivery across a Novel Human BBB Model" Pharmaceutics 15, no. 5: 1382. https://doi.org/10.3390/pharmaceutics15051382
APA StyleKaya, S., Callan, B., & Hawthorne, S. (2023). Non-Invasive, Targeted Nanoparticle-Mediated Drug Delivery across a Novel Human BBB Model. Pharmaceutics, 15(5), 1382. https://doi.org/10.3390/pharmaceutics15051382