
Preparation of Apixaban Solid Dispersion for the Enhancement of Apixaban Solubility and Permeability
 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. APX Solubility Study in Organic Solvents
2.2.2. APX Solubility Study with Hydrophilic Carriers
2.2.3. Preparation of SDs
2.2.4. Differential Scanning Calorimetry (DSC) Analysis
2.2.5. Fourier Transform Infrared Spectroscopy (FT-IR) Analysis
2.2.6. Powder X-ray Diffraction (PXRD) Analysis
2.2.7. Disc Intrinsic Dissolution Rate (DIDR) Study
2.2.8. Powder Dissolution Test
2.2.9. Ex Vivo Permeation Test
2.2.10. HPLC Analysis
2.2.11. In Vivo Pharmacokinetic Study
2.2.12. LC-MS/MS Analysis
2.2.13. Statistical Analysis
3. Results and Discussion
3.1. Preparation of Apixaban (APX) Solid Dispersions (SDs)
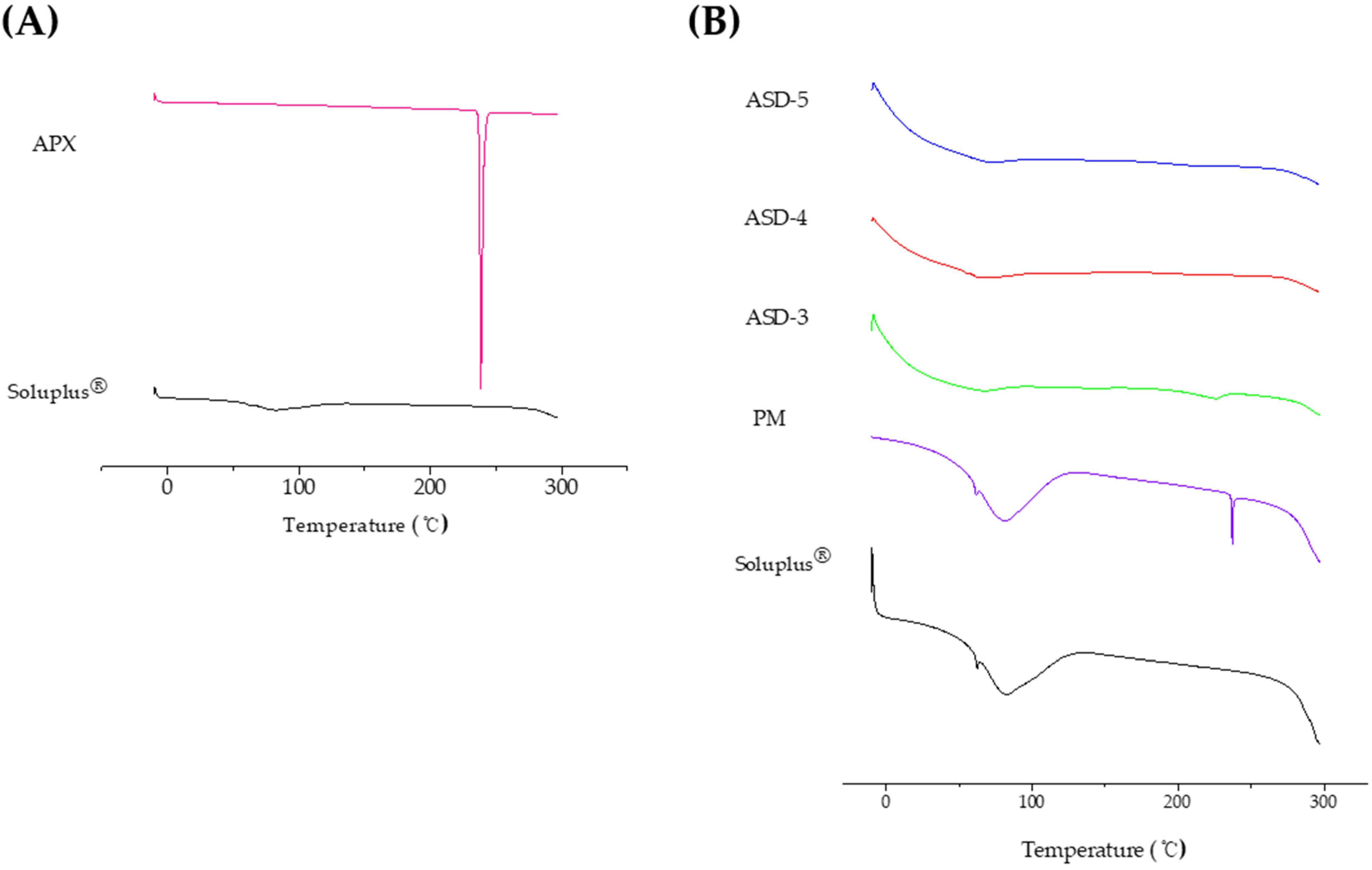
3.2. Differential Scanning Calorimetry (DSC) Analysis
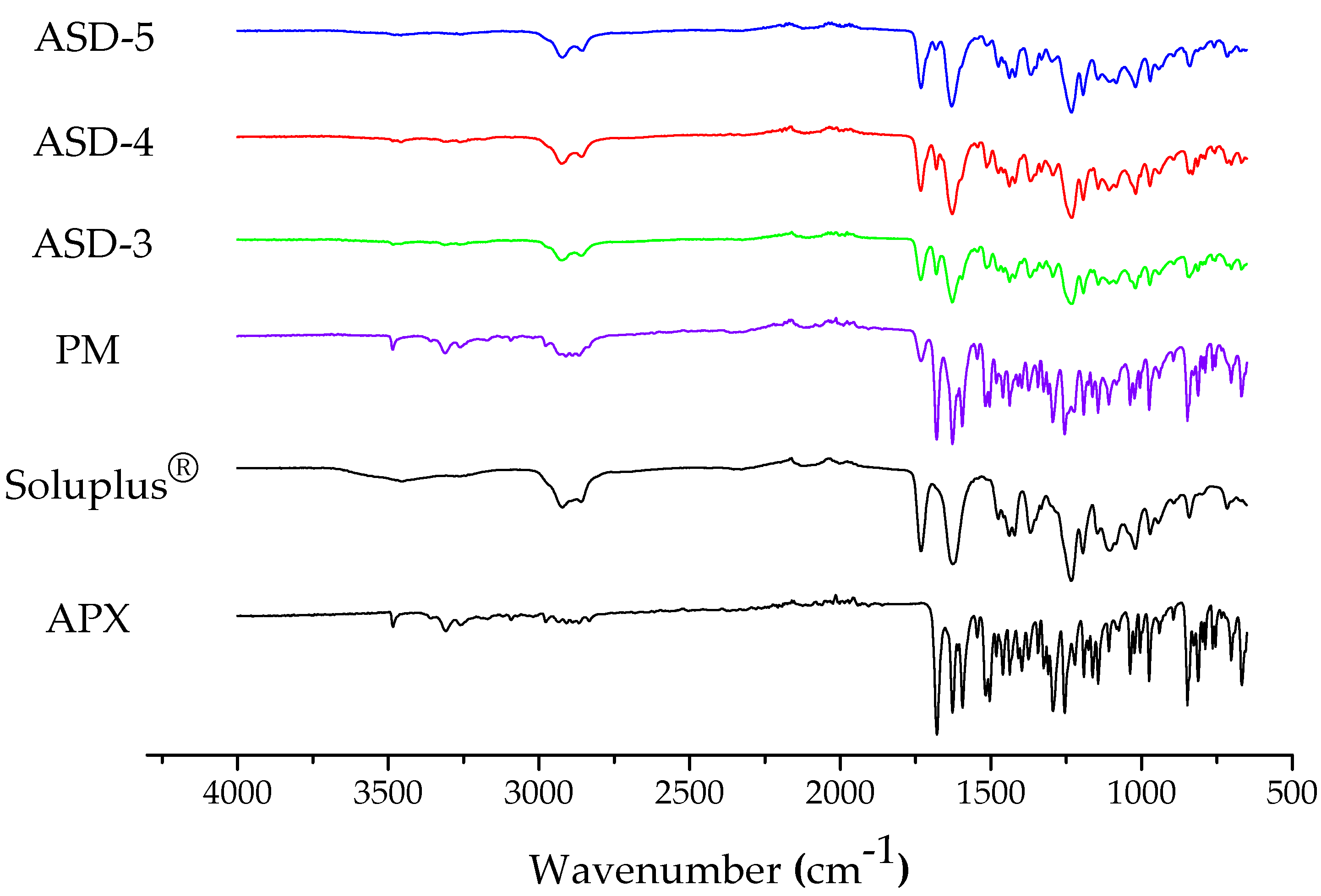
3.3. Fourier Transform Infrared Spectroscopy (FT-IR)
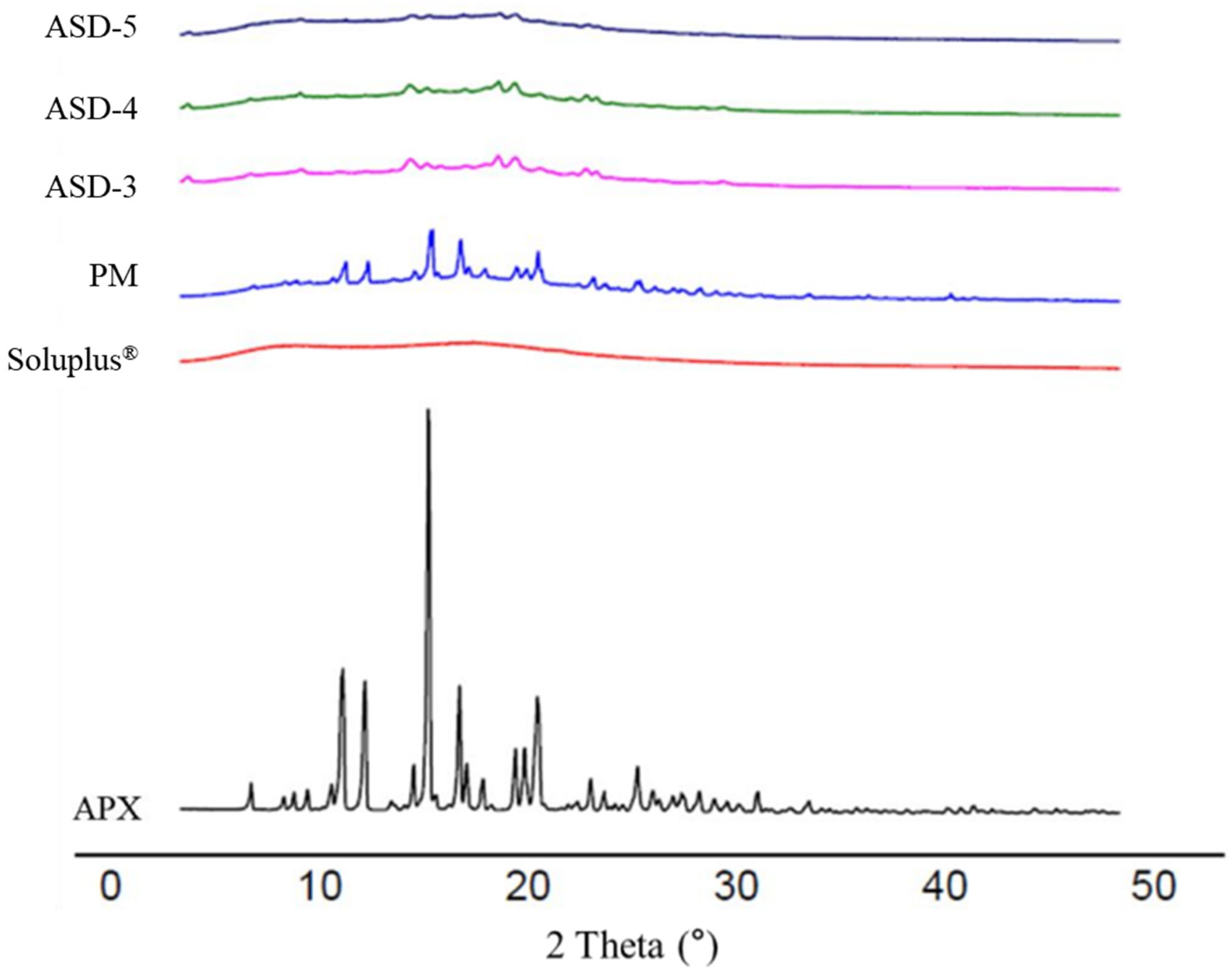
3.4. Powder X-ray Diffraction (PXRD)
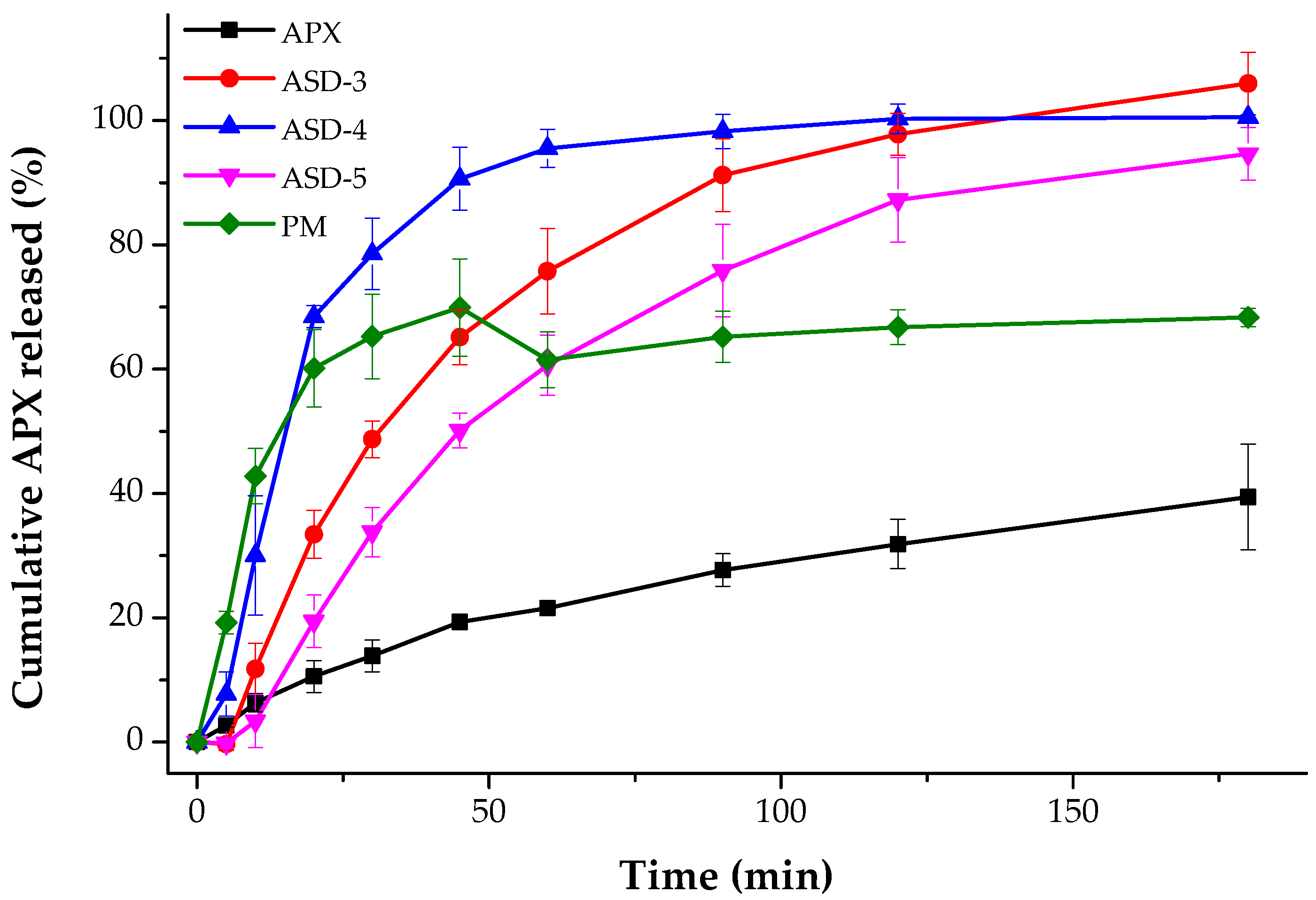
3.5. In Vitro Dissolution Studies
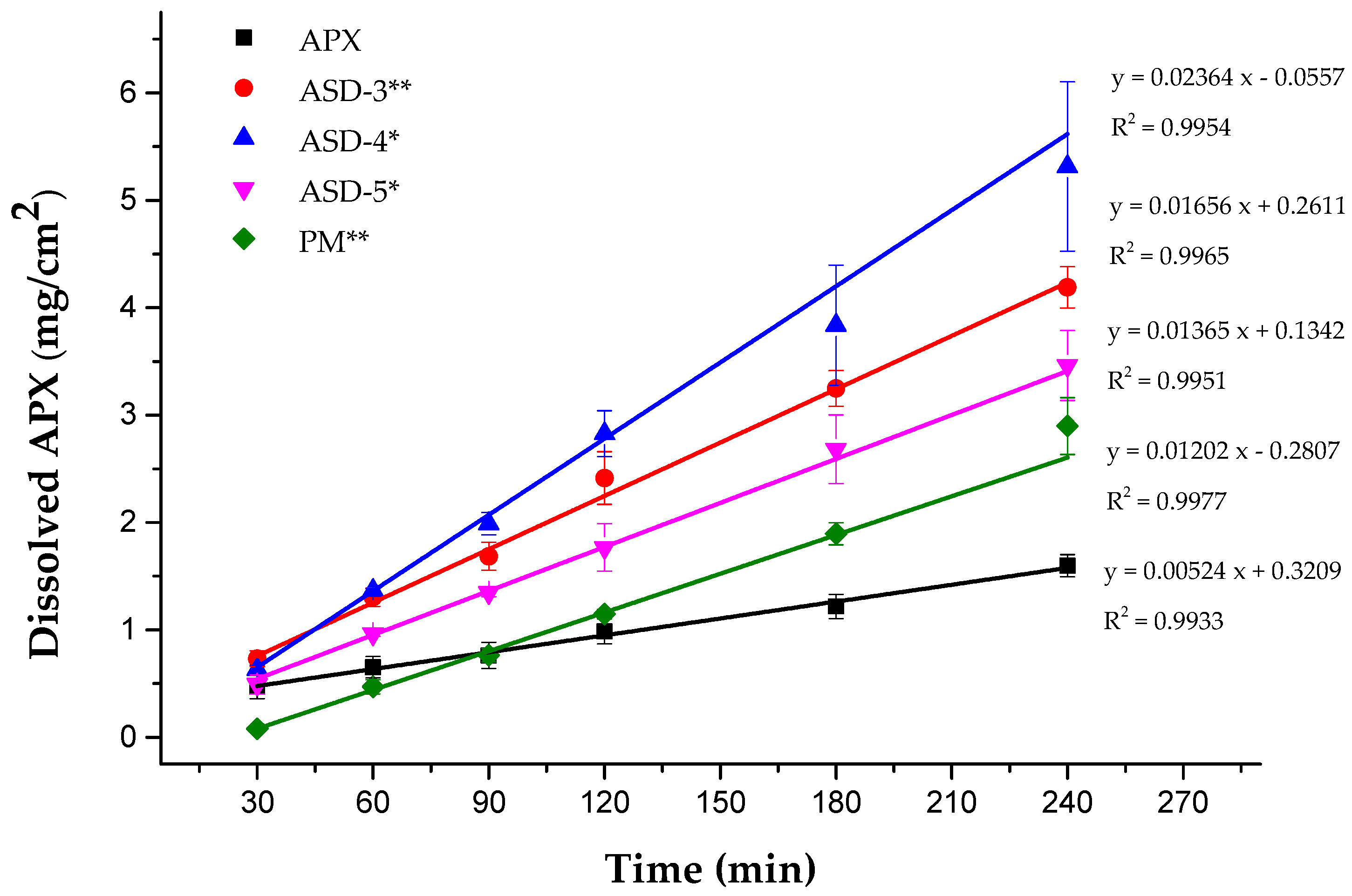
3.6. Ex Vivo Permeation Test
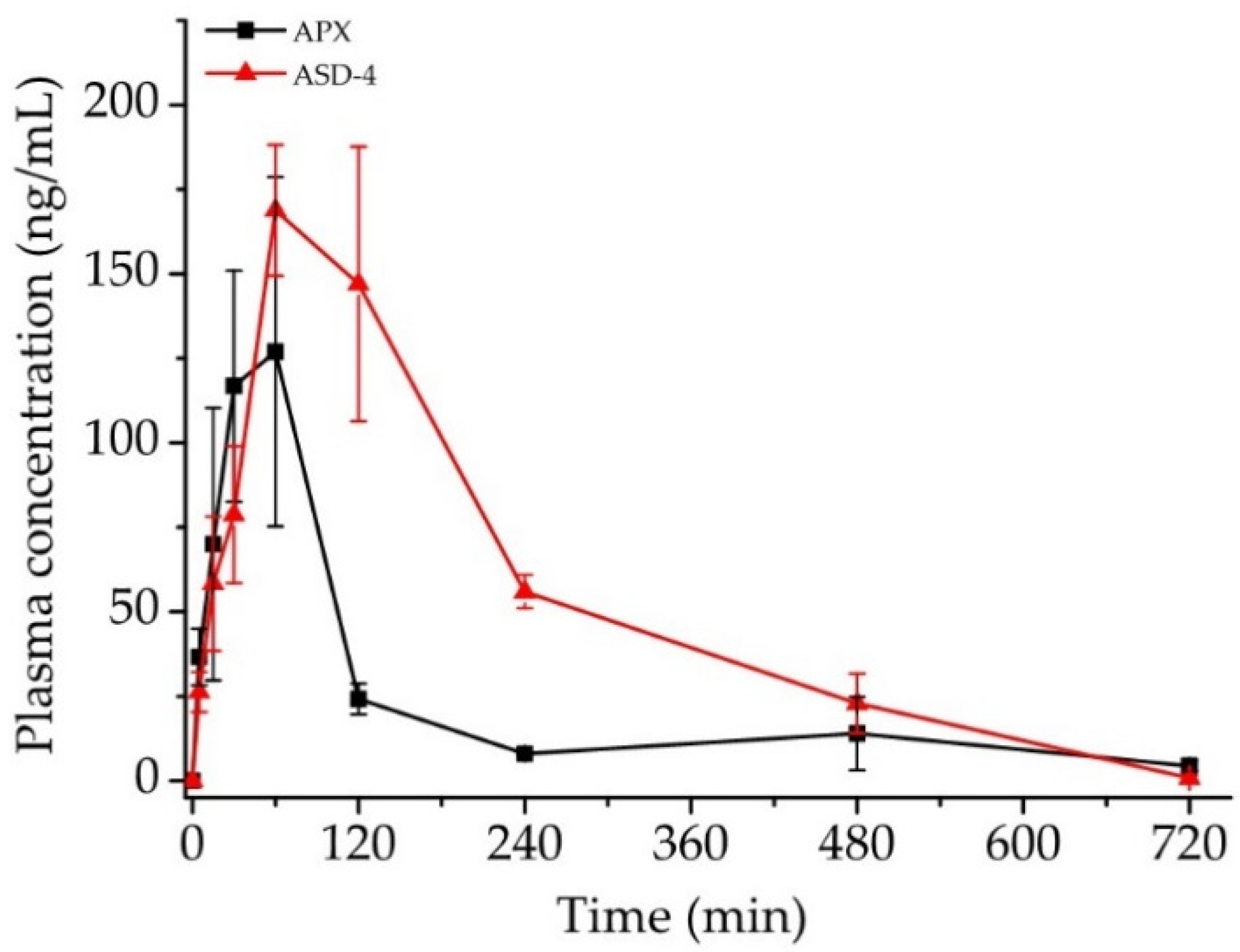
3.7. In Vivo Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, P.C.; Pinto, D.J.P.; Zhang, D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J. Thromb. Thrombolysis 2011, 31, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Frost, C.; Wang, J.; Nepal, S.; Schuster, A.; Barrett, Y.C.; Mosqueda-Garcia, R.; Reeves, R.A.; LaCreta, F. Apixaban, an oral, direct factor X a inhibitor: Single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br. J. Clin. Pharmacol. 2013, 75, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic Review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; He, K.; Herbst, J.J.; Kolb, J.; Shou, W.; Wang, L.; Balimane, P.V.; Han, Y.-H.; Gan, J.; Frost, C.E.; et al. Characterization of Efflux Transporters Involved in Distribution and Disposition of Apixaban. Drug Metab. Dispos. 2013, 41, 827–835. [Google Scholar] [CrossRef]
- Vakkalagadda, B.; Frost, C.; Byon, W.; Boyd, R.A.; Wang, J.; Zhang, D.; Yu, Z.; Dias, C.; Shenker, A.; LaCreta, F. Effect of Rifampin on the Pharmacokinetics of Apixaban, an Oral Direct Inhibitor of Factor Xa. Am. J. Cardiovasc. Drugs 2016, 16, 119–127. [Google Scholar] [CrossRef]
- ICH Harmonised Guideline. Biopharmaceutics Classification System-Based Biowaivers M9. Int. Counc. Harmon. Tech. Requir. Pharm. Hum. Use. 2019. Available online: https://database.ich.org/sites/default/files/M9_Guideline_Step4_2019_1116.pdf (accessed on 5 March 2023).
- Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutics Review(s). Application Number: 202155Orig1s000 (apixaban). Content current as of: 23 November 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202155Orig1s000ClinPharmR.pdf (accessed on 18 February 2023).
- Chen, Y.; Li, L.; Yao, J.; Ma, Y.-Y.; Chen, J.-M.; Lu, T.-B. Improving the Solubility and Bioavailability of Apixaban via Apixaban–Oxalic Acid Cocrystal. Cryst. Growth Des. 2016, 16, 2923–2930. [Google Scholar] [CrossRef]
- Yu, L.X.; Carlin, A.S.; Amidon, G.L.; Hussain, A.S. Feasibility studies of utilizing disk intrinsic dissolution rate to classify drugs. Int. J. Pharm. 2003, 270, 221–227. [Google Scholar] [CrossRef]
- Zhang, L.; Kong, D.; Wang, H.; Jiao, L.; Zhao, X.; Song, J.; Yang, D.; Yang, H.; Yang, S.; Du, G.; et al. Cocrystal of Apixaban–Quercetin: Improving Solubility and Bioavailability of Drug Combination of Two Poorly Soluble Drugs. Molecules 2021, 26, 2677. [Google Scholar] [CrossRef]
- Zhoye, Y.Y.; Yan, Q. A Kind of Apixaban Tablet and Preparation Method Thereof. Patent CN108096205, 1 June 2018. [Google Scholar]
- Asati, A.V.; Salunkhe, K.S.; Chavan, M.J.; Chintamani, R.B.; Singh, R.P. Solubility Enhancement of BCS Classified II/IV Drug—Solid Dispersion of Apixaban by Solvent Evaporation. Int. J. Pharm. Investig. 2020, 10, 430–436. [Google Scholar] [CrossRef]
- Siahi-Shadbad, M.R.; Ghanbarzadeh, S.; Barzegar-Jalali, M.; Valizadeh, H.; Taherpoor, A.; Mohammadi, G.; Barzegar-Jalali, A.; Adibkia, K. Development and Characterization of Solid Dispersion for Dissolution Improvement of Furosemide by Cogrinding Method. Adv. Pharm. Bull. 2014, 4, 391–399. [Google Scholar] [CrossRef]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef]
- Franchini, M.; Liumbruno, G.M.; Bonfanti, C.; Lippi, G. The evolution of anticoagulant therapy. Blood Transfus. 2016, 14, 175–184. [Google Scholar] [CrossRef]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.V.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Ave-zum, A.; et al. Apixaban versus Warfarin in Patients with Atrial Fibrillation. N. Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef]
- Hyun, S.-M.; Lee, B.J.; Abuzar, S.; Lee, S.; Joo, Y.; Hong, S.-H.; Kang, H.; Kwon, K.-A.; Velaga, S.; Hwang, S.-J. Preparation, characterization, and evaluation of celecoxib eutectic mixtures with adipic acid/saccharin for improvement of wettability and dissolution rate. Int. J. Pharm. 2018, 554, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Dinh, L.; Abuzar, S.M.; Lee, E.S.; Hwang, S.-J. Synthesis of Celecoxib-Eutectic Mixture Particles via Supercritical CO2 Process and Celecoxib Immediate Release Tablet Formulation by Quality by Design Approach. Pharmaceutics 2022, 14, 1549. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.L. A guide to Ussing chamber studies of mouse intestine. Am. J. Physiol. Liver Physiol. 2009, 296, G1151–G1166. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, A.; Alves, G.; Falcão, A.; Soares-Da-Silva, P. Evaluation of the permeability and P-glycoprotein efflux of carbamazepine and several derivatives across mouse small intestine by the Ussing chamber technique. Epilepsia 2012, 53, 529–538. [Google Scholar] [CrossRef]
- Sareen, S.; Joseph, L.; Mathew, G. Improvement in solubility of poor water-soluble drugs by solid dispersion. Int. J. Pharm. Investig. 2012, 2, 12–17. [Google Scholar] [CrossRef]
- Nguyen, T.-T.; Duong, V.-A.; Maeng, H.-J. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics 2021, 13, 1103. [Google Scholar] [CrossRef]
- Hou, P.; Ni, J.; Cao, S.; Lei, H.; Cai, Z.; Zhang, T.; Yu, F.; Tan, Q. Preparation and Evaluation of Solid Dispersions of A New Antitumor Compound Based on Early-Stage Preparation Discovery Concept. AAPS PharmSciTech 2013, 14, 629–638. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Sofroniou, C.; Baglioni, M.; Mamusa, M.; Resta, C.; Doutch, J.; Smets, J.; Baglioni, P. Self-Assembly of Soluplus in Aqueous Solutions: Characterization and Prospectives on Perfume Encapsulation. ACS Appl. Mater. Interfaces 2022, 14, 14791–14804. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Prezotti, F.; Araújo, F.; Lopes, C.; Loureiro, A.; Marques, S.; Sarmento, B. Third-generation solid dispersion combining Soluplus and poloxamer 407 enhances the oral bioavailability of resveratrol. Int. J. Pharm. 2021, 595, 120245. [Google Scholar] [CrossRef] [PubMed]
- Kanojiya, P.S.; Charde, Y.; Avari, J.G.; Wadetwar, R.N. Solid Dispersion of Lumefantrine Using Soluplus® by Solvent Evaporation Method: Formulation, Characterization and in-vitro Antimalarial Screening. Indian J. Pharm. Educ. Res. 2022, 56, 121–132. [Google Scholar] [CrossRef]
- Lavra, Z.M.M.; de Santana, D.P.; Ré, M.I. Solubility and dissolution performances of spray-dried solid dispersion of Efavirenz in Soluplus. Drug Dev. Ind. Pharm. 2016, 43, 42–54. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Result |
|---|---|
| Precursor Ion m/z | 460.2 [M + H] + |
| Product Ions m/z | 443.02 |
| Range | 1 to 100 ng/mL |
| Selectivity | No interferences |
| Carry-over | Not observed |
| Calibration curve | Y = 0.0164x − 0.0027, R2 = 0.9997 |
| Accuracy and Precision | Concentrations: 1, 40, 40 and 80 ng/mL Intra-batch RE *: −18.1%, −1.9%, 7.7%, 12.5% Inter-batch RE: 6.3%, 10.4%, −0.31%, −0.83% Intra-batch CV **: 9.5%, 2.3%, 3.5%, 5.5% Inter-batch CV: 3.6%, 8.9%, 4.8%, 4.0% |
| LLOQ | 1 ng/mL, S/N ratio = 94.63 |
| Matrix effect | Low concentration CV: 9.3% High concentration CV: 6.0% |
| Solvent | Saturation Solubility (mg/mL) |
|---|---|
| Chloroform | 13.87 |
| Water | 0.031 |
| Ethyl acetate | 0.06 |
| 2-Propanol | 0.13 |
| Ethanol | 0.43 |
| Acetone | 1.01 |
| Methanol | 2.21 |
| Acetonitrile (ACN) | 2.38 |
| Hydrophilic Carrier | Saturation Solubility (µg/mL) | |||
|---|---|---|---|---|
| Product | Description | 0.1% | 0.5% | 1% |
| Eudragit® L100 * | Methacrylic acid-methyl methacrylate (1:1) | 34.23 | 12.89 | 3.73 |
| Eudragit® S100 * | Methacrylic acid-methyl methacrylate (1:2) | 31.63 | 32.89 | 30.69 |
| Eudragit® E PO | Butyl methyl methacrylate, dimethyl aminoethyl methacrylate, methacrylate, methyl methacrylate | 33.11 | 33.97 | 34.85 |
| Kollidon® VA 64 | Povidone, vinylpyrrolidone-vinyl acetate | 38.75 | 46.37 | 57.42 |
| Kollidon® | Povidone (PVP) | 35.53 | 41.46 | 43.67 |
| Poly (vinyl alcohol) (PVA) | 34.89 | 36.94 | 38.60 | |
| Soluplus® * | Polyvinyl caprolactampolyvinyl acetate-polyethylene glycol | 41.01 | 59.61 | 53.61 |
| Kolliphor® P 407 * | Poloxamer 407 | 38.68 | 43.30 | 44.49 |
| Kolliphor® P 188 * | Poloxamer 188 | 36.62 | 42.55 | 43.72 |
| Kolliphor® TPGS * | D-alpha tocopheryl polyethylene glycol 1000 succinate | 39.63 | 58.98 | 80.35 |
| Preparation Method | Hydrophilic Carrier (APX-to-Carrier Ratio w/w) | Saturation Solubility (µg/mL) | |
|---|---|---|---|
| Kneading method | Kollidon® (1:5) | 40.83 | |
| Kolliphor® P 188 (1:5) | 46.78 | ||
| Kolliphor® P 407 (1:5) | 48.78 | ||
| Kollidon® VA 64 (1:5) | 55.66 | ||
| Soluplus® (1:5) | 57.83 | ||
| Kolliphor® TPGS (1:5) | 69.52 | ||
| Solvent evaporation method | Formulation | Hydrophilic carrier (APX-to-carrier ratio w/w) | Saturation solubility (µg/mL) |
| ASD-1 | Kolliphor® TPGS (1:5) | 85.7 | |
| ASD-2 | Kollidon® VA 64 (1:5) | 95.8 | |
| ASD-3 | Soluplus® (1:3) | 131.9 | |
| ASD-4 | Soluplus® (1:5) | 183.2 | |
| ASD-5 | Soluplus® (1:10) | 200.9 | |
| Donor Solution | Apparent Permeability Coefficient (×10−6 cm/s) | ||
|---|---|---|---|
| 0–1 h | 0–2 h | 0–3 h | |
| APX | 9.78 | 15.60 | 20.26 |
| ASD-4 | 24.88 | 18.03 | 17.86 |
| Enhancement ratio | 2.54 | 1.16 | 0.88 |
| Parameter | APX | ASD-4 |
|---|---|---|
| Cmax (ng/mL), Tmax (min) | Rat 1, 163.01, 15 Rat 2, 240.77, 60 Rat 3, 184.30, 30 | Rat 1, 249.81,120 Rat 2, 134.02, 60 Rat 3, 161.76, 60 |
| Mean Cmax (ng/mL) | 196.0 ± 40.18 | 178.9 ± 55.41 |
| Mean Tmax (h) | 0.58 ± 0.38 | 1.33 ± 0.58 |
| AUC0→12 (ng∙h/mL) | 16,962.6 ± 3601.48 | 39,192.1 ± 11,580.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Lee, J.-J.; Lee, S.; Dinh, L.; Oh, H.; Abuzar, S.M.; Ahn, J.-H.; Hwang, S.-J. Preparation of Apixaban Solid Dispersion for the Enhancement of Apixaban Solubility and Permeability. Pharmaceutics 2023, 15, 907. https://doi.org/10.3390/pharmaceutics15030907
Lee J, Lee J-J, Lee S, Dinh L, Oh H, Abuzar SM, Ahn J-H, Hwang S-J. Preparation of Apixaban Solid Dispersion for the Enhancement of Apixaban Solubility and Permeability. Pharmaceutics. 2023; 15(3):907. https://doi.org/10.3390/pharmaceutics15030907
Chicago/Turabian StyleLee, Juseung, Jong-Ju Lee, Seungyeol Lee, Linh Dinh, Hangyu Oh, Sharif Md Abuzar, Jun-Hyun Ahn, and Sung-Joo Hwang. 2023. "Preparation of Apixaban Solid Dispersion for the Enhancement of Apixaban Solubility and Permeability" Pharmaceutics 15, no. 3: 907. https://doi.org/10.3390/pharmaceutics15030907
APA StyleLee, J., Lee, J.-J., Lee, S., Dinh, L., Oh, H., Abuzar, S. M., Ahn, J.-H., & Hwang, S.-J. (2023). Preparation of Apixaban Solid Dispersion for the Enhancement of Apixaban Solubility and Permeability. Pharmaceutics, 15(3), 907. https://doi.org/10.3390/pharmaceutics15030907