
Building a Human Physiologically Based Pharmacokinetic Model for Aflatoxin B1 to Simulate Interactions with Drugs

 ,
,  ,
,  and
and
Abstract
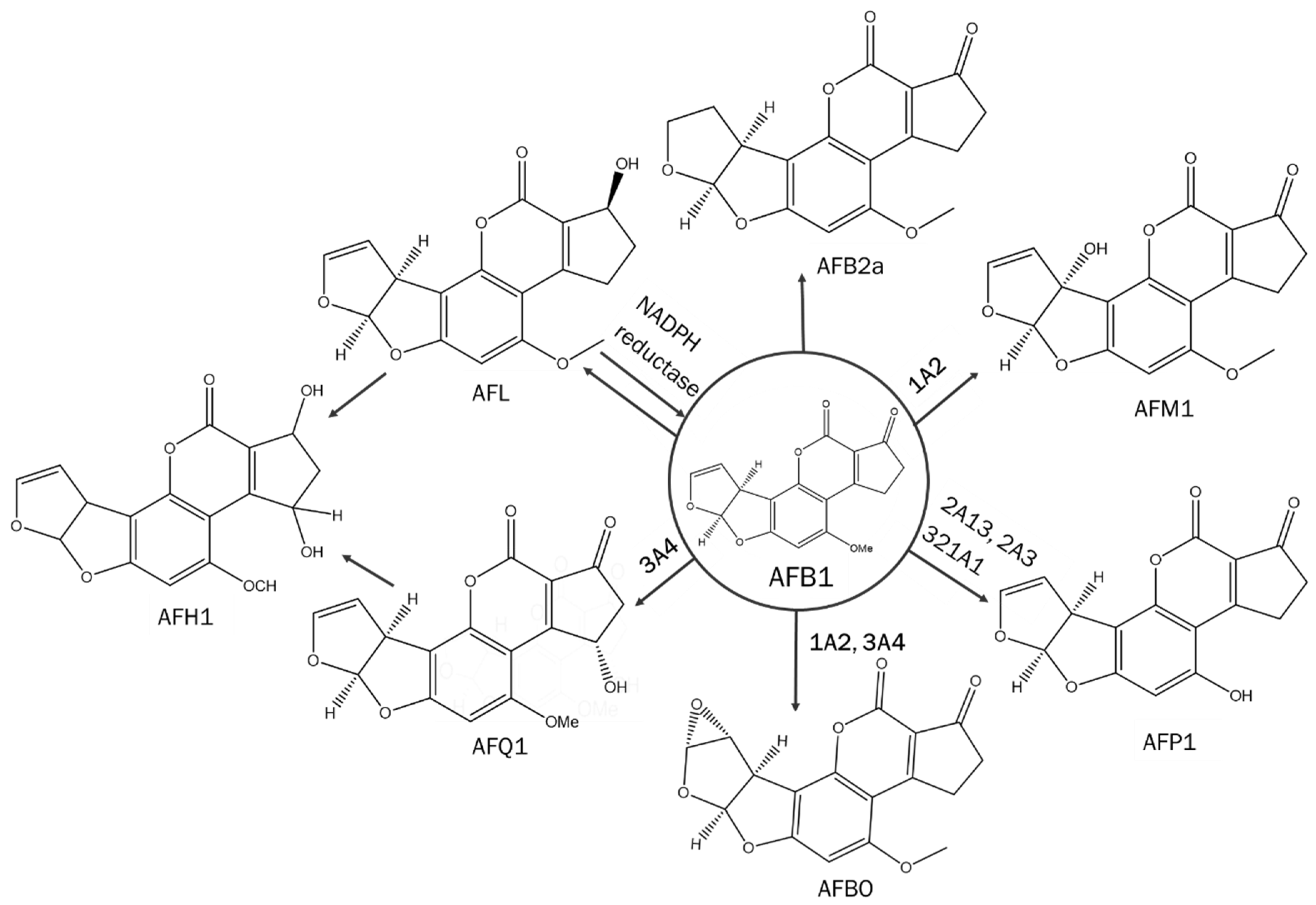
1. Introduction
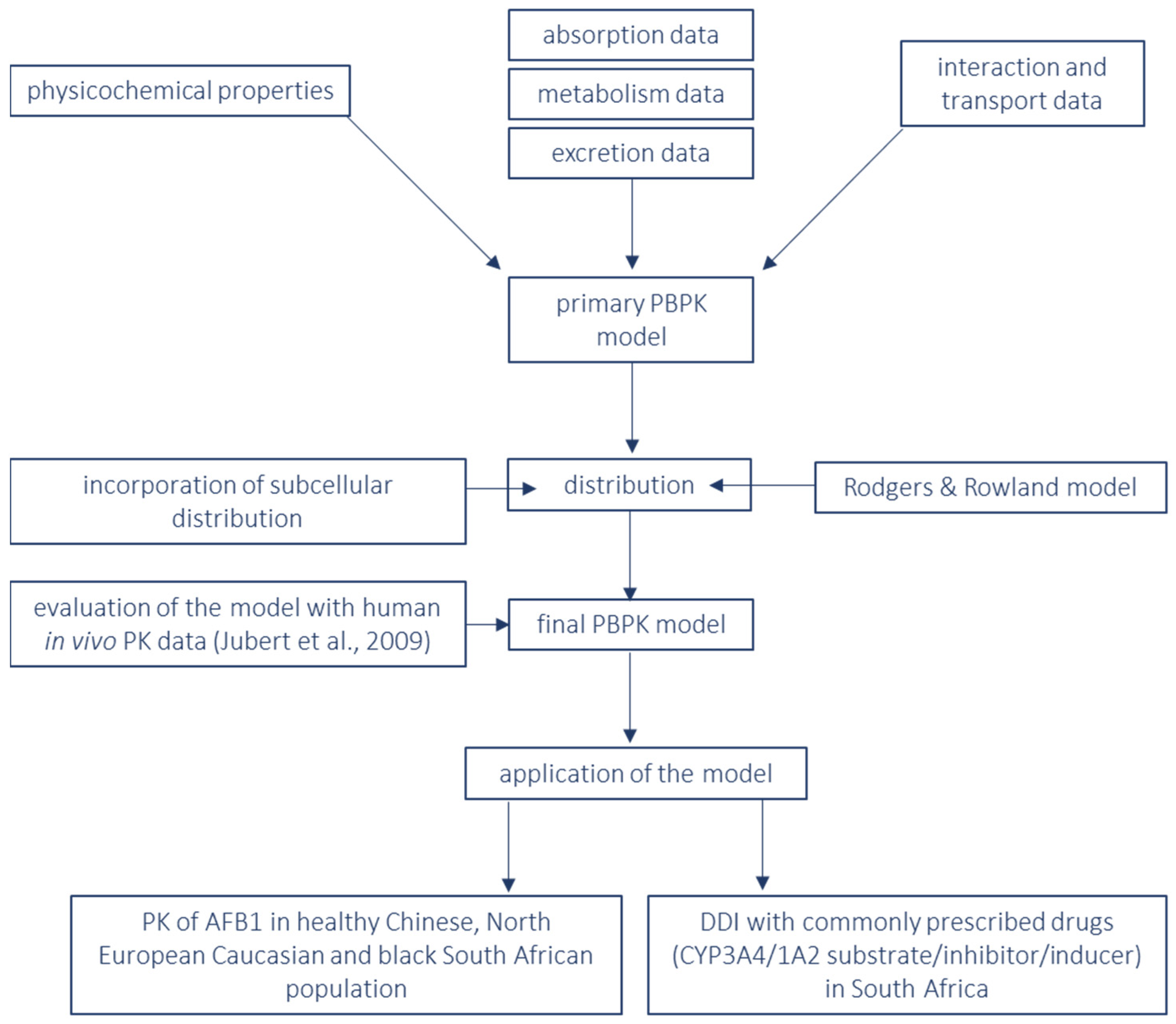
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. “Mycotoxins”. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/mycotoxins (accessed on 18 August 2022).
- IARC. “Monograph IARC Aflatoxins”. 2002. Available online: https://monographs.iarc.fr/wp-content/uploads/2018/06/mono100F-23.pdf (accessed on 25 May 2020).
- Smith, M.-C.; Madec, S.; Coton, E.; Hymery, N. Natural Co-Occurrence of Mycotoxins in Foods and Feeds and Their in vitro Combined Toxicological Effects. Toxins 2016, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Rotimi, O.A.; Rotimi, S.O.; Goodrich, J.M.; Adelani, I.B.; Agbonihale, E.; Talabi, G. Time-Course Effects of Acute Aflatoxin B1 Exposure on Hepatic Mitochondrial Lipids and Oxidative Stress in Rats. Front. Pharmacol. 2019, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Claeys, L.; Romano, C.; De Ruyck, K.; Wilson, H.; Fervers, B.; Korenjak, M.; Zavadil, J.; Gunter, M.J.; De Saeger, S.; De Boevre, M.; et al. Mycotoxin exposure and human cancer risk: A systematic review of epidemiological studies. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1449–1464. [Google Scholar] [CrossRef]
- Lewis, C.; Smith, J.; Anderson, J.; Freshney, R. Increased cytotoxicity of food-borne mycotoxins toward human cell lines in vitro via enhanced cytochrome p450 expression using the MTT bioassay. Mycopathologia 1999, 148, 97–102. [Google Scholar] [CrossRef]
- He, X.-Y.; Tang, L.; Wang, S.-L.; Cai, Q.-S.; Wang, J.-S.; Hong, J.-Y. Efficient activation of aflatoxin B1 by cytochrome P450 2A13, an enzyme predominantly expressed in human respiratory tract. Int. J. Cancer 2006, 118, 2665–2671. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Lu, H.-Y.; Li, Z.-Y.; Bian, Q.; Qiu, L.-L.; Li, Z.; Liu, Q.; Li, J.; Wang, X.; Wang, S.-L. Cytochrome P450 2A13 mediates aflatoxin B1-induced cytotoxicity and apoptosis in human bronchial epithelial cells. Toxicology 2012, 300, 138–148. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, L.; Zhang, N.-Y.; Karrow, N.A.; Krumm, C.S.; Qi, D.-S.; Sun, L.-H. Aflatoxin B1 metabolism: Regulation by phase I and II metabolizing enzymes and chemoprotective agents. Mutat. Res. Mol. Mech. Mutagen. 2018, 778, 79–89. [Google Scholar] [CrossRef]
- Gallagher, L.C.; Wienkers, P.L.; Stapleton, K.L.; Kunze, E.; Eaton, D.L. Role of Human Microsomal and Human Complementary DNA-Expressed Cytochromes P4501A2 and P4503A4 in the Bioactivation of Aflatoxin 1. 1994. Available online: https://cancerres.aacrjournals.org/content/canres/54/1/101.full.pdf (accessed on 28 May 2020).
- Kumar, V.; Bahuguna, A.; Ramalingam, S.; Dhakal, G.; Shim, J.-J.; Kim, M. Recent technological advances in mechanism, toxicity, and food perspectives of enzyme-mediated aflatoxin degradation. Crit. Rev. Food Sci. Nutr. 2021, 62, 5395–5412. [Google Scholar] [CrossRef]
- Kamdem, L.K.; Meineke, I.; Gödtel-Armbrust, U.; Brockmöller, J.; Wojnowski, L. Dominant Contribution of P450 3A4 to the Hepatic Carcinogenic Activation of Aflatoxin B1. Chem. Res. Toxicol. 2006, 19, 577–586. [Google Scholar] [CrossRef]
- DEfsa Panel on Contaminants in the Food Chain (Contam); Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.; et al. Risk assessment of aflatoxins in food. EFSA J. 2020, 18, e06040. [Google Scholar] [CrossRef]
- Salhab, A.S.; Abramson, F.P.; Geelhoed, G.W.; Edwards, G.S. Aflatoxicol M1, A New Metabolite of Aflatoxicol. Xenobiotica 1977, 7, 401–408. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Aflatoxins in food | EFSA. 2006. Available online: https://www.efsa.europa.eu/en/topics/topic/aflatoxins-food (accessed on 17 January 2023).
- Gomez, K.S.; Roldán, E.C.; Sosa, R.; Munguía-Pérez, R. Mycotoxins and Climate Change. In The Impact of Climate Change on Fungal Diseases; Springer: Berlin/Heidelberg, Germany, 2022; pp. 239–256. [Google Scholar] [CrossRef]
- Zain, M.E. Impact of mycotoxins on humans and animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef]
- FAO. Worldwide Regulations for Mycotoxins in Food and Feed in 2003. 2003. Available online: http://www.fao.org/docrep/007/y5499e/y5499e06.htm#bm06.1 (accessed on 12 April 2016).
- PUdomkun, P.; Wiredu, A.N.; Nagle, M.; Bandyopadhyay, R.; Müller, J.; Vanlauwe, B. Mycotoxins in Sub-Saharan Africa: Present situation, socio-economic impact, awareness, and outlook. Food Control 2017, 72, 110–122. [Google Scholar] [CrossRef]
- Azziz-Baumgartner, E.; Lindblade, K.; Gieseker, K.; Rogers, H.S.; Kieszak, S.; Njapau, H.; Schleicher, R.L.; McCoy, L.F.; Misore, A.; Decock, K.M.; et al. Case–Control Study of an Acute Aflatoxicosis Outbreak, Kenya, 2004. Environ. Health Perspect. 2005, 113, 1779–1783. [Google Scholar] [CrossRef] [PubMed]
- Darwish, W.S.; Ikenaka, Y.; Nakayama, S.M.; Ishizuka, M. An Overview on Mycotoxin Contamination of Foods in Africa. J. Vet. Med. Sci. 2014, 76, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Kamala, A.; Shirima, C.; Jani, B.; Bakari, M.; Sillo, H.; Rusibamayila, N.; De Saeger, S.; Kimanya, M.; Gong, Y.; Simba, A.; et al. Outbreak of an acute aflatoxicosis in Tanzania during 2016. World Mycotoxin J. 2018, 11, 311–320. [Google Scholar] [CrossRef]
- WHO. Health Emergency Information and Risk Assessment. 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/326465/OEW33-1218082019.pdf (accessed on 24 March 2020).
- Zeng, D.; Lin, Z.; Zeng, Z.; Fang, B.; Li, M.; Cheng, Y.-H.; Sun, Y. Assessing Global Human Exposure to T-2 Toxin via Poultry Meat Consumption Using a Lifetime Physiologically Based Pharmacokinetic Model. J. Agric. Food Chem. 2019, 67, 1563–1571. [Google Scholar] [CrossRef]
- Fæste, C.K.; Ivanova, L.; Sayyari, A.; Hansen, U.; Sivertsen, T.; Uhlig, S. Prediction of deoxynivalenol toxicokinetics in humans by in vitro-to-in vivo extrapolation and allometric scaling of in vivo animal data. Arch. Toxicol. 2018, 92, 2195–2216. [Google Scholar] [CrossRef] [PubMed]
- Gilbert-Sandoval, I.; Wesseling, S.; Rietjens, I.M.C.M. Predicting the Acute Liver Toxicity of Aflatoxin B1 in Rats and Humans by an In Vitro–In Silico Testing Strategy. Mol. Nutr. Food Res. 2020, 64, e2000063. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Jubert, C.; Mata, J.; Bench, G.; Dashwood, R.; Pereira, C.; Tracewell, W.; Turteltaub, K.; Williams, D.; Bailey, G. Effects of Chlorophyll and Chlorophyllin on Low-Dose Aflatoxin B1 Pharmacokinetics in Human Volunteers. Cancer Prev. Res. 2009, 2, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Aflatoxin B1 | C17H12O6–PubChem. National Center for Biotechnology Information. PubChem Database. 2017. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Aflatoxin-B1#section=Other-Experimental-Properties (accessed on 10 August 2022).
- Varma, M.V.; Steyn, S.J.; Allerton, C.; El-Kattan, A.F. Predicting Clearance Mechanism in Drug Discovery: Extended Clearance Classification System (ECCS). Pharm. Res. 2015, 32, 3785–3802. [Google Scholar] [CrossRef] [PubMed]
- Lootens, O.; De Boevre, M.; Gasthuys, E.; Van Bocxlaer, J.; Vermeulen, A.; De Saeger, S. Unravelling the pharmacokinetics of aflatoxin B1: In vitro determination of Michaelis–Menten constants, intrinsic clearance and the metabolic contribution of CYP1A2 and CYP3A4 in pooled human liver microsomes. Front. Microbiol. 2022, 13, 3258. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, T.; Ufuk, A.; Cantrill, C.; Kosa, R.E.; Bi, Y.-A.; Niosi, M.; Modi, S.; Rodrigues, A.D.; Tremaine, L.M.; Varma, M.V.S.; et al. Predicting Human Clearance of Organic Anion Transporting Polypeptide Substrates Using Cynomolgus Monkey: In Vitro–In Vivo Scaling of Hepatic Uptake Clearance. Drug Metab. Dispos. 2018, 46, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Loe, D.W.; Stewart, R.K.; Massey, T.E.; Deeley, R.G.; Cole, S.P.C. ATP-Dependent Transport of Aflatoxin B1 and Its Glutathione Conjugates by the Product of the Multidrug Resistance Protein (MRP) Gene. Mol. Pharmacol. 1997, 51, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Vildhede, A.; Wiśniewski, J.R.; Norén, A.; Karlgren, M.; Artursson, P. Comparative Proteomic Analysis of Human Liver Tissue and Isolated Hepatocytes with a Focus on Proteins Determining Drug Exposure. J. Proteome Res. 2015, 14, 3305–3314. [Google Scholar] [CrossRef]
- Barter, Z.E.; Tucker, G.T.; Rowland-Yeo, K. Differences in Cytochrome P450-Mediated Pharmacokinetics Between Chinese and Caucasian Populations Predicted by Mechanistic Physiologically Based Pharmacokinetic Modelling. Clin. Pharmacokinet. 2013, 52, 1085–1100. [Google Scholar] [CrossRef]
- DuBois, D.; DuBois, E.F. A formula to estimate the approximate surface area if height and weight be known. Nutrition 1989, 5, 303–311; discussion 312–313. [Google Scholar]
- O Nwoye, L. Body surface area of Africans: A study based on direct measurements of Nigerian males. Hum. Biol. 1989, 61, 439–457. [Google Scholar]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A New Equation to Estimate Glomerular Filtration Rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Rajman, I.; Knapp, L.; Morgan, T.; Masimirembwa, C. African Genetic Diversity: Implications for Cytochrome P450-mediated Drug Metabolism and Drug Development. Ebiomedicine 2017, 17, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.; Dolgos, H. Requirements to Establishing Confidence in Physiologically Based Pharmacokinetic (PBPK) Models and Overcoming Some of the Challenges to Meeting Them. Clin. Pharmacokinet. 2019, 58, 1355–1371. [Google Scholar] [CrossRef] [PubMed]
- Page, K.M. Validation of Early Human Dose Prediction: A Key Metric for Compound Progression in Drug Discovery. Mol. Pharm. 2016, 13, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Persuad, N. EMLs Around the World. WHO Bulletin. 2019. Available online: https://global.essentialmeds.org/dashboard/medicines/215 (accessed on 15 August 2022).
- Food and Drug Administration. Compliance Policy Guide Sec. 570.375 Aflatoxins in Peanuts and Peanut Products: Guidance for FDA Staff. June 2021. Available online: https://www.fda.gov/media/72073/download (accessed on 10 August 2022).
- EUR-Lex–02006R1881-20100701–EN–EUR-Lex. 2006. Available online: https://eur-lex.europa.eu/eli/reg/2006/1881/2010-07-01 (accessed on 14 February 2023).
- EFSA Scientific Committee Statement on the applicability of the Margin of Exposure approach for the safety assessment of impurities which are both genotoxic and carcinogenic in substances added to food/feed. EFSA J. 2012, 10, 2578. [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties and Blood Binding | ||
|---|---|---|
| Parameter | Model Input Value | Reference |
| Mw (g/moL) | 312.27 | [29] |
| LogPo:w | 1.60 | [29] |
| Compound type | neutral | [29] |
| ECCS | Class 2 | [30] |
| B:P | 1.03 | Predicted * |
| fu,p | 0.17 | [26] |
| Absorption | ||
| fa | 0.99 | Predicted * |
| ka | 2.39 | Predicted * |
| Peff,man (10−4 cm/s) | 5.47 | predicted * |
| Ptrans,0 (10−6 cm/s) | 135.8 | Predicted * |
| Absorption and Metabolism (ADAM) Model | ||
| Formulation—Diffusion Layer Model—Aqueous Phase Solubility—Solid State 1 | ||
| S0 | 1.24 | Predicted * |
| Distribution | ||
| Full PBPK model | ||
| Vss (L/kg) | 0.33 | prediction method 3 |
| Tissue: plasma partition coefficients/Kp scalar = 1 | ||
| Adipose | 0.44 | predicted * |
| Bone | 0.15 | predicted * |
| Brain | 0.55 | predicted * |
| Gut | 0.36 | predicted * |
| Pancreas | 0.26 | predicted * |
| Heart | 0.37 | predicted * |
| Kidney | 0.36 | predicted * |
| Liver | 0.44 | predicted * |
| Lung | 0.33 | predicted * |
| Muscle | 0.23 | predicted * |
| Skin | 0.28 | predicted * |
| Spleen | 0.44 | predicted * |
| Elimination | ||
| Enzyme kinetics | ||
| CYPs | Recombinant | |
| CYP3A4 Km | 49.60 µM | Experimental [31] |
| CYP3A4 Vmax | 88.10 pmol/min/pmol CYP | Experimental [31] |
| CYP3A4 ISEF | 0.50 | Experimental [31] |
| CYP1A2 Km | 58.20 µM | Experimental [31] |
| CYP1A2 Vmax | 199.00 pmol/min/pmol CYP | Experimental [31] |
| ISEF | 1.42 | Experimental [31] |
| Interaction | ||
| CYP1A2 Ki | 10.2 µM | Experimental [31] |
| Transport | ||
| Using permeability limited liver model | ||
| CLPD (mL/min/million hepatocytes) | 0.05 | derived from [32] |
| fu,IW | 0.35 | predicted * |
| fu,EW | 0.17 | predicted * |
| Drug concentration for passive permeability: unbound (ionized and unionized) Sinusoidal: Efflux: ABCC3 (MRP3) | ||
| Jmax (pmol/min/million cells) | 180.00 | [33] |
| Km (µM) | 0.19 | |
| fu,inc | 1.00 | |
| RAF/REF | 2.50 | [34] |
| Height | |||
| MALE | FEMALE | ||
| C0 | 161.780 | C0 | 155.376 |
| C1 | 0.359 | C1 | 0.207 |
| C2 | −0.00429 | C2 | −0.00268 |
| CV (%) | 7.33 | CV (%) | 5.83 |
| Weight | |||
| MALE | FEMALE | ||
| C0 | 2.97 | C0 | 3.19 |
| C1 | 0.007 | C1 | 0.007 |
| CV (%) | 21.1 | CV (%) | 26.38 |
| Black South African | Sim-NEur Caucasian | Sim-Chinese Healthy Volunteer | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CYP450 Enzyme | Abundance (pmol/mg Protein)/CV | Phenotype Frequency | Abundance (pmol/mg Protein)/CV | Phenotype Frequency | Abundance (pmol/mg Protein)/CV | Phenotype Frequency | |||
| EM | PM | EM | PM | EM | PM | ||||
| CYP1A2 | 52/67% | 1 | 0 | 52/67% | 1 | 0 | 42/50% | 1 | 0 |
| CYP2B6 | 6.9/122% | 0.85 | 0.15 | 21.6/68% | 0.40 | 0.10 | 6.7/63% | 0.52 | 0.07 |
| CYP2C9 | 73/ 54% | 0.98 | 0.02 | 77.7/64% | 0.66 | 0.019 | 87.6/55% | 0.93 | 0.003 |
| CYP2C19 | 14/106% | 0.96 | 0.04 | 4.4/52% | 0.42 | 0.023 | 4.4/52% | 0.40 | 0.13 |
| CYP2D6 | 8/61% | 0.97 | 0.03 | 9.4/65% | 0.57 | 0.08 | 10.47/65% | 0.60 | 0.003 |
| CYP3A4 | 137/41% | 1 | 0 | 137/41% | 1 | 0 | 120/33% | 1 | 0 |
| CYP3A5 | 71/78% | 0.82 | 0.18 | 103/65% | 0.17 | 0.83 | 82.3/68% | 0.42 | 0.58 |
| CYP3A7 | 35.4/61% | 0.12 | 0.88 | 35.4/61% | 0.12 | 0.88 | 14/71% | 0.12 | 0.88 |
| parameter | parameter value | CV (%) | parameter value | CV (%) | parameter value | CV (%) | |||
| LV (L) | 1.924 | 12 | 1.651 | 12 | 1.403 | 12 | |||
| MPPGL (mg/g) | 39.79 | N.A. | 39.79 | N.A. | 39.45 | N.A. | |||
| LD (g/L) | 1080 | N.A. | 1080 | N.A. | 1080 | N.A. | |||
| Hematocrit (%) (male) | 43 | 6.51 | 43 | 6.5 | 45.3 | 9.5 | |||
| Hematocrit (%) (female) | 38 | 7.13 | 38 | 7.1 | 40.5 | 10.9 | |||
| AGP (g/L) (male) | 0.811 | 15 | 0.739 | 23 | 0.683 | 23 | |||
| AGP (g/L) (female) | 0.791 | 13 | 0.715 | 24 | 0.575 | 24 | |||
| HSA (g/L) (male) | 50.34 | 10 | 50.34 | 10 | 50.34 | 10 | |||
| HSA (g/L) (female) | 49.38 | 10 | 49.38 | 10 | 49.38 | 10 | |||
| Weibull α (male) | 1.47 | N.A. | 5.47 | N.A. | 1.5 | N.A. | |||
| Weibull β (male) | 30.17 | N.A. | 66.5 | N.A. | 19 | N.A. | |||
| Weibull α (female) | 1.47 | N.A. | 5.22 | N.A. | 4.48 | N.A. | |||
| Weibull β (female) | 32.8 | N.A. | 68.57 | N.A. | 53.4 | N.A. | |||
| Drug | Dose (mg) QD | CYP3A4/CYP1A2 Substrate/Inhibitor/Inducer | Drug Class |
|---|---|---|---|
| artemether | 20 | CYP3A4 substrate | antimalarial |
| atazanavir | 200 | CYP3A4 substrate CYP3A4 inhibitor | protease inhibitor |
| carbamazepine | 200 | CYP3A4 substrate CYP3A4 inducer | anticonvulsant |
| ciprofloxacin | 250 | CYP1A2 inhibitor | quinolone antibiotics |
| efavirenz | 600 | CYP3A4 inducer | non-nucleoside reverse transcriptase inhibitor |
| ethinylestradiol | 0.035 | CYP3A4 substrate CYP1A2 inhibitor | estrogen |
| phenobarbital | 100 | CYP3A4 and CYP1A2 inducer | barbiturate |
| phenytoin | 100 | CYP3A4 and CYP1A2 inducer | anticonvulsant |
| fluconazole | 50 | CYP3A4 inhibitor | triazole antifungal |
| fluoxetine | 20 | CYP3A4 inhibitor | selective serotonin reuptake inhibitor |
| midazolam | 5 | CYP3A4 substrate | benzodiazepine |
| nifedipine | 20 | CYP3A4 substrate | calcium channel blocker |
| rifampicin | 600 | CYP1A2 inducer CYP3A4 inducer | antimycobacterial |
| ritonavir | 600 BID | CYP3A4 substrate CYP3A4 inhibitor | protease inhibitor |
| simvastatin | 20 | CYP3A4 substrate | statins |
| Observed Data (Mean ± SD) | Predicted Data (Mean ± SD) | Predicted/Observed Ratio | |
|---|---|---|---|
| Cmax (pg/mL) | 0.941 ± 0.154 | 1.02 ± 0.035 | 1.08 |
| AUC0–24 h (pg/mL.h) | 12.4 ± 1.8 | 9.87 ± 0.825 | 0.80 |
| Tmax (h) | 1.02 ± 0.31 h | 1.64 ± 0.075 h | 1.61 |
| AFE on Cp | 1.12 | ||
| AAFE on Cp | 1.35 |
| Sim-Chinese Healthy Volunteers | North European Caucasian | Black South African | |
|---|---|---|---|
| mean Cmax (pg/mL) | 0.967 | 0.740 | 0.755 |
| mean Tmax (h) | 1.92 | 1.67 | 1.64 |
| mean AUC0–24 h (pg/mL.h) | 9.85 | 6.78 | 6.24 |
| mean CL (L/h) | 4.62 | 6.52 | 8.78 |
| AFB1 Alone | AFB1 + Drug | Ratio of PK Parameters (with Drug/without Drug) | |||
|---|---|---|---|---|---|
| ME | +Atazanavir (200 mg) QD | ME | |||
| Cmax (pg/mL) | 1.19 | 0.076 | 1.69 | 0.12 | 1.39 |
| Tmax (h) | 0.96 | 0.055 | 1.09 | 0.07 | 1.14 |
| AUC0-inf (pg/mL.h) | 11.7 | 1.21 | 23.7 | 2.54 | 2.09 |
| Cmin (pg/mL) | 0.18 | 0.0091 | 0.55 | 0.0055 | 3.10 |
| CL (L/h) | 5.82 | 0.58 | 3.12 | 0.31 | 0.54 |
| +carbamazepine (200 mg) QD | |||||
| Cmax (pg/mL) | 1.21 | 0.08 | 1.00 | 0.063 | 0.83 |
| Tmax (h) | 0.96 | 0.06 | 0.92 | 0.055 | 0.96 |
| AUC0-inf (pg/mL.h) | 12.09 | 1.24 | 8.92 | 0.91 | 0.74 |
| Cmin (pg/mL) | 0.19 | 0.00096 | 0.12 | 0.00048 | 0.62 |
| CL (L/h) | 5.69 | 0.57 | 7.28 | 0.72 | 1.28 |
| +ciprofloxacin (250 mg) QD | |||||
| Cmax (pg/mL) | 1.20 | 0.075 | 1.52 | 0.093 | 1.27 |
| Tmax (h) | 0.95 | 0.055 | 1.28 | 0.08 | 1.35 |
| AUC0-inf (pg/mL.h) | 11.8 | 1.22 | 17.42 | 1.68 | 1.47 |
| Cmin (pg/mL) | 0.19 | 0.00092 | 0.28 | 0.00188 | 1.51 |
| CL (L/h) | 5.86 | 0.58 | 3.21 | 0.31 | 0.55 |
| +efavirenz (600 mg) QD | |||||
| Cmax (pg/mL) | 1.21 | 0.082 | 0.80 | 0.0053 | 0.66 |
| Tmax (h) | 0.95 | 0.06 | 0.71 | 0.045 | 0.75 |
| AUC0-inf (pg/mL.h) | 12.20 | 1.25 | 4.41 | 0.44 | 0.36 |
| Cmin (pg/mL) | 0.20 | 0.0090 | 0.03 | 0.00006 | 0.16 |
| CL (L/h) | 5.91 | 0.59 | 15.52 | 1.62 | 2.63 |
| +phenobarbital (100 mg) QD | |||||
| Cmax (pg/mL) | 1.21 | 0.078 | 0.78 | 0.055 | 0.64 |
| Tmax (h) | 0.95 | 0.06 | 0.80 | 0.05 | 0.84 |
| AUC0-inf (pg/mL.h) | 12.1 | 1.25 | 5.39 | 0.56 | 0.45 |
| Cmin (pg/mL) | 0.20 | 0.009 | 0.05 | 0.00047 | 0.25 |
| CL (L/h) | 5.91 | 0.59 | 13.60 | 1.38 | 2.30 |
| +phenytoin (100 mg) QD | |||||
| Cmax (pg/mL) | 1.21 | 0.08 | 0.95 | 0.063 | 0.79 |
| Tmax (h) | 0.96 | 0.06 | 0.89 | 0.055 | 0.93 |
| AUC0-inf (pg/mL.h) | 12.2 | 1.26 | 8.09 | 0.82 | 0.66 |
| Cmin (pg/mL) | 0.20 | 0.0095 | 0.11 | 0.00284 | 0.55 |
| CL (L/h) | 5.78 | 0.57 | 8.78 | 0.87 | 1.52 |
| +fluconazole (50 mg) QD | |||||
| Cmax (pg/mL) | 1.20 | 0.064 | 1.35 | 0.07 | 1.13 |
| Tmax (h) | 0.95 | 0.06 | 1.00 | 0.06 | 1.05 |
| AUC0-inf (pg/mL.h) | 12.0 | 0.8 | 15.0 | 0.95 | 1.25 |
| Cmin (pg/mL) | 0.19 | 0.00113 | 0.267 | 0.00201 | 1.41 |
| CL (L/h) | 5.88 | 0.92 | 4.75 | 0.79 | 0.81 |
| +rifampicin (600 mg) QD | |||||
| Cmax (pg/mL) | 1.19 | 0.08 | 0.64 | 0.051 | 0.54 |
| Tmax (h) | 0.96 | 0.055 | 0.71 | 0.045 | 0.74 |
| AUC0-inf (pg/mL.h) | 11.7 | 1.21 | 3.02 | 0.31 | 0.26 |
| Cmin (pg/mL) | 0.18 | 0.0090 | 0.013 | 0.00001 | 0.07 |
| CL (L/h) | 5.82 | 0.575 | 24.03 | 2.47 | 4.13 |
| + ritonavir (600 mg) BID | |||||
| Cmax (pg/mL) | 1.21 | 0.078 | 1.95 | 0.15 | 1.56 |
| Tmax (h) | 0.95 | 0.055 | 1.10 | 0.07 | 1.16 |
| AUC0-inf (pg/mL.h) | 12.2 | 1.21 | 29.0 | 3.1 | 2.50 |
| Cmin (pg/mL) | 0.20 | 0.0090 | 0.75 | 0.078 | 3.75 |
| CL (L/h) | 5.91 | 0.575 | 2.78 | 0.27 | 0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lootens, O.; De Boevre, M.; Ning, J.; Gasthuys, E.; Van Bocxlaer, J.; De Saeger, S.; Vermeulen, A. Building a Human Physiologically Based Pharmacokinetic Model for Aflatoxin B1 to Simulate Interactions with Drugs. Pharmaceutics 2023, 15, 894. https://doi.org/10.3390/pharmaceutics15030894
Lootens O, De Boevre M, Ning J, Gasthuys E, Van Bocxlaer J, De Saeger S, Vermeulen A. Building a Human Physiologically Based Pharmacokinetic Model for Aflatoxin B1 to Simulate Interactions with Drugs. Pharmaceutics. 2023; 15(3):894. https://doi.org/10.3390/pharmaceutics15030894
Chicago/Turabian StyleLootens, Orphélie, Marthe De Boevre, Jia Ning, Elke Gasthuys, Jan Van Bocxlaer, Sarah De Saeger, and An Vermeulen. 2023. "Building a Human Physiologically Based Pharmacokinetic Model for Aflatoxin B1 to Simulate Interactions with Drugs" Pharmaceutics 15, no. 3: 894. https://doi.org/10.3390/pharmaceutics15030894
APA StyleLootens, O., De Boevre, M., Ning, J., Gasthuys, E., Van Bocxlaer, J., De Saeger, S., & Vermeulen, A. (2023). Building a Human Physiologically Based Pharmacokinetic Model for Aflatoxin B1 to Simulate Interactions with Drugs. Pharmaceutics, 15(3), 894. https://doi.org/10.3390/pharmaceutics15030894