
Cannabimimetic N-Stearoylethanolamine as “Double-Edged Sword” in Anticancer Chemotherapy: Proapoptotic Effect on Tumor Cells and Suppression of Tumor Growth versus Its Bio-Protective Actions in Complex with Polymeric Carrier on General Toxicity of Doxorubicin In Vivo

 , , , , ,
, , , , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Part
Synthesis of the Polymeric Carrier (PC)
2.2. Biological Part
2.2.1. Materials
2.2.2. Cell Culture and Cytotoxicity Measurements
2.2.3. Apoptosis Ana Lysis
2.2.4. Cell Cycle Analysis
2.2.5. Intracellular ROS Content
2.2.6. Studies of the Functional Status of Mitochondria
2.2.7. FACS Analysis of Intracellular Dx Accumulation
2.2.8. Microscopic Uptake Studies
2.2.9. Immunoblot Analysis of Cellular Proteins
2.2.10. Animals
2.2.11. Murine Tumor Models and Drug Treatment Schemes
2.2.12. Blood Cell and Serum Analysis in Mice
2.2.13. Analysis of Lipid Profile in Heart Tissue of Mice under Treatment with Dx and NSE
2.2.14. Statistical Analyses
3. Results
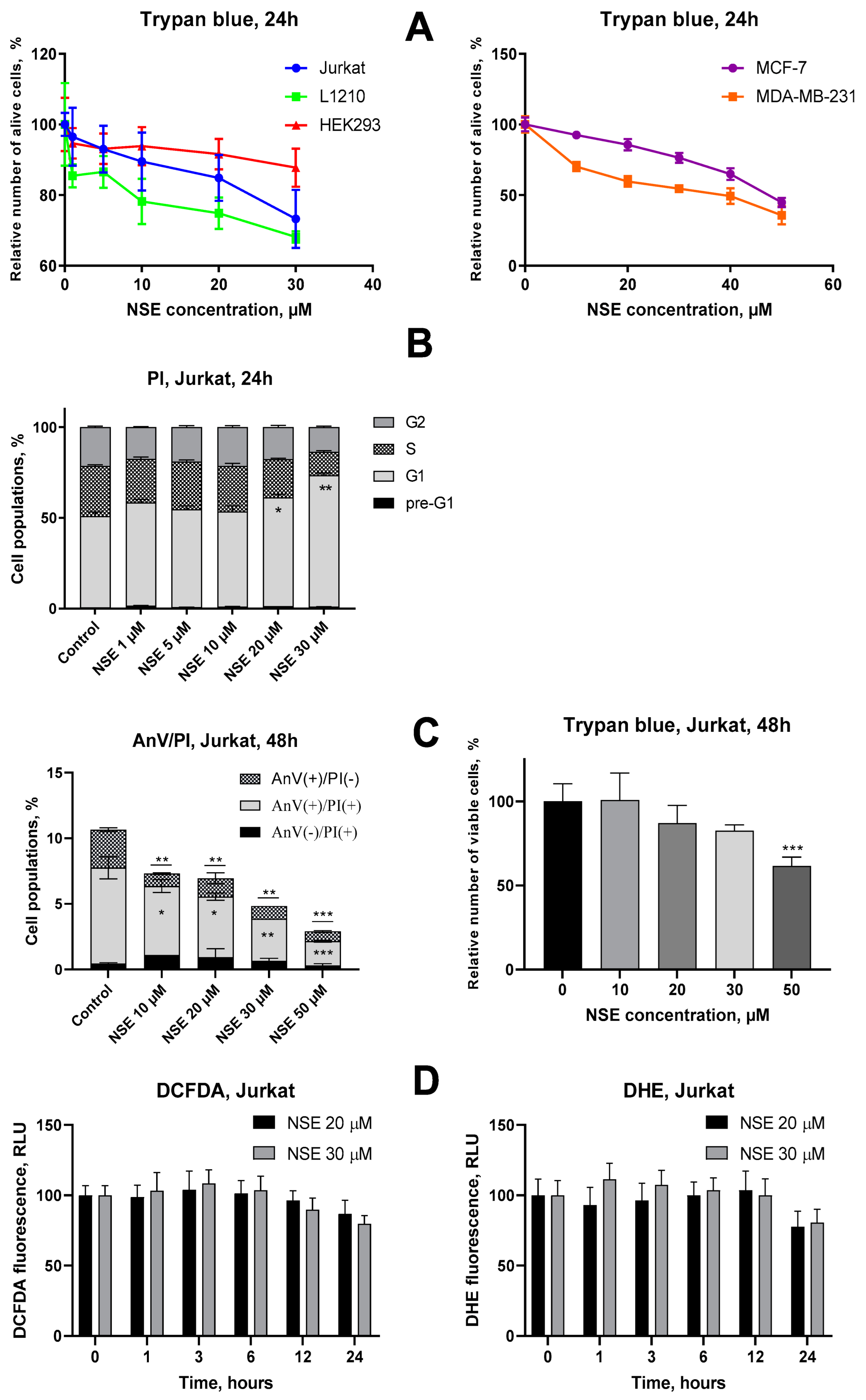
3.1. Assessment of Cytotoxic Activities of NSE In Vitro
3.2. Impact of NSE Pre-Treatment on the Cytotoxic Activity of Dx In Vitro
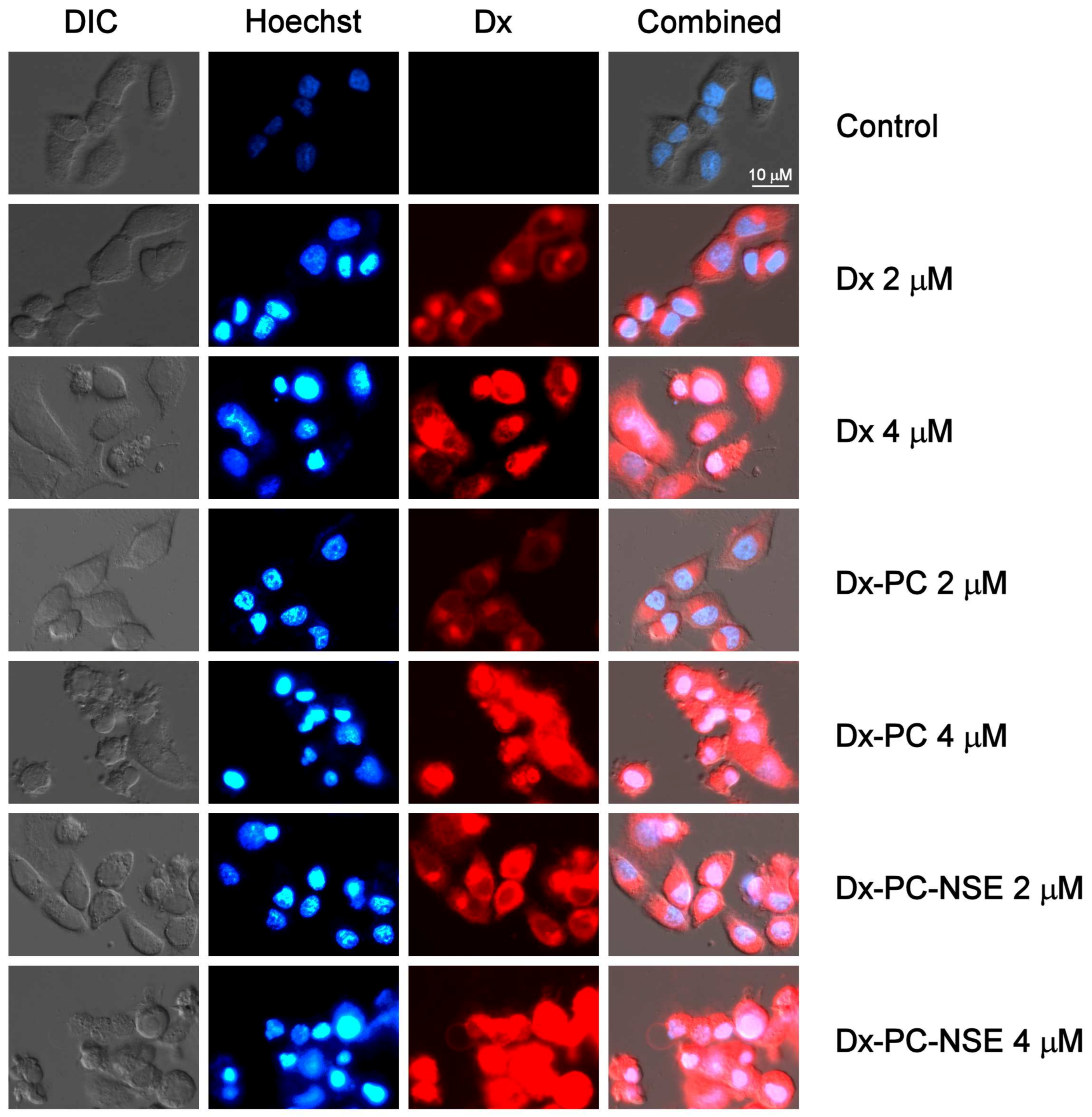
3.3. Modulation of the Cytotoxic Effect of Dx towards Tumor Cells In Vitro by NSE Immobilized on PC
3.4. Impact of Co-Immobilization of Dx and NSE on PC on ROS and Induction of Cell Death in Tumor Cells In Vitro
3.5. Activity of PC with Immobilized Dx and NSE in Tumor-Bearing Mouse Models
3.6. Bio-Protective Effects of NSE Conjugated to PC on General Toxicity of Dx In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef]
- Menna, P.; Paz, O.G.; Chello, M.; Covino, E.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2012, 11 (Suppl. 1), S21–S36. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nature Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Aryal, S.; Zhang, L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther. Deliv. 2010, 1, 323–334. [Google Scholar] [CrossRef]
- Su, Z.; Dong, S.; Zhao, S.-C.; Liu, K.; Tan, Y.; Jiang, X.; Assaraf, Y.G.; Qin, B.; Chen, Z.-S.; Zou, C. Novel nanomedicines to overcome cancer multidrug resistance. Drug Resist. Updat. 2021, 58, 100777. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 12320–12364. [Google Scholar] [CrossRef] [PubMed]
- Senkiv, Y.; Riabtseva, A.; Heffeter, P.; Boiko, N.; Kowol, C.R.; Jungwith, U.; Shlyakhtina, Y.; Garasevych, S.G.; Mitina, N.; Berger, W.; et al. Enhanced anticancer activity and circumvention of resistance mechanisms by novel polymeric/phospholipidic nanocarriers of doxorubicin. J. Biomed. Nanotechnol. 2014, 10, 1369–1381. [Google Scholar] [CrossRef]
- Mock, E.D.; Gagestein, B.; van der Stelt, M. Anandamide and other N-acylethanolamines: A class of signaling lipids with therapeutic opportunities. Prog. Lipid Res. 2023, 89, 101194. [Google Scholar] [CrossRef]
- Voitychuk, O.I.; Asmolkova, V.S.; Gula, N.M.; Sotkis, G.V.; Galadari, S.; Howarth, F.C.; Oz, M.; Shuba, Y.M. Modulation of excitability, membrane currents and survival of cardiac myocytes by N-acylethanolamines. Biochim. Et Biophys. Acta 2012, 1821, 1167–1176. [Google Scholar] [CrossRef]
- Berdyshev, A.G.; Kosiakova, H.V.; Onopchenko, O.V.; Panchuk, R.R.; Stoika, R.S.; Hula, N.M. N-Stearoylethanolamine suppresses the pro-inflammatory cytokines production by inhibition of NF-kappaB translocation. Prostaglandins Other Lipid Mediat. 2015, 121, 91–96. [Google Scholar] [CrossRef]
- Hula, N.M.; Khmel, T.O.; Klimashevs’kyi, V.M.; Kulik, H.I.; Todor, I.M. N-stearoylethanolamine inhibits growth and metastasis of the Lewis carcinoma and modulates lipid composition of the lung during tumorogenesis in mice. Ukr. Biokhimichnyi Zhurnal 2006, 78, 135–142. [Google Scholar]
- Voronov, S.A.; Kiselyov, E.M.; Minko, S.S.; Budishevska, O.G.; Roiter, Y.V. Structure and reactivity of peroxide monomers. J. Polym. Sci. Part A Polym. Chem. 1996, 34, 2507–2511. [Google Scholar] [CrossRef]
- Riabtseva, A.; Mitina, N.; Grytsyna, I.; Boiko, N.; Garamus, V.M.; Stryhanyuk, H.; Stoika, R.; Zaichenko, A. Functional micelles formed by branched polymeric surfactants: Synthesis, characteristics, and application as nanoreactors and carriers. Eur. Polym. J. 2016, 75, 406–422. [Google Scholar] [CrossRef]
- Braum, D.; Cherobon, H.; Rehahn, M.; Riher, H.; Voits, B. Polymer Synthesis Theory and Practice, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Steyermark, A. Quantitative Organic Microanalysis; Elsevier: Amsterdam, The Netherlands, 1961; p. 665. [Google Scholar] [CrossRef]
- Crompton, T.R. Functional Groups in Polymers. In Practical Polymer Analysis; Crompton, T.R., Ed.; Springer: Boston, MA, USA, 1993; pp. 241–255. [Google Scholar] [CrossRef]
- Hudz, I.A.; Chernyshenko, V.O.; Kasatkina, L.O.; Urvant, L.P.; Klimashevskyi, V.M.; Tkachenko, O.S.; Kosiakova, H.V.; Hula, N.M.; Platonova, T.M. N-Stearoylethanolamine Inhibits Integrin-Mediated Activation, Aggregation, and Adhesion of Human Platelets. J. Pharmacol. Exp. Ther. 2022, 383, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.R.; Kwast-Welfeld, J.; Gourdeau, H.; Leblanc, J.; Neugebauer, W.; Sikorska, M. Relationship between Apoptosis and the Cell Cycle in Lymphocytes: Roles of Protein Kinase C, Tyrosine Phosphorylation, and AP1. Exp. Cell Res. 1993, 207, 142–151. [Google Scholar] [CrossRef]
- Peterson, G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977, 83, 346–356. [Google Scholar] [CrossRef]
- Geran, R.I. Protocols for screening chemical agents and natural products against animal tumors and other biological systems. Cancer Chemother. Rep. 1972, 3, 51–61. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Svetashev, V.I.; Vaskovsky, V.E. A simplified technique for thin-layer microchromatography of lipids. J. Chromatogr. A 1972, 67, 376–378. [Google Scholar] [CrossRef]
- Kates, M. Techniques of Lipidology: Isolation, analysis and identification of lipids. In Laboratory Techniques in Biochemistry and Molecular Biology; Work, T.S., Work, E., Eds.; Elsevier: Amsterdam, The Netherlands, 1972; Volume 3, p. 267. [Google Scholar]
- Vaskovsky, V.E.; Kostetsky, E.Y.; Vasendin, I.M. A universal reagent for phospholipid analysis. J. Chromatogr. A 1975, 114, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Bifulco, M. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 34, 273–282. [Google Scholar] [CrossRef]
- Gulaya, N.M.; Kuzmenko, A.I.; Margitich, V.M.; Govseeva, N.M.; Melnichuk, S.D.; Goridko, T.M.; Zhukov, A.D. Long-chain N-acylethanolamines inhibit lipid peroxidation in rat liver mitochondria under acute hypoxic hypoxia. Chem. Phys. Lipids 1998, 97, 49–54. [Google Scholar] [CrossRef]
- Hula, N.; Chumak, A.; Berdyshev, A.; Mehed, O.; Horid’ko, T.; Kindruk, N.; Kosiakova, H.; Zhukov, O. Anti-inflammatory effect of N-stearoylethanolamine in experimental burn injury in rats. Ukr. Biokhimichnyi Zhurnal 2009, 81, 107–116. [Google Scholar]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.M.; Fiebig, H.-H. Chapter 16-screening using animal systems. In Anticancer Drug Development; Baguley, B.C., Kerr, D.J., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 285–299. [Google Scholar] [CrossRef]
- Panchuk, R.R.; Boiko, N.M.; Lootsik, M.D.; Stoika, R.S. Changes in cytokine production and morphology of murine lymphoma NK/Ly cells in course of tumor development. Cent. Eur. J. Biol. 2007, 2, 71. [Google Scholar] [CrossRef]
- Panchuk, R.R.; Boiko, N.M.; Lootsik, M.D.; Stoika, R.S. Changes in signaling pathways of cell proliferation and apoptosis during NK/Ly lymphoma aging. Cell Biol. Int. 2008, 32, 1057–1063. [Google Scholar] [CrossRef]
- Swystun, L.L.; Shin, L.Y.; Beaudin, S.; Liaw, P.C. Chemotherapeutic agents doxorubicin and epirubicin induce a procoagulant phenotype on endothelial cells and blood monocytes. J. Thromb. Haemost. 2009, 7, 619–626. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M. Understanding the causes of multidrug resistance in cancer: A comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Audet-Delage, Y.; Annis, M.G.; Xue, Y.; Vernier, M.; Zhao, K.; St-Louis, C.; Minarrieta, L.; Patten, D.A.; Morin, G.; et al. Resistance to different anthracycline chemotherapeutics elicits distinct and actionable primary metabolic dependencies in breast cancer. eLife 2021, 10, e65150. [Google Scholar] [CrossRef]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. Breast 2012, 21, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Owattanapanich, W.; Owattanapanich, N.; Kungwankiattichai, S.; Ungprasert, P.; Ruchutrakool, T. Efficacy and Toxicity of Idarubicin versus High-dose Daunorubicin for Induction Chemotherapy in Adult Acute Myeloid Leukemia: A Systematic Review and Meta-analysis. Clin. Lymphoma Myeloma Leuk. 2018, 18, 814–821. [Google Scholar] [CrossRef]
- Lao, J.; Madani, J.; Puertolas, T.; Alvarez, M.; Hernandez, A.; Pazo-Cid, R.; Artal, A.; Anton Torres, A. Liposomal Doxorubicin in the treatment of breast cancer patients: A review. J. Drug Deliv. 2013, 2013, 456409. [Google Scholar] [CrossRef] [PubMed]
- Hudz’ Ie, A.; Hula, N.M.; Khmel, T.O.; Horid’ko, T.M.; Berdyshev, A.H. Antioxidative effect of the N-stearoylethanolamine in the heart tissue and blood plasma of rats under doxorubicin treatment. Ukr. Biokhimichnyi Zhurnal 2011, 83, 86–91. [Google Scholar]
- Hudz’ Ie, A.; Hula, N.M.; Horid’ko, T.M.; Bashta Iu, M.; Voieĭkov, A.I. Organ-specific antitoxic effects of N-stearoylethanolamine in male mice with Lewis carcinoma under doxorubicin intoxication. Ukr. Biokhimichnyi Zhurnal 2013, 85, 45–51. [Google Scholar]
- Bhagat, A.; Shrestha, P.; Jeyabal, P.; Peng, Z.; Watowich, S.S.; Kleinerman, E.S. Doxorubicin-induced cardiotoxicity is mediated by neutrophils through release of neutrophil elastase. Front. Oncol. 2022, 12, 947604. [Google Scholar] [CrossRef]
- Hudz’ Ie, A.; Hula, N.M.; Horid’ko, T.M.; Bashta Iu, M.; Voieĭkov, A.I.; Berdyshev, A.H.; Kosiakova, H.V.; Panchuk, R.R.; Stoĭka, R.S.; Riabtseva, A.O.; et al. Antitoxic and antioxidant effects of N-stearoylethanolamin in the content of nanocomposite complex with doxorubicin in organs of mice with Lewis carcinoma. Ukr. Biokhimichnyi Zhurnal 2013, 85, 97–104. [Google Scholar]
- Hudz’ Ie, A.; Hula, N.M.; Khmel, T.O.; Horid’ko, T.M.; Bashta Iu, M.; Panchuk, R.R.; Stoĭka, R.S.; Riabtseva, A.O.; Zaichenko, O.S. Protective effect of N-stearoylethanolamine in suspension and in nanocomposite complex in the organs of mice with the Lewis carcinoma under doxorubicin intoxication administration. Ukr. Biokhimichnyi Zhurnal 2012, 84, 61–69. [Google Scholar]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition of the Water Dispersion mg/mL | DLS Z-Average Hydrodynamic Diameter (nm) | Polydispersity Index (PDI) | Zeta Potential, (mV) | ||
|---|---|---|---|---|---|---|
| [PC] | [NSE] | [Dx] | ||||
| PC | 10 | - | - | 51 ± 16 | 0.35 | −0.11 |
| PC + NSE | 10 | 0.3 | - | 420 ± 120 | 0.25 | +4.25 |
| PC + Dx | 10 | - | 0.3 | 40 ± 9 | 0.06 | +1.57 |
| PC + NSE + Dx | 10 | 0.3 | 0.3 | 680 ± 110 | 0.17 | +6.10 |
| Compound | LC50 Values of Compounds for Cell Line, µM (M ± SD) | |||||||
|---|---|---|---|---|---|---|---|---|
| Jurkat | MCF-7 | L1210 | HeLa | SW1573 | SW1573/ 2R160 | HL-60 | HL-60/adr | |
| Dx | 0.52 ± 0.09 | 0.71 ± 0.04 | 0.49 ± 0.04 | 1.03 ± 0.15 | 1.03 ± 0.06 | 11.02 ± 0.12 | 0.17 ± 0.02 | 37.99 ± 0.56 |
| Dx-PC | 0.45 ± 0.13 | 0.42 ± 0.05 | 0.32 ± 0.05 | 0.98 ± 0.18 | 0.13 ± 0.01 | 9.32 ± 0.11 | 0.19 ± 0.03 | 11.11 ± 0.21 |
| Dx-PC-NSE | 0.32 ± 0.07 | 0.20 ± 0.01 | 0.02 ± 0.01 | 0.53 ± 0.09 | 0.05 ± 0.005 | 0.85 ± 0.07 | 0.09 ± 0.01 | 6.24 ± 0.09 |
| Phospholipid | Untreated | NSE | Dx | Dx + NSE | NSE, Dx | |
|---|---|---|---|---|---|---|
| Phosphatidylcholine | PCh | 39.146 ± 0.391 | 36.895 ± 1.564 | 43.485 ± 2.403 | 39.24 ± 1.757 | 41.119 ± 2.622 |
| Phosphatidylethanolamine | PE | 31.246 ± 1.909 | 33.492 ± 1.659 | 35.177 ± 2.029 | 32.676 ± 1.814 | 34.733 ± 2.91 |
| Diphosphoglycerol | DPG | 11.345 ± 0.852 | 12.227 ± 0.309 | 6.351 ± 1.08 *@ | 11.493 ± 0.928 # | 11.471 ± 1.186 # |
| Sphingomyelin | SM | 5.163 ± 1.036 | 6.385 ± 0.643 | 3.755 ± 0.861 @ | 7.904 ± 1.208 # | 2.978 ± 0.935 @$ |
| Phosphatidylinositol | PI | 3.528 ± 0.328 | 4.551 ± 1.236 | 6.56 ± 1.133 * | 2.01 ± 0.208 *# | 3.933 ± 1.007 |
| Phosphatidylserine | PS | 2.65 ± 0.486 | 3.916 ± 0.337 | 8.977 ± 1.858 *@ | 4.008 ± 0.83 # | 3.064 ± 0.601 # |
| Lysophosphatidylcholine | LPC | 0.924 ± 0.039 | 1.55 ± 0.351 | 3.667 ± 0.333 *@ | 2.343 ± 0.087 *# | 2.788 ± 0.693 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panchuk, R.; Skorokhyd, N.; Chumak, V.; Lehka, L.; Kosiakova, H.; Horid’ko, T.; Hudz, I.; Hula, N.; Riabtseva, A.; Mitina, N.; et al. Cannabimimetic N-Stearoylethanolamine as “Double-Edged Sword” in Anticancer Chemotherapy: Proapoptotic Effect on Tumor Cells and Suppression of Tumor Growth versus Its Bio-Protective Actions in Complex with Polymeric Carrier on General Toxicity of Doxorubicin In Vivo. Pharmaceutics 2023, 15, 835. https://doi.org/10.3390/pharmaceutics15030835
Panchuk R, Skorokhyd N, Chumak V, Lehka L, Kosiakova H, Horid’ko T, Hudz I, Hula N, Riabtseva A, Mitina N, et al. Cannabimimetic N-Stearoylethanolamine as “Double-Edged Sword” in Anticancer Chemotherapy: Proapoptotic Effect on Tumor Cells and Suppression of Tumor Growth versus Its Bio-Protective Actions in Complex with Polymeric Carrier on General Toxicity of Doxorubicin In Vivo. Pharmaceutics. 2023; 15(3):835. https://doi.org/10.3390/pharmaceutics15030835
Chicago/Turabian StylePanchuk, Rostyslav, Nadiya Skorokhyd, Vira Chumak, Lilya Lehka, Halyna Kosiakova, Tetyana Horid’ko, Iehor Hudz, Nadiya Hula, Anna Riabtseva, Nataliya Mitina, and et al. 2023. "Cannabimimetic N-Stearoylethanolamine as “Double-Edged Sword” in Anticancer Chemotherapy: Proapoptotic Effect on Tumor Cells and Suppression of Tumor Growth versus Its Bio-Protective Actions in Complex with Polymeric Carrier on General Toxicity of Doxorubicin In Vivo" Pharmaceutics 15, no. 3: 835. https://doi.org/10.3390/pharmaceutics15030835
APA StylePanchuk, R., Skorokhyd, N., Chumak, V., Lehka, L., Kosiakova, H., Horid’ko, T., Hudz, I., Hula, N., Riabtseva, A., Mitina, N., Zaichenko, A., Heffeter, P., Berger, W., & Stoika, R. (2023). Cannabimimetic N-Stearoylethanolamine as “Double-Edged Sword” in Anticancer Chemotherapy: Proapoptotic Effect on Tumor Cells and Suppression of Tumor Growth versus Its Bio-Protective Actions in Complex with Polymeric Carrier on General Toxicity of Doxorubicin In Vivo. Pharmaceutics, 15(3), 835. https://doi.org/10.3390/pharmaceutics15030835