


Amorphous Solid Dispersions Layered onto Pellets—An Alternative to Spray Drying?



Abstract
1. Introduction
2. Materials and Methods
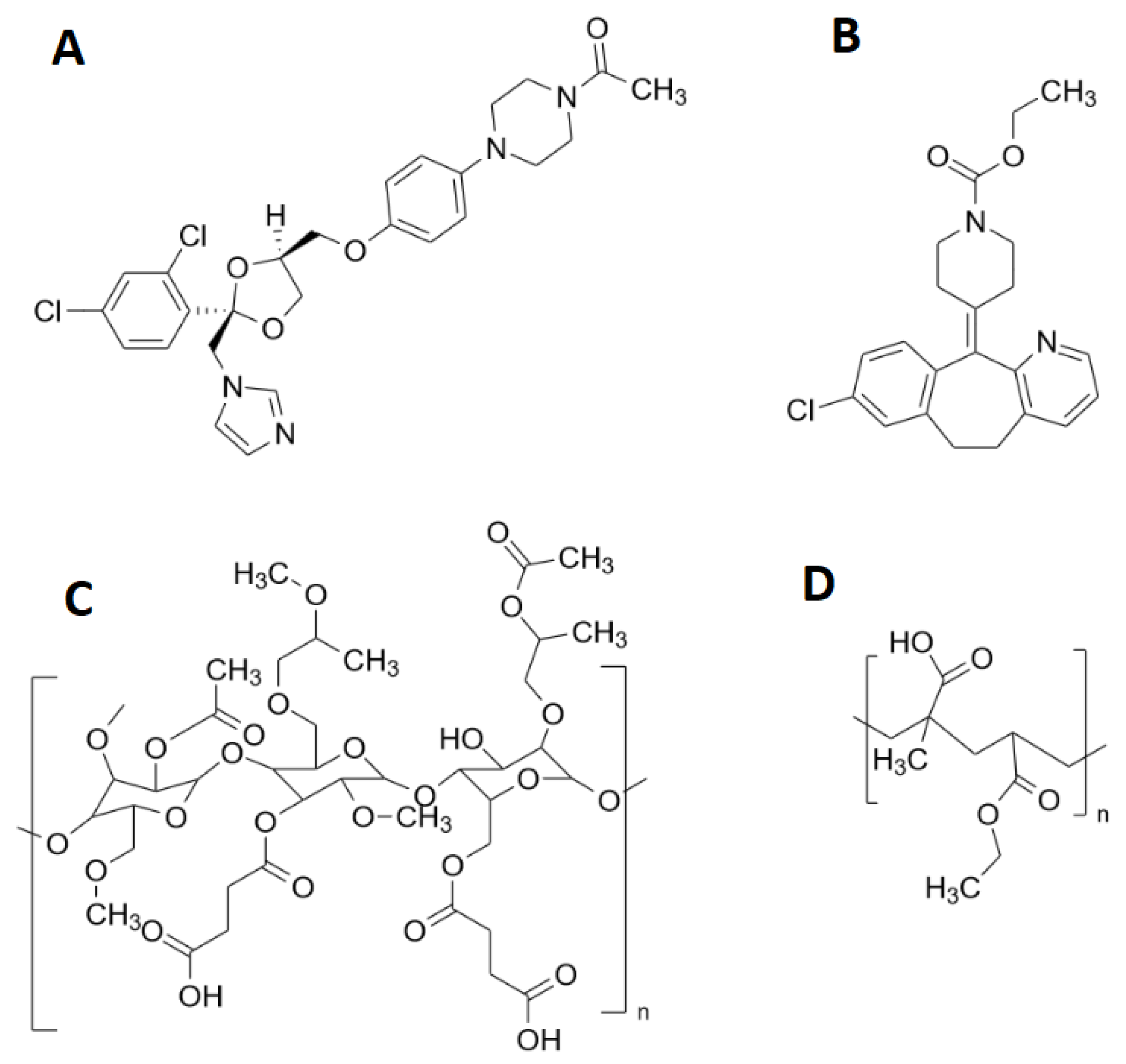
2.1. Materials
2.2. Methods
2.2.1. Spray Drying (SD)
2.2.2. Pellet Coating (PC)
2.2.3. Gas Chromatography (GC)
2.2.4. High Performance Liquid Chromatography (HPLC) Analysis
2.2.5. Dynamic Image Analysis
2.2.6. Pycnometric Density
2.2.7. Fourier-Transform Infrared Spectroscopy (FT-IR)
2.2.8. Scanning Electron Microscopy (SEM)
2.2.9. X-ray Powder Diffraction (XRPD)
2.2.10. Differential Scanning Calorimetry (DSC)
2.2.11. Determination of Solubility
2.2.12. Non-Sink Dissolution
2.2.13. Physical Stability
3. Results
3.1. Sample Preparation
3.2. Sample Characterization
3.2.1. Particle Characterization
3.2.2. Solid State Characterization
3.3. Non-Sink Dissolution Testing
3.4. Stability
4. Discussion
4.1. Process
4.2. Sample Characteristics
4.3. Solid State
4.4. Non-Sink Dissolution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Dhote, V.; Bhargava, A.; Jain, D.K.; Mishra, P.K. Amorphous solid dispersion technique for improved drug delivery: Basics to clinical applications. Drug Deliv. Transl. Res. 2015, 5, 552–565. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Chatterji, A.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Shah, N. Evaluation of solid state properties of solid dispersions prepared by hot-melt extrusion and solvent co-precipitation. Int. J. Pharm. 2008, 355, 141–149. [Google Scholar] [CrossRef]
- Potter, C.; Tian, Y.; Walker, G.; McCoy, C.; Hornsby, P.; Donnelly, C.; Jones, D.S.; Andrews, G.P. Novel Supercritical Carbon Dioxide Impregnation Technique for the Production of Amorphous Solid Drug Dispersions: A Comparison to Hot Melt Extrusion. Mol. Pharm. 2015, 12, 1377–1390. [Google Scholar] [CrossRef]
- Schönfeld, B.V.; Westedt, U.; Wagner, K.G. Compression of amorphous solid dispersions prepared by hot-melt extrusion, spray drying and vacuum drum drying. Int. J. Pharm. X 2021, 3, 100102. [Google Scholar] [CrossRef]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H. Chapter 3 Overview of Amorphous Solid Dispersion Technologies. In Amorphous Solid Dispersions: Theory and Practice; Malick, A.W., Ed.; Advances in Delivery Science and Technology; Springer: New York, NY, USA, 2014; pp. 91–122. ISBN 978-1-4939-1597-2. [Google Scholar]
- Sóti, P.L.; Bocz, K.; Pataki, H.; Eke, Z.; Farkas, A.; Verreck, G.; Kiss, É.; Fekete, P.; Vigh, T.; Wagner, I.; et al. Comparison of spray drying, electroblowing and electrospinning for preparation of Eudragit E and itraconazole solid dispersions. Int. J. Pharm. 2015, 494, 23–30. [Google Scholar] [CrossRef]
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Gilis, P.M.V.; Conde, V.F.V.D.; Vandecruys, R.P.G. Beads Having a Core Coated with an Antifungal and a Polymer. U.S. Patent US5344657A, 6 September 1994. [Google Scholar]
- Vervaet, C.; Remon, J.P. Continuous granulation in the pharmaceutical industry. Chem. Eng. Sci. 2005, 60, 3949–3957. [Google Scholar] [CrossRef]
- Crowley, M.M.; Zhang, F.; Repka, M.A.; Thumma, S.; Upadhye, S.B.; Kumar Battu, S.; McGinity, J.W.; Martin, C. Pharmaceutical Applications of Hot-Melt Extrusion: Part I. Drug Dev. Ind. Pharm. 2007, 33, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Sebhatu, T.; Alderborn, G. Relationships between the effective interparticulate contact area and the tensile strength of tablets of amorphous and crystalline lactose of varying particle size. Eur. J. Pharm. Sci. 1999, 8, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ghebre-Sellassie, I. Pellets: A General Overview. In Pharmaceutical Pelletization Technology; Drugs and the Pharmaceutical Sciences; CRC Press Taylor & Francis Group: New York, NY, USA, 2010; Volume 37, p. 35. ISBN 978-0-8247-8085-2. [Google Scholar]
- Ghanam, D.; Kleinebudde, P. Suitability of a flat die press for the manufacture of pharmaceutical pellets by extrusion/spheronization. Drug Dev. Ind. Pharm. 2011, 37, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.P.; Patel, J.K.; Patel, R.R.; Patel, M.P. Pellets: A General Overview. Int. J. Curr. Res. Rev. 2010, 2, 21–31. [Google Scholar]
- Nagane, K.; Kimura, S.; Ukai, K.; Takahashi, C.; Ogawa, N.; Yamamoto, H. Application of spherical silicate to prepare solid dispersion dosage forms with aqueous polymers. Int. J. Pharm. 2015, 493, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.-O.; Su, H.-L.; Tsai, T.; Sheu, M.-T. The preparation and characterization of solid dispersions on pellets using a fluidized-bed system. Int. J. Pharm. 1996, 139, 223–229. [Google Scholar] [CrossRef]
- Sun, N.; Wei, X.; Wu, B.; Chen, J.; Lu, Y.; Wu, W. Enhanced dissolution of silymarin/polyvinylpyrrolidone solid dispersion pellets prepared by a one-step fluid-bed coating technique. Powder Technol. 2008, 182, 72–80. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, N.; Wu, B.; Lu, Y.; Guan, T.; Wu, W. Physical characterization of lansoprazole/PVP solid dispersion prepared by fluid-bed coating technique. Powder Technol. 2008, 182, 480–485. [Google Scholar] [CrossRef]
- Dereymaker, A.; Scurr, D.J.; Steer, E.D.; Roberts, C.J.; Van den Mooter, G. Controlling the Release of Indomethacin from Glass Solutions Layered with a Rate Controlling Membrane Using Fluid-Bed Processing. Part 1: Surface and Cross-Sectional Chemical Analysis. Mol. Pharm. 2017, 14, 959–973. [Google Scholar] [CrossRef]
- Miller, D.A.; DiNunzio, J.C.; Yang, W.; McGinity, J.W.; Williams, R.O. Enhanced In Vivo Absorption of Itraconazole via Stabilization of Supersaturation Following Acidic-to-Neutral pH Transition. Drug Dev. Ind. Pharm. 2008, 34, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Monschke, M.; Wagner, K.G. Amorphous solid dispersions of weak bases with pH-dependent soluble polymers to overcome limited bioavailability due to gastric pH variability—An in-vitro approach. Int. J. Pharm. 2019, 564, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Zecevic, D.E.; Meier, R.; Daniels, R.; Wagner, K.-G. Site specific solubility improvement using solid dispersions of HPMC-AS/HPC SSL—Mixtures. Eur. J. Pharm. Biopharm. 2014, 87, 264–270. [Google Scholar] [CrossRef] [PubMed]
- WHO. Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products; WHO: Geneva, Switzerland, 2018; Volume 52, pp. 309–351. [Google Scholar]
- Janssens, S.; den Mooter, G.V. Review: Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- EMA. ICH Guideline Q3C (R6) on Impurities: Guidline for Residual Solvents; EMA: Amsterdam, The Netherlands, 2017; p. 39.
- Wulff, R.; Leopold, C.S. Coatings of Eudragit® RL and L-55 Blends: Investigations on the Drug Release Mechanism. AAPS PharmSciTech 2016, 17, 493–503. [Google Scholar] [CrossRef]
- Vinjamuri, B.P.; Haware, R.V.; Stagner, W.C. Inhalable Ipratropium Bromide Particle Engineering with Multicriteria Optimization. AAPS PharmSciTech 2017, 18, 1925–1935. [Google Scholar] [CrossRef]
- LeClair, D.A.; Cranston, E.D.; Xing, Z.; Thompson, M.R. Optimization of Spray Drying Conditions for Yield, Particle Size and Biological Activity of Thermally Stable Viral Vectors. Pharm. Res. 2016, 33, 2763–2776. [Google Scholar] [CrossRef]
- Año, G.; Esquisabel, A.; Pastor, M.; Talavera, A.; Cedré, B.; Fernández, S.; Sifontes, S.; Aranguren, Y.; Falero, G.; García, L.; et al. A new oral vaccine candidate based on the microencapsulation by spray-drying of inactivated Vibrio cholerae. Vaccine 2011, 29, 5758–5764. [Google Scholar] [CrossRef]
- Ziaee, A.; Albadarin, A.B.; Padrela, L.; Femmer, T.; O’Reilly, E.; Walker, G. Spray drying of pharmaceuticals and biopharmaceuticals: Critical parameters and experimental process optimization approaches. Eur. J. Pharm. Sci. 2019, 127, 300–318. [Google Scholar] [CrossRef]
- Couto, R.O.; Martins, F.S.; Chaul, L.T.; Conceição, E.C.; Freitas, L.A.P.; Bara, M.T.F.; Paula, J.R. Spray Drying of Eugenia Dysenterica Extract: Effects of in-Process Parameters on Product Quality. Rev. Bras. Farmacogn. 2013, 23, 115–123. [Google Scholar] [CrossRef]
- Zecevic, D.E.; Wagner, K.G. Rational Development of Solid Dispersions via Hot-Melt Extrusion Using Screening, Material Characterization, and Numeric Simulation Tools. J. Pharm. Sci. 2013, 102, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Shao, Z.J. Process Development, Optimization, and Scale-Up. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2017; pp. 777–792. ISBN 978-0-12-802447-8. [Google Scholar]
- Zhang, X.; Rao, Q.; Qiu, Z.; Lin, Y.; Zhang, L.; Hu, Q.; Chen, T.; Ma, Z.; Gao, H.; Luo, D.; et al. Using Acetone/Water Binary Solvent to Enhance the Stability and Bioavailability of Spray Dried Enzalutamide/HPMC-AS Solid Dispersions. J. Pharm. Sci. 2021, 110, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Alhalaweh, A.; Alzghoul, A.; Bergström, C.A.S. Molecular Drivers of Crystallization Kinetics for Drugs in Supersaturated Aqueous Solutions. J. Pharm. Sci. 2019, 108, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Monschke, M.; Kayser, K.; Wagner, K.G. Influence of Particle Size and Drug Load on Amorphous Solid Dispersions Containing pH-Dependent Soluble Polymers and the Weak Base Ketoconazole. AAPS PharmSciTech 2021, 22, 44. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.A.; Whitney, W.R. The Rate of Solution of Solid Substances in Their Own Solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Han, Y.R.; Ma, Y.; Lee, P.I. Impact of phase separation morphology on release mechanism of amorphous solid dispersions. Eur. J. Pharm. Sci. 2019, 136, 104955. [Google Scholar] [CrossRef] [PubMed]
- Saboo, S.; Mugheirbi, N.A.; Zemlyanov, D.Y.; Kestur, U.S.; Taylor, L.S. Congruent release of drug and polymer: A “sweet spot” in the dissolution of amorphous solid dispersions. J. Control. Release 2019, 298, 68–82. [Google Scholar] [CrossRef]
- Sarode, A.L.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H. Hot melt extrusion (HME) for amorphous solid dispersions: Predictive tools for processing and impact of drug–polymer interactions on supersaturation. Eur. J. Pharm. Sci. 2013, 48, 371–384. [Google Scholar] [CrossRef]
- Liu, X.; Ma, X.; Kun, E.; Guo, X.; Yu, Z.; Zhang, F. Influence of lidocaine forms (salt vs. freebase) on properties of drug–eudragit® L100-55 extrudates prepared by reactive melt extrusion. Int. J. Pharm. 2018, 547, 291–302. [Google Scholar] [CrossRef]
- Borodkin, S.; Yunker, M.H. Interaction of Amine Drugs with a Polycarboxylic Acid Ion-Exchange Resin. J. Pharm. Sci. 1970, 59, 481–486. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pellet Properties | |
|---|---|
| d50 (xc min) [µm] | 1123.44 (±7.36) |
| SPAN | 0.166 (±0.002) |
| b/l | 0.893 (±0.000) |
| SPHT | 0.956 (±0.001) |
| Particle density [g/cm3] | 1.452 (±0.016) |
| Sm [cm2/g] | 36.41 (±0.29) |
| Sample ID | API | Polymer | Comp. API/Polymer | Production Technique | Batch Size [g] (Solids) | Drug-Load Achieved [% (w/w)] | Assay [% (w/w)] |
|---|---|---|---|---|---|---|---|
| KCZ_HPMC-AS_SD | KCZ | HPMC-AS LG | 20/80 | SD | 100 | 19.12 ± 0.37 | 96.97 ± 3.11 |
| KCZ_HPMC-AS_PC | KCZ | HPMC-AS LG | 20/80 | PC | 50 | 9.32 ± 0.50 | 93.18 ± 5.52 |
| KCZ_EL100-55_SD | KCZ | Eudragit L100-55 | 20/80 | SD | 100 | 19.13 ± 0.11 | 95.72 ± 3.56 |
| KCZ_EL100-55_PC | KCZ | Eudragit L100-55 | 20/80 | PC | 50 | 9.34 ± 0.55 | 93.44 ± 5.04 |
| LRD_HPMC-AS_SD | LRD | HPMC-AS LG | 20/80 | SD | 100 | 19.06 ± 0.73 | 95.30 ± 3.63 |
| LRD_HPMC-AS_PC | LRD | HPMC-AS LG | 20/80 | PC | 50 | 9.56 ± 0.28 | 95.56 ± 2.81 |
| LRD_EL100-55_SD | LRD | Eudragit L100-55 | 20/80 | SD | 100 | 19.42 ± 0.72 | 97.09 ± 3.88 |
| LRD_EL100-55_PC | LRD | Eudragit L100-55 | 20/80 | PC | 50 | 9.01 ± 0.45 | 92.43 ± 4.36 |
| Preparation Technique | Average Process Time [h/100 g] | Average Process Yield [% (w/w)] |
|---|---|---|
| Spray drying | 4.1 ± 0.2 | 54.45 ± 2.41 |
| Pellet coating | 9.2 ± 0.2 | 99.67 ± 1.16 |
| Sample ID | Sieve Fraction [µm] | d50 [µm] | SPAN | l/b-Ratio | SPHT | PD [g/cm3] |
Sm [cm2/g] | Residual Solvent [ppm] | Tg [°C] |
|---|---|---|---|---|---|---|---|---|---|
| KCZ_HPMC-AS_SD | 1000–2000 | 1491.1 ± 2.4 | 0.766 ± 0.003 | 0.644 ± 0.001 | 0.743 ± 0.004 | 1.287 ± 0.022 | 64.46 ± 0.83 | 3100 ± 790 | 91.92 ± 1.20 * |
| 710–1000 | 870.9 ± 0.9 | 0.618 ± 0.005 | 0.643 ± 0.001 | 0.744 ± 0.004 | 93.19 ± 1.05 | ||||
| 500–710 | 605.4 ± 1.2 | 1.019 ± 0.010 | 0.641 ± 0.001 | 0.721 ± 0.005 | 157.68 ± 1.48 | ||||
| KCZ_HPMC-AS_PC | - | 1422.9 ± 1.5 | 0.193 ± 0.003 | 0.909 ± 0.000 | 0.877 ± 0.000 | 1.357 ± 0.014 | 42.27 ± 0.35 | 2680 ± 220 | 91.86 ± 0.48 * |
| KCZ_EL100-55_SD | 1000–2000 | 1515.6 ± 1.0 | 0.639 ± 0.001 | 0.660 ± 0.000 | 0.774 ± 0.003 | 1.249 ± 0.031 | 50.02 ± 0.49 | 3220 ± 880 | 110.13 ± 0.99 * |
| 710–1000 | 882.4 ± 0.9 | 0.487 ± 0.006 | 0.665 ± 0.001 | 0.781 ± 0.003 | 82.73 ± 0.93 | ||||
| 500–710 | 626.4 ± 1.1 | 0.488 ± 0.002 | 0.663 ± 0.001 | 0.782 ± 0.003 | 99.55 ± 1.54 | ||||
| KCZ_EL100-55_PC | - | 1506.8 ± 1.3 | 0.153 ± 0.002 | 0.912 ± 0.000 | 0.945 ± 0.000 | 1.312 ± 0.037 | 40.74 ± 0.86 | 3220 ± 400 | 109.72 ± 0.82 * |
| LRD_HPMC-AS_SD | 1000–2000 | 1508.8 ± 2.6 | 0.820 ± 0.008 | 0.631 ± 0.001 | 0.741 ± 0.006 | 1.267 ± 0.047 | 40.73 ± 0.38 | 3520 ± 690 | 89.86 ± 2.47 * |
| 710–1000 | 905.2 ± 3.2 | 0.612 ± 0.005 | 0.631 ± 0.000 | 0.754 ± 0.005 | 64.07 ± 1.84 | ||||
| 500–710 | 641.2 ± 1.5 | 0.620 ± 0.010 | 0.627 ± 0.001 | 0.755 ± 0.005 | 94.84 ± 1.43 | ||||
| LRD_HPMC-AS_PC | - | 1552.6 ± 1.2 | 0.249 ± 0.005 | 0.898 ± 0.000 | 0.867 ± 0.000 | 1.332 ± 0.016 | 40.23 ± 0.94 | 3240 ± 660 | 91.78 ± 2.33 * |
| LRD_EL100-55_SD | 1000–2000 | 1510.1 ± 1.8 | 0.635 ± 0.003 | 0.673 ± 0.002 | 0.772 ± 0.002 | 1.228 ± 0.037 | 44.95 ± 0.68 | 3210 ± 330 | 113.00 ± 0.79 * |
| 710–1000 | 884.4 ± 1.6 | 0.460 ± 0.004 | 0.671 ± 0.000 | 0.772 ± 0.003 | 74.74 ± 0.43 | ||||
| 500–710 | 610.0 ± 2.3 | 0.541 ± 0.001 | 0.673 ± 0.001 | 0.781 ± 0.004 | 139.60 ± 1.59 | ||||
| LRD_EL100-55_PC | - | 1562.5 ± 1.6 | 0.188 ± 0.007 | 0.898 ± 0.000 | 0.942 ± 0.000 | 1.307 ± 0.034 | 38.80 ± 0.34 | 3380 ± 290 | 112.08 ± 3.59 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuwirth, M.; Kappes, S.K.; Hartig, M.U.; Wagner, K.G. Amorphous Solid Dispersions Layered onto Pellets—An Alternative to Spray Drying? Pharmaceutics 2023, 15, 764. https://doi.org/10.3390/pharmaceutics15030764
Neuwirth M, Kappes SK, Hartig MU, Wagner KG. Amorphous Solid Dispersions Layered onto Pellets—An Alternative to Spray Drying? Pharmaceutics. 2023; 15(3):764. https://doi.org/10.3390/pharmaceutics15030764
Chicago/Turabian StyleNeuwirth, Marius, Sebastian K. Kappes, Michael U. Hartig, and Karl G. Wagner. 2023. "Amorphous Solid Dispersions Layered onto Pellets—An Alternative to Spray Drying?" Pharmaceutics 15, no. 3: 764. https://doi.org/10.3390/pharmaceutics15030764
APA StyleNeuwirth, M., Kappes, S. K., Hartig, M. U., & Wagner, K. G. (2023). Amorphous Solid Dispersions Layered onto Pellets—An Alternative to Spray Drying? Pharmaceutics, 15(3), 764. https://doi.org/10.3390/pharmaceutics15030764