
Physiologically Based Pharmacokinetic Modeling in Neonates: Current Status and Future Perspectives


Abstract
:1. Introduction
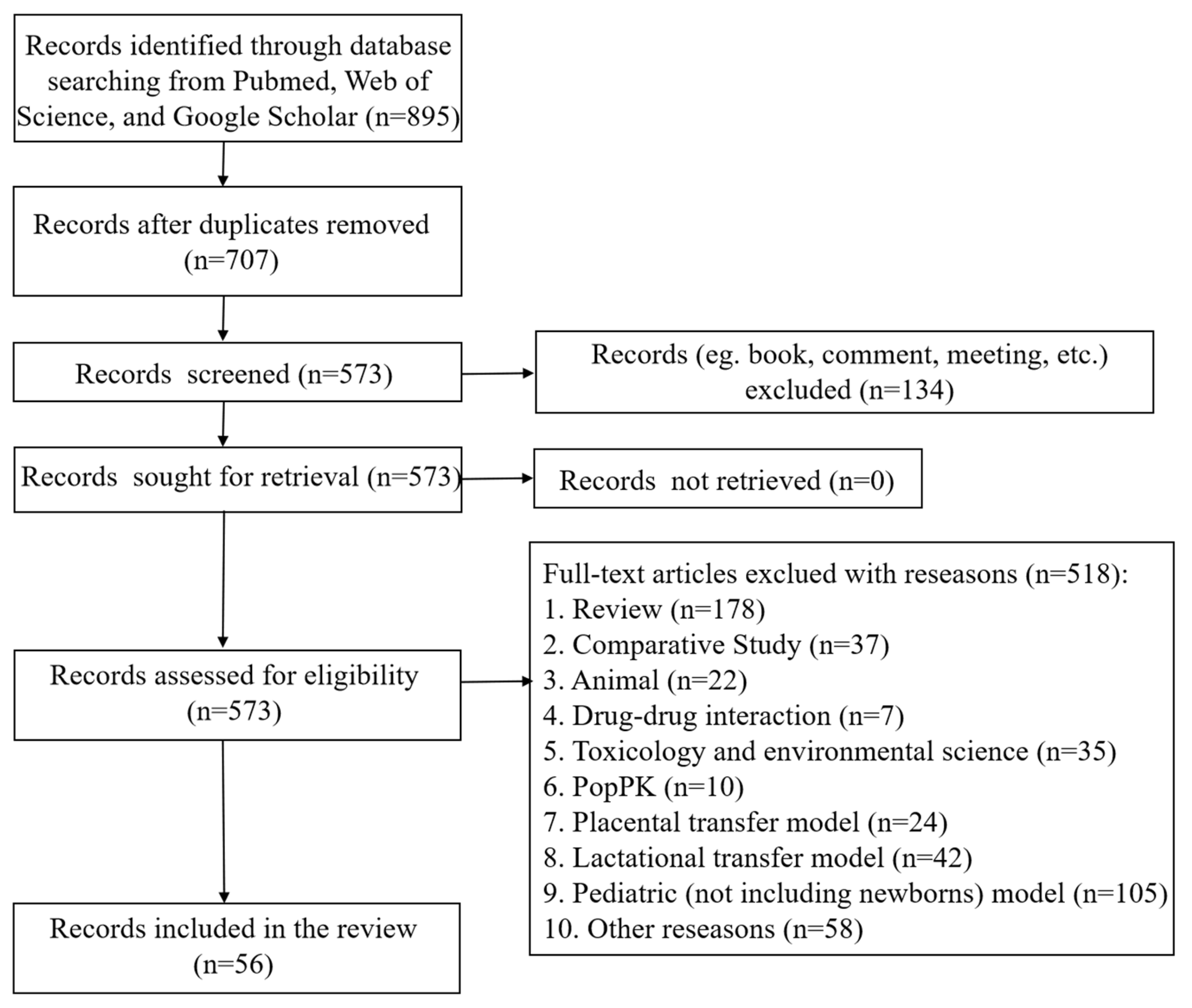
2. Methods
2.1. Literature Search
2.2. Inclusion and Exclusion Criteria
2.3. Article Selection
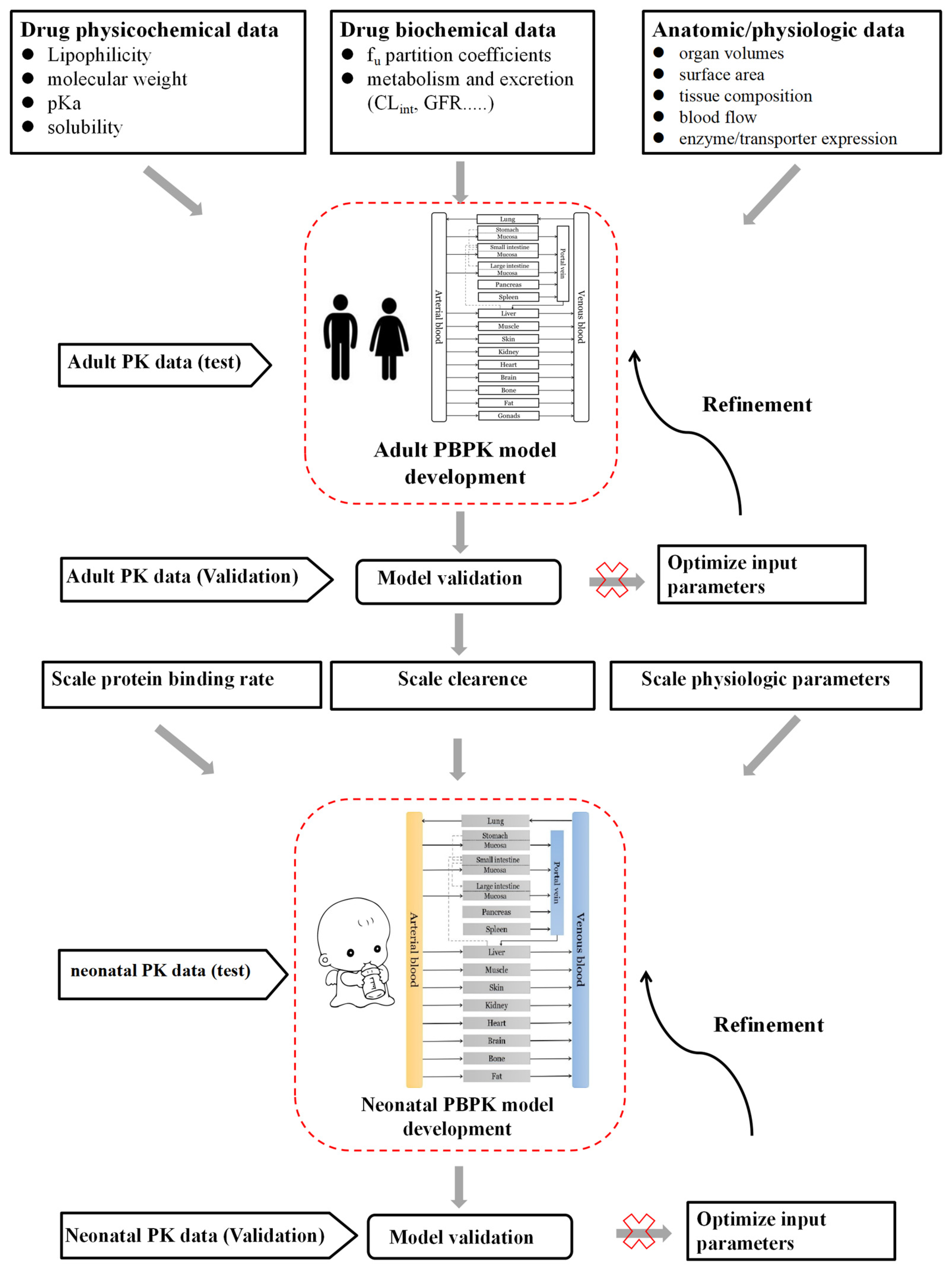
2.4. Neonatal PBPK Modeling Mechanism and Workflow
3. Results
3.1. Neonatal Physiologic Changes Affecting the Drug Disposition Process
3.1.1. Drug Absorption
3.1.2. Drug Distribution
3.1.3. Drug Metabolism

{kind=link}
{kind=link}
{kind=link}
| Enzymes | Measure Methods | Age-Related Changes |
|---|---|---|
| CYP1A1 | Western blot | 1. Detectable in fetal liver during 11–20 weeks, undetectable in adults [103]. |
| Gene expression | 1. Detectable in human embryonic livers (GW 6–12), and decreases with increasing age [104,105]. | |
| CYP1A2 | Western blot | 1. No expression in fetal and neonatal livers, and its levels increased in infants aged 1–3 months to reach 50% of the adult value by 1 year of age [106]. |
| Gene expression | 1. Expression was only found in adult livers [104]. | |
| CYP1B1 | Gene expression | 1. Detectable in fetal liver (GW 12–19) [107]. 2. Undetectable in either fetal or adult livers [108]. |
| CYP2A6 | Immunohistochemistry | 1. Expression approaches adult levels by 1 year of age [109]. |
| Gene expression | 1. Undetectable in fetal liver, increases with age [110]. | |
| CYP2B6 | Immunohistochemistry | 1. Approximately 10% of adult levels within the 1st year of life [109]. |
| Gene expression | 1. Undetectable in fetal liver at GW 11–24 [110]. | |
| CYP2C8 | Western blot | 1. Undetected in fetal livers and matures in the first few weeks after birth, not related to GA [111]. |
| Gene expression | 1. Low expression in fetuses, approximately 10% of the adult values [110,111]. | |
| CYP2C9 | Quantitative proteomics | 1. Increases linearly over age and reaches adult level in the pediatric period [92]. |
| Gene expression | 1. Undetectable in fetal samples, comparable in pediatric population and adults [92]. | |
| CYP2C19 | Quantitative proteomics | 1. Expression peaked during the pediatric period (>2-fold higher compared to adult) [92]. |
| Western blot | 1. Expression in children was 140% of that in adult liver [112]. | |
| Gene expression | 1. Undetectable in fetuses, higher expression in the pediatric population than in adults [92]. | |
| CYP2D6 | Western blot | 1. Undetectable in fetal livers (GW 11–13) [113]. 2. Expression in fetal liver (>GW 30) was comparable to that of newborns aged 1–7 days; increased significantly after birth and reached 50 to 75% of adult level during the neonatal period [103]. |
| Gene expression | 1. Expression peaked in newborns and declined [113]. | |
| CYP2E1 | Western blot | 1. CYP2E1 was detectable in the liver as early as the second trimester; its expression in neonates was lower than that of infants 31 to 90 days less than that of older infants, children, and young adults [88]. 2. Expression increased gradually, reaching 30 to 40% of adult levels by one year and approaching adult levels by 10 years [114]. 3. Expression in fetal liver (GW 16) was about 10 to 30% of adult levels, and remained stable for up to 24 weeks [115]. |
| Gene expression | 1. Low expression in fetal livers and increased after birth [116]. | |
| CYP2J2 | Western blot | 1. Expression in fetal liver (GW 13–18) was comparable to the adult level [117]. |
| CYP3A4 | Quantitative proteomics | 1. Increased after birth and reached adult levels around 1 year of age [92]. |
| Western blot | 1. Expression in fetal livers was low, and increased after birth to reach 30%–40% of adult levels [85]. 2. Expression increased with age [118]. 3. Expression in children was 60% of adult levels [112]. | |
| Gene expression | 1. Low expression in fetuses, increased during childhood, and then became comparable with adults in pediatric period [92]. 2. Expression increased rapidly after birth and reached a plateau at the first week of age [85]. 3. Only detectable after birth and was highly variable (10-fold) among adults [119]. 4. Expression exhibited a 29–fold increase after a postnatal surge [118]. | |
| CYP3A5 | Quantitative proteomics | 1. Comparable from fetuses to adults [92]. |
| Gene expression | 1. Expression remained stable in fetuses, pediatrics, and adults [92]. 2. Detectable in all the fetal and 23% of adult samples [119]. | |
| CYP3A7 | Quantitative proteomics | 1. Very high expression in fetal samples, then decreased in the pediatrics and adults [92]. |
| Western blot | 1. High expression in the fetal livers; its activity peaked in the first week after birth, then decreased [85]. | |
| Gene expression | 1. High expression in fetal livers and decreased with age to be undetectable in adults [92]. 2. Detectable in fetal livers at GA 50–60 days, continued to be expressed at a significant levels during the perinatal period, then decreased after first week of age until undetectable by 1 year old [85]. 3. Expression in adults was proven [119]. | |
| CYP4A1 | Western blot | 1. Expression in fetal livers reached 40% of the adult levels and continued to increase during the first week after birth [120]. |
| Carboxylesterases | Western blot | 1. No significant difference in expression of carboxylesterases between infants (2–24 months) and adults (20–36 years) [90]. 2. Expression was age-dependent: adult > children > fetuses [91]. |
| Gene expression | 1. The liver expressed two major carboxylesterases: HCE1 and HCE2. Expression was age-dependent: adult > children > fetuses [91]. | |
| FMO1 | Quantitative proteomics | 1. High expression in fetuses, and undetectable in pediatrics and adults [92]. |
| Western blot | 1. Highest expression in the embryo (GW 8–15) and suppressed within 3 days after birth [121]. | |
| Gene expression | 1. Higher in fetuses, decreased with age [92]. | |
| FMO3 | Quantitative proteomics | 1. A linear increase from the fetal period into adulthood [92]. |
| Western blot | 1. Low expression in embryo, undetectable in the fetus, increased to be detectable by 1–2 years of age, continued to increase up to 18 years of age [121]. 2. Higher expression in children 2–8 years of age than in adults [112]. | |
| Gene expression | 1. Undetectable in fetuses, increased with age [92]. | |
| ADH1A | Western blot | 1. High expression in the fetus, particularly in the first trimester, decreased in the last trimester, and finally undetected in neonates and adults [122]. |
| ADH1B | Western blot | 1. Detectable in the fetal liver at 17th GW, and dominated by week 36 [122]. |
| ADH1C | Western blot | 1. Detectable in the fetal liver at 19th GW [122]. |
| ADH2 | Gene expression | 1. Detected only in fetal livers at concentrations equivalent to adults [123]. |
| ADH3 | Gene expression | 1. Widely distributed in fetal tissues at concentrations equivalent to adults [123]. |
| ADH5 | Gene expression | 1. Detected only in fetal livers at concentrations equivalent to adults [123]. |
| EPHX1 | Immunocytochemistry | 1. Expression in fetal livers was approximately 25–50% of adult levels, and its activity was detected in fetal livers at GW 6 and increased linearly with age [124,125]. |
| PON1 | Gene expression | 1. Detectable in fetal livers [126]. |
| PON2 | Gene expression | 1. Detectable in fetal livers [126]. |
| AOX | Western blot | 1. Detectable in infants > 4 months old; Undetectable in infants of 13 days old and 2 months old [127]. |
| UGT1A1 | Quantitative proteomics | 1. Neonatal abundances of UGT1A1 were 12.2% of adult levels whereas infant abundances (% of adult abundance) were 43; UGT1A1 is the most abundant of the UGT1As in neonates [95]. |
| Gene expression | 1. Undetected in the fetal liver (GW 20) and stayed stable after 6 months of age [128]. | |
| UGT1A3 | Gene expression | 1. Undetected in the fetal liver (GW 20) and stayed stable after 6 months of age [128]. |
| UGT1A4 | Quantitative proteomics | 1. Neonatal abundances of UGT1A4 were 1.8% of adult levels whereas infant abundances (% of adult abundance) were 16 [95]. |
| Gene expression | 1. Undetected in the fetal liver (GW 20) and stayed stable after 6 months of age [128]. | |
| UGT1A6 | Quantitative proteomics | 1. Neonatal abundances of UGT1A6 were 2.9% of adult levels whereas infant abundances (% of adult abundance) were 15 [95]. |
| Gene expression | 1. Undetected in the fetal liver (GW 20) and stayed stable after 6 months of age [128]. | |
| UGT1A9 | Quantitative proteomics | 1. Neonatal abundances of UGT1A9 were 3.0% of adult levels whereas infant abundances (% of adult abundance) were 24 [95]. |
| Gene expression | 1. Undetected in the fetal liver (GW 20), increased with age from 6 to 24 months, reaching 70% of the adult levels [128]. | |
| UGT2B4 | Gene expression | 1. Undetectable in the fetal liver (GW 20), increased progressively [128]. |
| UGT2B7 | Quantitative proteomics | 1. Neonatal abundances of UGT2B7 were 13.0% of adult levels whereas infant abundances (% of adult abundance) were 41 [95]. |
| Western blot | 1. Low protein levels and activity at <1 year of age, increased progressively with age, but still less than adult levels at 17 years of age [129]. | |
| Gene expression | 1. Undetectable in the fetal liver (GW 20) and reached adult levels at 6 months of age [128]. | |
| UGT2B15 | Quantitative proteomics | 1. Neonatal abundances of UGT2B15 were 38.6% of adult levels whereas infant abundances (% of adult abundance) were 60 [95]. |
| UGT2B17 | Quantitative proteomics | 1. Undetectable in children under 9 years, and increased by about 10-fold to reach adult levels during pubertal development [130]. |
| SULT1A1 | Western blot | 1. Detectable in fetuses at GW 10 and remained stable in fetal and postnatal periods, then increased [99,131,132]. |
| SULT1A3 | Western blot | 1. High expression in early fetal stage and decreased substantially in late fetal stage, then reached low levels in adults [132]. |
| Gene expression | 1. High expression in early fetal stage and decreased substantially in late fetal stage, then reached low levels in adults [132]. | |
| SULT1C1 | Gene expression | 1. Low expression in fetal livers and undetectable in adult livers [133]. |
| SULT1E1 | Western blot | 1. Expression peaked in the earliest gestation period [99]. 2. Higher expression in fetal livers than in adults [132]. |
| Gene expression | 1. Detectable in fetuses at GW 10–14, slightly higher than in adults [132]. | |
| SULT2A1 | Western blot | 1. Low expression in fetuses at GW 25, increased to approach adult levels in neonates [99]. |
| GSTA1/2 | Starch gel electrophoresis | 1. Undetectable in fetal livers before GW 30, and steadily increased to reach adult levels by PNA 1–2 years [134]. |
| Western blot | 1. Detectable at GW 8 and rapidly increased at GW 13 [135]. | |
| GSTM | Immunohistochemistry | 1. Detectable in fetal livers (GW 10–30) and then increased rapidly to adult levels by 42 weeks after birth [136]. 2. GSTM levels remained constant over pre- and postnatal period [137]. |
| Western blot | 1. Detectable at GW 8 and slightly decreased at GW 13 [135]. | |
| GSTP1 | Immunohistochemistry | 1. Expression peaked in early fetal stages at GW 10–22, then decreased in the second and third trimesters, remained detectable in neonates [136]. |
| Western blot | 1. Detectable in embryo livers at GW 8 and slightly increased at GW 13 [135]. | |
| GSTZ1 | Western blot | 1. Undetectable in fetal livers, increased with age until 7 years of age, remained stable between 7 and 74 years of age [138]. |
3.1.4. Drug Excretion
3.1.5. Transporters
3.2. Neonatal Pathological Changes Affecting the Drug Disposition
3.3. Literature Search Results
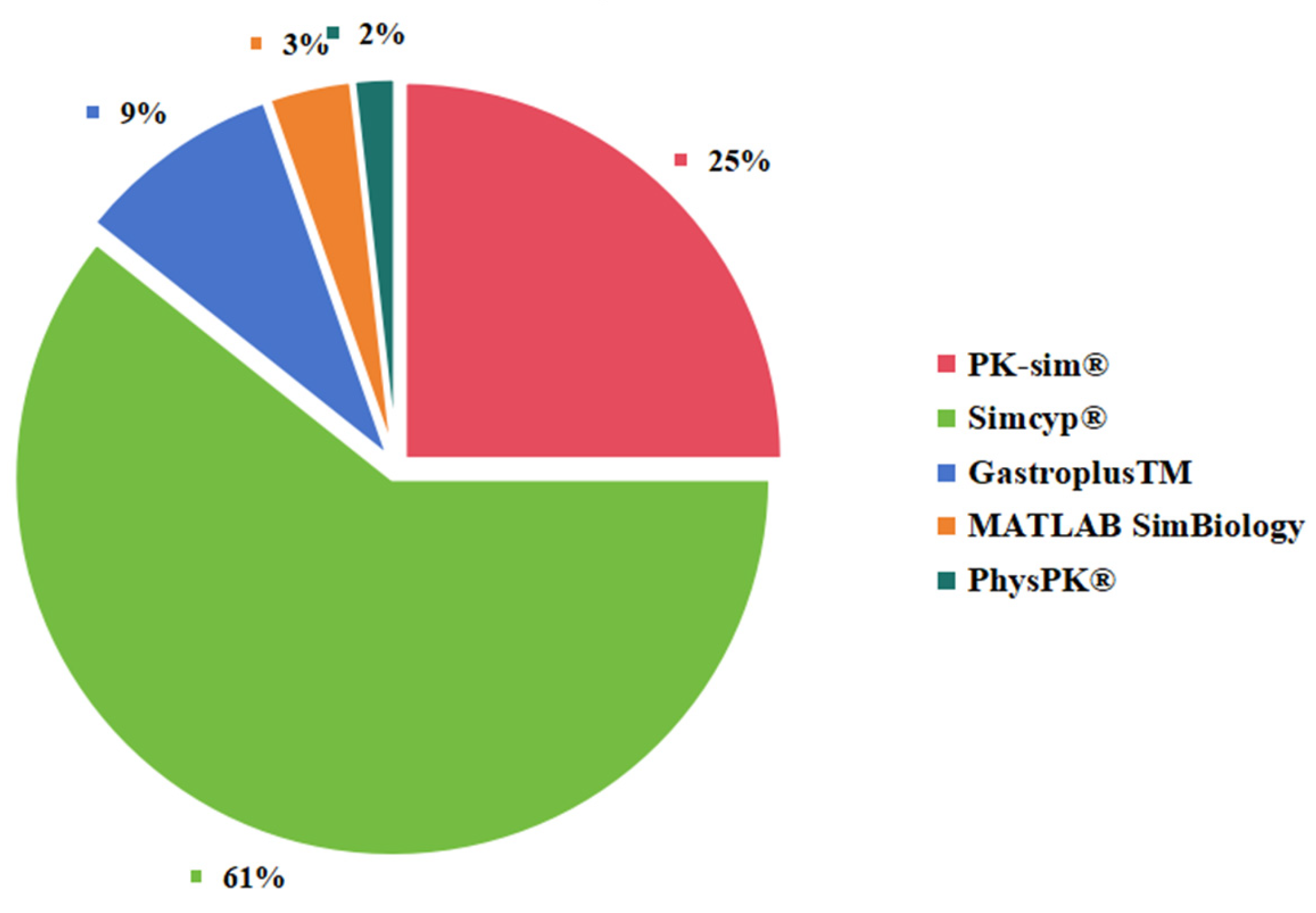
3.4. Neonatal PBPK Modeling Platform
3.5. Application of Neonatal-PBPK Modeling for Dose Optimization/Regimens/Selection
3.5.1. Antiretroviral Drugs
3.5.2. Antibiotics
3.5.3. Cardiovascular Drugs
3.5.4. Antiepileptic Drugs
3.5.5. Other Drugs
3.6. How Pediatricians Can Benefit from PBPK Modeling and Simulation
4. Conclusions/Future Directions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Laughon, M.M.; Avant, D.; Tripathi, N.; Hornik, C.P.; Cohen-Wolkowiez, M.; Clark, R.H.; Smith, P.B.; Rodriguez, W. Drug labeling and exposure in neonates. JAMA Pediatr. 2014, 168, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Dun, J.; Chen, X.; Xiang, B.; Dang, Y.; Cao, D. Predicting the correct dose in children: Role of computational Pediatric Physiological-based pharmacokinetics modeling tools. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Knibbe, C.; Danhof, M.; Della Pasqua, O. What is the right dose for children? Br. J. Clin. Pharmacol. 2010, 70, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental pharmacology--drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Kaneria, N.S.; Tuleu, C.; Ernest, T. Opportunities for enteral drug delivery for neonates, infants, and toddlers: A critical exploration. Expert Opin. Drug Deliv. 2022, 19, 475–519. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.; Annaert, P.; Allegaert, K. Drug disposition and clinical practice in neonates: Cross talk between developmental physiology and pharmacology. Int. J. Pharm. 2013, 452, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; Abbasi, M.Y.; Michelet, R.; Olafuyi, O. The Impact of Low Cardiac Output on Propofol Pharmacokinetics across Age Groups-An Investigation Using Physiologically Based Pharmacokinetic Modelling. Pharmaceutics 2022, 14, 1957. [Google Scholar] [CrossRef]
- Johnson, T.N.; Bonner, J.J.; Tucker, G.T.; Turner, D.B.; Jamei, M. Development and applications of a physiologically-based model of paediatric oral drug absorption. Eur. J. Pharm. Sci. 2018, 115, 57–67. [Google Scholar] [CrossRef]
- Kohlmann, P.; Stillhart, C.; Kuentz, M.; Parrott, N. Investigating Oral Absorption of Carbamazepine in Pediatric Populations. AAPS J. 2017, 19, 1864–1877. [Google Scholar] [CrossRef]
- Lu, H.; Rosenbaum, S. Developmental pharmacokinetics in pediatric populations. J. Pediatr. Pharmacol. Ther. 2014, 19, 262–276. [Google Scholar] [CrossRef]
- Badee, J.; Qiu, N.; Parrott, N.; Collier, A.C.; Schmidt, S.; Fowler, S. Optimization of Experimental Conditions of Automated Glucuronidation Assays in Human Liver Microsomes Using a Cocktail Approach and Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry. Drug Metab. Dispos. 2019, 47, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Badee, J.; Fowler, S.; de Wildt, S.N.; Collier, A.C.; Schmidt, S.; Parrott, N. The Ontogeny of UDP-glucuronosyltransferase Enzymes, Recommendations for Future Profiling Studies and Application Through Physiologically Based Pharmacokinetic Modelling. Clin. Pharmacokinet. 2019, 58, 189–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jiang, K.; Wei, X.; Li, Y.; Wang, T.; Song, Y. Physiologically Based Pharmacokinetic Models Are Effective Support for Pediatric Drug Development. AAPS PharmSciTech 2021, 22, 208. [Google Scholar] [CrossRef] [PubMed]
- Van den Anker, J.; Reed, M.D.; Allegaert, K.; Kearns, G.L. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J. Clin. Pharmacol. 2018, 58 (Suppl. 10), S10–S25. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Prediction of Drug Clearance in Premature and Mature Neonates, Infants, and Children ≤ 2 Years of Age: A Comparison of the Predictive Performance of 4 Allometric Models. J. Clin. Pharmacol. 2016, 56, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Prediction of drug clearance in children: A review of different methodologies. Expert Opin. Drug Metab. Toxicol. 2015, 11, 573–587. [Google Scholar] [CrossRef]
- Mahmood, I.; Staschen, C.M.; Goteti, K. Prediction of drug clearance in children: An evaluation of the predictive performance of several models. AAPS J. 2014, 16, 1334–1343. [Google Scholar] [CrossRef]
- Mahmood, I. Dosing in children: A critical review of the pharmacokinetic allometric scaling and modelling approaches in paediatric drug development and clinical settings. Clin. Pharmacokinet. 2014, 53, 327–346. [Google Scholar] [CrossRef]
- Edginton, A.N.; Shah, B.; Sevestre, M.; Momper, J.D. The integration of allometry and virtual populations to predict clearance and clearance variability in pediatric populations over the age of 6 years. Clin. Pharmacokinet. 2013, 52, 693–703. [Google Scholar] [CrossRef]
- Mansoor, N.; Ahmad, T.; Alam Khan, R.; Sharib, S.M.; Mahmood, I. Prediction of Clearance and Dose of Midazolam in Preterm and Term Neonates: A Comparative Study Between Allometric Scaling and Physiologically Based Pharmacokinetic Modeling. Am. J. Ther. 2019, 26, e32–e37. [Google Scholar] [CrossRef]
- Johnson, T.N.; Rostami-Hodjegan, A.; Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 2006, 45, 931–956. [Google Scholar] [CrossRef] [PubMed]
- Leong, R.; Vieira, M.L.; Zhao, P.; Mulugeta, Y.; Lee, C.S.; Huang, S.M.; Burckart, G.J. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 2012, 91, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N. The problems in scaling adult drug doses to children. Arch. Dis. Child. 2008, 93, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Manolis, E.; Osman, T.E.; Herold, R.; Koenig, F.; Tomasi, P.; Vamvakas, S.; Saint Raymond, A. Role of modeling and simulation in pediatric investigation plans. Paediatr. Anaesth. 2011, 21, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Gardner, I.B.; Collard, W.T.; Stanley, P.J.; Oxley, P.; Hosea, N.A.; Plowchalk, D.; Gernhardt, S.; Lin, J.; Dickins, M.; et al. Simulation of human intravenous and oral pharmacokinetics of 21 diverse compounds using physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 2011, 50, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Parrott, N.; Jorga, K.; Lave, T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharmacokinet. 2006, 45, 511–542. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, S. Reduction and lumping of physiologically based pharmacokinetic models: Prediction of the disposition of fentanyl and pethidine in humans by successively simplified models. J. Pharmacokinet. Pharmacodyn. 2003, 30, 285–307. [Google Scholar] [CrossRef]
- Edginton, A.N.; Theil, F.P.; Schmitt, W.; Willmann, S. Whole body physiologically-based pharmacokinetic models: Their use in clinical drug development. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1143–1152. [Google Scholar] [CrossRef]
- Nestorov, I. Whole-body physiologically based pharmacokinetic models. Expert Opin. Drug Metab. Toxicol. 2007, 3, 235–249. [Google Scholar] [CrossRef]
- Edginton, A.N.; Schmitt, W.; Voith, B.; Willmann, S. A mechanistic approach for the scaling of clearance in children. Clin. Pharmacokinet. 2006, 45, 683–704. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Rev. Esp. Cardiol. 2021, 74, 790–799. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Edginton, A.N. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e150. [Google Scholar] [CrossRef]
- Ganguly, S.; Edginton, A.N.; Gerhart, J.G.; Cohen-Wolkowiez, M.; Greenberg, R.G.; Gonzalez, D.; Best Pharmaceuticals for Children Act-Pediatric Trials Network Steering Committee. Physiologically Based Pharmacokinetic Modeling of Meropenem in Preterm and Term Infants. Clin. Pharmacokinet. 2021, 60, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Carry, M.R.; Ringel, S.P.; Starcevich, J.M. Distribution of capillaries in normal and diseased human skeletal muscle. Muscle Nerve 1986, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Tayman, C.; Rayyan, M.; Allegaert, K. Neonatal pharmacology: Extensive interindividual variability despite limited size. J. Pediatr. Pharmacol. Ther. 2011, 16, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Afsar, F.S. Physiological skin conditions of preterm and term neonates. Clin. Exp. Dermatol. 2010, 35, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Sussman, H.H.; Benitz, W.E. Plasma concentrations after rectal administration of acetaminophen in preterm neonates. Paediatr. Anaesth. 1997, 7, 457–459. [Google Scholar] [CrossRef]
- Burri, P.H. Fetal and postnatal development of the lung. Annu. Rev. Physiol. 1984, 46, 617–628. [Google Scholar] [CrossRef]
- El-Gendy, N.; Kaviratna, A.; Berkland, C.; Dhar, P. Delivery and performance of surfactant replacement therapies to treat pulmonary disorders. Ther. Deliv. 2013, 4, 951–980. [Google Scholar] [CrossRef]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef]
- Butranova, O.I.; Ushkalova, E.A.; Zyryanov, S.K.; Chenkurov, M.S. Developmental Pharmacokinetics of Antibiotics Used in Neonatal ICU: Focus on Preterm Infants. Biomedicines 2023, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Avery, G.B.; Randolph, J.G.; Weaver, T. Gastric acidity in the first day of life. Pediatrics 1966, 37, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Bornhorst, G.M.; Henrick, B.M.; German, J.B. Protein Digestion of Baby Foods: Study Approaches and Implications for Infant Health. Mol. Nutr. Food Res. 2018, 62, 1700231. [Google Scholar] [CrossRef]
- Indrio, F.; Neu, J.; Pettoello-Mantovani, M.; Marchese, F.; Martini, S.; Salatto, A.; Aceti, A. Development of the Gastrointestinal Tract in Newborns as a Challenge for an Appropriate Nutrition: A Narrative Review. Nutrients 2022, 14, 1405. [Google Scholar] [CrossRef] [PubMed]
- Lebenthal, E.; Lee, P.C.; Heitlinger, L.A. Impact of development of the gastrointestinal tract on infant feeding. J. Pediatr. 1983, 102, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Verscheijden, L.F.M.; Koenderink, J.B.; Johnson, T.N.; de Wildt, S.N.; Russel, F.G.M. Physiologically-based pharmacokinetic models for children: Starting to reach maturation? Pharmacol. Ther. 2020, 211, 107541. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; Mian, P.; van den Anker, J.N. Developmental Pharmacokinetics in Neonates: Maturational Changes and Beyond. Curr. Pharm. Des. 2017, 23, 5769–5778. [Google Scholar] [CrossRef]
- Bourlieu, C.; Menard, O.; Bouzerzour, K.; Mandalari, G.; Macierzanka, A.; Mackie, A.R.; Dupont, D. Specificity of infant digestive conditions: Some clues for developing relevant in vitro models. Crit. Rev. Food Sci. Nutr. 2014, 54, 1427–1457. [Google Scholar] [CrossRef]
- van Kalken, C.K.; Giaccone, G.; van der Valk, P.; Kuiper, C.M.; Hadisaputro, M.M.; Bosma, S.A.; Scheper, R.J.; Meijer, C.J.; Pinedo, H.M. Multidrug resistance gene (P-glycoprotein) expression in the human fetus. Am. J. Pathol. 1992, 141, 1063–1072. [Google Scholar]
- Mooij, M.G.; Schwarz, U.I.; de Koning, B.A.; Leeder, J.S.; Gaedigk, R.; Samsom, J.N.; Spaans, E.; van Goudoever, J.B.; Tibboel, D.; Kim, R.B.; et al. Ontogeny of human hepatic and intestinal transporter gene expression during childhood: Age matters. Drug Metab. Dispos. 2014, 42, 1268–1274. [Google Scholar] [CrossRef]
- Linakis, M.W.; Roberts, J.K.; Lala, A.C.; Spigarelli, M.G.; Medlicott, N.J.; Reith, D.M.; Ward, R.M.; Sherwin, C.M. Challenges Associated with Route of Administration in Neonatal Drug Delivery. Clin. Pharmacokinet. 2016, 55, 185–196. [Google Scholar] [CrossRef]
- Barbero, G.J.; Runge, G.; Fischer, D.; Crawford, M.N.; Torres, F.E.; Gyorgy, P. Investigations on the bacterial flora, pH, and sugar content in the intestinal tract of infants. J. Pediatr. 1952, 40, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Fredrikzon, B.; Olivecrona, T. Decrease of lipase and esterase activities in intestinal contents of newborn infants during test meals. Pediatr. Res. 1978, 12, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.M.; Bouzom, F.; Hugues, C.; Ungell, A.L. Oral drug absorption in pediatrics: The intestinal wall, its developmental changes and current tools for predictions. Biopharm. Drug Dispos. 2017, 38, 209–230. [Google Scholar] [CrossRef] [PubMed]
- van Elburg, R.M.; Fetter, W.P.; Bunkers, C.M.; Heymans, H.S. Intestinal permeability in relation to birth weight and gestational and postnatal age. Arch. Dis. Child. Fetal Neonatal Ed. 2003, 88, F52–F55. [Google Scholar] [CrossRef] [PubMed]
- Scholtens, P.A.; Oozeer, R.; Martin, R.; Amor, K.B.; Knol, J. The early settlers: Intestinal microbiology in early life. Annu. Rev. Food Sci. Technol. 2012, 3, 425–447. [Google Scholar] [CrossRef] [PubMed]
- de Waal, T.; Brouwers, J.; Mols, R.; Hoffman, I.; Rayyan, M.; Augustijns, P. Characterization of neonatal and infant enterostomy fluids. Int. J. Pharm. 2023, 639, 122943. [Google Scholar] [CrossRef]
- Lebenthal, A.; Lebenthal, E. The ontogeny of the small intestinal epithelium. JPEN J. Parenter. Enteral Nutr. 1999, 23 (Suppl. 5), S3–S6. [Google Scholar] [CrossRef]
- Lebenthal, E.; Lee, P.C. Development of functional responses in human exocrine pancreas. Pediatrics 1980, 66, 556–560. [Google Scholar] [CrossRef]
- Carriere, F.; Barrowman, J.A.; Verger, R.; Laugier, R. Secretion and contribution to lipolysis of gastric and pancreatic lipases during a test meal in humans. Gastroenterology 1993, 105, 876–888. [Google Scholar] [CrossRef]
- Lindower, J.B. Water balance in the fetus and neonate. Semin. Fetal Neonatal Med. 2017, 22, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.M.; Benjamin, D.; Barrett, J.S.; Allegaert, K.; Portman, R.; Davis, J.M.; Turner, M.A. Safety, dosing, and pharmaceutical quality for studies that evaluate medicinal products (including biological products) in neonates. Pediatr. Res. 2017, 81, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Brown, L.K.; Ennis, S.; Beattie, R.M.; Johnson, M.J. Total body water in full-term and preterm newborns: Systematic review and meta-analysis. Arch. Dis. Child. Fetal Neonatal Ed. 2021, 106, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; Cossey, V.; van den Anker, J.N. Dosing Guidelines of Aminoglycosides in Neonates: A Balance Between Physiology and Feasibility. Curr. Pharm. Des. 2015, 21, 5699–5704. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; van den Anker, J.N. Perinatal and neonatal use of paracetamol for pain relief. Semin. Fetal Neonatal Med. 2017, 22, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Gonzalez, D.; Derendorf, H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2010, 99, 1107–1122. [Google Scholar] [CrossRef]
- Weaving, G.; Batstone, G.F.; Jones, R.G. Age and sex variation in serum albumin concentration: An observational study. Ann. Clin. Biochem. 2016, 53 Pt 1, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Hammo, B.; Barry, J.; Radhakrishnan, K. Overview of Albumin Physiology and its Role in Pediatric Diseases. Curr. Gastroenterol. Rep. 2021, 23, 11. [Google Scholar] [CrossRef]
- Anell-Olofsson, M.; Ahmadi, S.; Lonnqvist, P.A.; Eksborg, S.; von Horn, H.; Bartocci, M. Plasma concentrations of alpha-1-acid glycoprotein in preterm and term newborns: Influence of mode of delivery and implications for plasma protein binding of local anaesthetics. Br. J. Anaesth. 2018, 121, 427–431. [Google Scholar] [CrossRef]
- Nau, H.; Luck, W.; Kuhnz, W. Decreased serum protein binding of diazepam and its major metabolite in the neonate during the first postnatal week relate to increased free fatty acid levels. Br. J. Clin. Pharmacol. 1984, 17, 92–98. [Google Scholar] [CrossRef]
- Notarianni, L.J. Plasma protein binding of drugs in pregnancy and in neonates. Clin. Pharmacokinet. 1990, 18, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Yanni, S.B.; Smith, P.B.; Benjamin, D.K., Jr.; Augustijns, P.F.; Thakker, D.R.; Annaert, P.P. Higher clearance of micafungin in neonates compared with adults: Role of age-dependent micafungin serum binding. Biopharm. Drug Dispos. 2011, 32, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Pokorna, P.; Wildschut, E.D.; Vobruba, V.; van den Anker, J.N.; Tibboel, D. The Impact of Hypothermia on the Pharmacokinetics of Drugs Used in Neonates and Young Infants. Curr. Pharm. Des. 2015, 21, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.K.; White, C.A.; Cummings, B.S.; Hines, R.N.; Muralidhara, S.; Bruckner, J.V. Ontogeny of plasma proteins, albumin and binding of diazepam, cyclosporine, and deltamethrin. Pediatr. Res. 2016, 79, 409–415. [Google Scholar] [CrossRef] [PubMed]
- McNamara, P.J.; Alcorn, J. Protein binding predictions in infants. AAPS PharmSci 2002, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, J.; McNamara, P.J. Ontogeny of hepatic and renal systemic clearance pathways in infants: Part I. Clin. Pharmacokinet. 2002, 41, 959–998. [Google Scholar] [CrossRef] [PubMed]
- Sharer, J.E.; Wrighton, S.A. Identification of the human hepatic cytochromes P450 involved in the in vitro oxidation of antipyrine. Drug Metab. Dispos. 1996, 24, 487–494. [Google Scholar]
- Engel, G.; Hofmann, U.; Heidemann, H.; Cosme, J.; Eichelbaum, M. Antipyrine as a probe for human oxidative drug metabolism: Identification of the cytochrome P450 enzymes catalyzing 4-hydroxyantipyrine, 3-hydroxymethylantipyrine, and norantipyrine formation. Clin. Pharmacol. Ther. 1996, 59, 613–623. [Google Scholar] [CrossRef]
- Allegaert, K.; Simons, S.H.P.; Tibboel, D.; Krekels, E.H.; Knibbe, C.A.; van den Anker, J.N. Non-maturational covariates for dynamic systems pharmacology models in neonates, infants, and children: Filling the gaps beyond developmental pharmacology. Eur. J. Pharm. Sci. 2017, 109S, S27–S31. [Google Scholar] [CrossRef]
- Hines, R.N. Ontogeny of human hepatic cytochromes P450. J. Biochem. Mol. Toxicol. 2007, 21, 169–175. [Google Scholar] [CrossRef]
- Mahmood, I.; Ahmad, T.; Mansoor, N.; Sharib, S.M. Prediction of Clearance in Neonates and Infants (≤3 Months of Age) for Drugs That Are Glucuronidated: A Comparative Study Between Allometric Scaling and Physiologically Based Pharmacokinetic Modeling. J. Clin. Pharmacol. 2017, 57, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Rostami-Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: Parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr. Anaesth. 2011, 21, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Brzezinski, M.R.; Fantel, A.G.; Juchau, M.R. Catalysis of drug oxidation during embryogenesis in human hepatic tissues using imipramine as a model substrate. Drug Metab. Dispos. 1999, 27, 1306–1308. [Google Scholar] [PubMed]
- Hakkola, J.; Raunio, H.; Purkunen, R.; Saarikoski, S.; Vahakangas, K.; Pelkonen, O.; Edwards, R.J.; Boobis, A.R.; Pasanen, M. Cytochrome P450 3A expression in the human fetal liver: Evidence that CYP3A5 is expressed in only a limited number of fetal livers. Biol. Neonate 2001, 80, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, D.; Sonnier, M.; Moncion, A.; Cheron, G.; Cresteil, T. Expression of CYP3A in the human liver--evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur. J. Biochem. 1997, 247, 625–634. [Google Scholar] [CrossRef] [PubMed]
- de Wildt, S.N.; Kearns, G.L.; Leeder, J.S.; van den Anker, J.N. Cytochrome P450 3A: Ontogeny and drug disposition. Clin. Pharmacokinet. 1999, 37, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Ince, I.; Knibbe, C.A.; Danhof, M.; de Wildt, S.N. Developmental changes in the expression and function of cytochrome P450 3A isoforms: Evidence from in vitro and in vivo investigations. Clin. Pharmacokinet. 2013, 52, 333–345. [Google Scholar] [CrossRef]
- Johnsrud, E.K.; Koukouritaki, S.B.; Divakaran, K.; Brunengraber, L.L.; Hines, R.N.; McCarver, D.G. Human hepatic CYP2E1 expression during development. J. Pharmacol. Exp. Ther. 2003, 307, 402–407. [Google Scholar] [CrossRef]
- Maxwell, D.M. The specificity of carboxylesterase protection against the toxicity of organophosphorus compounds. Toxicol. Appl. Pharmacol. 1992, 114, 306–312. [Google Scholar] [CrossRef]
- Pope, C.N.; Karanth, S.; Liu, J.; Yan, B. Comparative carboxylesterase activities in infant and adult liver and their in vitro sensitivity to chlorpyrifos oxon. Regul. Toxicol. Pharmacol. 2005, 42, 64–69. [Google Scholar] [CrossRef]
- Yang, D.; Pearce, R.E.; Wang, X.; Gaedigk, R.; Wan, Y.J.; Yan, B. Human carboxylesterases HCE1 and HCE2: Ontogenic expression, inter-individual variability and differential hydrolysis of oseltamivir, aspirin, deltamethrin and permethrin. Biochem. Pharmacol. 2009, 77, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Zane, N.R.; Chen, Y.; Wang, M.Z.; Thakker, D.R. Cytochrome P450 and flavin-containing monooxygenase families: Age-dependent differences in expression and functional activity. Pediatr. Res. 2018, 83, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.N.; Wu, A.H.; Hill, D.W. Alcohol dehydrogenase and catalase content in perinatal infant and adult livers: Potential influence on neonatal alcohol metabolism. Toxicol. Lett. 2007, 169, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Lin, Z. Exploring the genetic architecture of neonatal hyperbilirubinemia. Semin. Fetal Neonatal Med. 2010, 15, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.K.; Mehrotra, A.; Gaedigk, A.; Chapa, R.; Basit, A.; Zhang, H.; Choudhari, P.; Boberg, M.; Pearce, R.E.; Gaedigk, R.; et al. Age- and Genotype-Dependent Variability in the Protein Abundance and Activity of Six Major Uridine Diphosphate-Glucuronosyltransferases in Human Liver. Clin. Pharmacol. Ther. 2019, 105, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Allegaert, K.; Vanhaesebrouck, S.; Verbesselt, R.; van den Anker, J.N. In vivo glucuronidation activity of drugs in neonates: Extensive interindividual variability despite their young age. Ther. Drug Monit. 2009, 31, 411–415. [Google Scholar] [CrossRef]
- Levy, G.; Khanna, N.N.; Soda, D.M.; Tsuzuki, O.; Stern, L. Pharmacokinetics of acetaminophen in the human neonate: Formation of acetaminophen glucuronide and sulfate in relation to plasma bilirubin concentration and D-glucaric acid excretion. Pediatrics 1975, 55, 818–825. [Google Scholar] [CrossRef]
- McRorie, T.I.; Lynn, A.M.; Nespeca, M.K.; Opheim, K.E.; Slattery, J.T. The maturation of morphine clearance and metabolism. Am. J. Dis. Child. 1992, 146, 972–976. [Google Scholar] [CrossRef]
- Duanmu, Z.; Weckle, A.; Koukouritaki, S.B.; Hines, R.N.; Falany, J.L.; Falany, C.N.; Kocarek, T.A.; Runge-Morris, M. Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre- and postnatal human liver. J. Pharmacol. Exp. Ther. 2006, 316, 1310–1317. [Google Scholar] [CrossRef]
- Nies, A.S.; Shand, D.G.; Wilkinson, G.R. Altered hepatic blood flow and drug disposition. Clin. Pharmacokinet. 1976, 1, 135–155. [Google Scholar] [CrossRef]
- Bjorkman, S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: Theophylline and midazolam as model drugs. Br. J. Clin. Pharmacol. 2005, 59, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Gow, P.J.; Ghabrial, H.; Smallwood, R.A.; Morgan, D.J.; Ching, M.S. Neonatal hepatic drug elimination. Pharmacol. Toxicol. 2001, 88, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Wakamiya, N.; Ueng, Y.F.; Guengerich, F.P.; Inui, Y. Characterization of microsomal cytochrome P450 enzymes involved in the oxidation of xenobiotic chemicals in human fetal liver and adult lungs. Drug Metab. Dispos. 1996, 24, 515–522. [Google Scholar] [PubMed]
- Yang, H.Y.; Namkung, M.J.; Juchau, M.R. Expression of functional cytochrome P4501A1 in human embryonic hepatic tissues during organogenesis. Biochem. Pharmacol. 1995, 49, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Omiecinski, C.J.; Redlich, C.A.; Costa, P. Induction and developmental expression of cytochrome P450IA1 messenger RNA in rat and human tissues: Detection by the polymerase chain reaction. Cancer Res. 1990, 50, 4315–4321. [Google Scholar] [PubMed]
- Sonnier, M.; Cresteil, T. Delayed ontogenesis of CYP1A2 in the human liver. Eur. J. Biochem. 1998, 251, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar] [PubMed]
- Hakkola, J.; Pasanen, M.; Pelkonen, O.; Hukkanen, J.; Evisalmi, S.; Anttila, S.; Rane, A.; Mantyla, M.; Purkunen, R.; Saarikoski, S.; et al. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis 1997, 18, 391–397. [Google Scholar] [CrossRef]
- Tateishi, T.; Nakura, H.; Asoh, M.; Watanabe, M.; Tanaka, M.; Kumai, T.; Takashima, S.; Imaoka, S.; Funae, Y.; Yabusaki, Y.; et al. A comparison of hepatic cytochrome P450 protein expression between infancy and postinfancy. Life Sci. 1997, 61, 2567–2574. [Google Scholar] [CrossRef]
- Hakkola, J.; Pasanen, M.; Purkunen, R.; Saarikoski, S.; Pelkonen, O.; Maenpaa, J.; Rane, A.; Raunio, H. Expression of xenobiotic-metabolizing cytochrome P450 forms in human adult and fetal liver. Biochem. Pharmacol. 1994, 48, 59–64. [Google Scholar] [CrossRef]
- Treluyer, J.M.; Gueret, G.; Cheron, G.; Sonnier, M.; Cresteil, T. Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: In-vivo/in-vitro correlation and inducibility. Pharmacogenetics 1997, 7, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Yanni, S.B.; Annaert, P.P.; Augustijns, P.; Ibrahim, J.G.; Benjamin, D.K., Jr.; Thakker, D.R. In vitro hepatic metabolism explains higher clearance of voriconazole in children versus adults: Role of CYP2C19 and flavin-containing monooxygenase 3. Drug Metab. Dispos. 2010, 38, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Treluyer, J.M.; Jacqz-Aigrain, E.; Alvarez, F.; Cresteil, T. Expression of CYP2D6 in developing human liver. Eur. J. Biochem. 1991, 202, 583–588. [Google Scholar] [CrossRef]
- Vieira, I.; Sonnier, M.; Cresteil, T. Developmental expression of CYP2E1 in the human liver. Hypermethylation control of gene expression during the neonatal period. Eur. J. Biochem. 1996, 238, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.P.; Lasker, J.M.; Raucy, J.L. Expression, induction, and catalytic activity of the ethanol-inducible cytochrome P450 (CYP2E1) in human fetal liver and hepatocytes. Mol. Pharmacol. 1996, 49, 260–268. [Google Scholar] [PubMed]
- Boutelet-Bochan, H.; Huang, Y.; Juchau, M.R. Expression of CYP2E1 during embryogenesis and fetogenesis in human cephalic tissues: Implications for the fetal alcohol syndrome. Biochem. Biophys. Res. Commun. 1997, 238, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Su, T.; Chen, Y.; Zhang, Q.Y.; Ding, X. Expression of biotransformation enzymes in human fetal olfactory mucosa: Potential roles in developmental toxicity. Toxicol. Appl. Pharmacol. 2000, 165, 158–162. [Google Scholar] [CrossRef]
- Chen, Y.T.; Trzoss, L.; Yang, D.; Yan, B. Ontogenic expression of human carboxylesterase-2 and cytochrome P450 3A4 in liver and duodenum: Postnatal surge and organ-dependent regulation. Toxicology 2015, 330, 55–61. [Google Scholar] [CrossRef]
- Schuetz, J.D.; Beach, D.L.; Guzelian, P.S. Selective expression of cytochrome P450 CYP3A mRNAs in embryonic and adult human liver. Pharmacogenetics 1994, 4, 11–20. [Google Scholar] [CrossRef]
- Cresteil, T. Onset of xenobiotic metabolism in children: Toxicological implications. Food Addit. Contam. 1998, 15, 45–51. [Google Scholar] [CrossRef]
- Koukouritaki, S.B.; Simpson, P.; Yeung, C.K.; Rettie, A.E.; Hines, R.N. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr. Res. 2002, 51, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Hopkinson, D.A.; Harris, H. Developmental changes and polymorphism in human alcohol dehydrogenase. Ann. Hum. Genet. 1971, 34, 251–271. [Google Scholar] [CrossRef]
- Estonius, M.; Svensson, S.; Hoog, J.O. Alcohol dehydrogenase in human tissues: Localisation of transcripts coding for five classes of the enzyme. FEBS Lett. 1996, 397, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Omiecinski, C.J.; Aicher, L.; Swenson, L. Developmental expression of human microsomal epoxide hydrolase. J. Pharmacol. Exp. Ther. 1994, 269, 417–423. [Google Scholar] [PubMed]
- Cresteil, T.; Beaune, P.; Kremers, P.; Celier, C.; Guengerich, F.P.; Leroux, J.P. Immunoquantification of epoxide hydrolase and cytochrome P-450 isozymes in fetal and adult human liver microsomes. Eur. J. Biochem. 1985, 151, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Mackness, B.; Beltran-Debon, R.; Aragones, G.; Joven, J.; Camps, J.; Mackness, M. Human tissue distribution of paraoxonases 1 and 2 mRNA. IUBMB Life 2010, 62, 480–482. [Google Scholar] [CrossRef]
- Tayama, Y.; Sugihara, K.; Sanoh, S.; Miyake, K.; Kitamura, S.; Ohta, S. Developmental changes of aldehyde oxidase activity and protein expression in human liver cytosol. Drug Metab. Pharmacokinet. 2012, 27, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, C.P.; Strassburg, A.; Kneip, S.; Barut, A.; Tukey, R.H.; Rodeck, B.; Manns, M.P. Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut 2002, 50, 259–265. [Google Scholar] [CrossRef]
- Zaya, M.J.; Hines, R.N.; Stevens, J.C. Epirubicin glucuronidation and UGT2B7 developmental expression. Drug Metab. Dispos. 2006, 34, 2097–2101. [Google Scholar] [CrossRef]
- Bhatt, D.K.; Basit, A.; Zhang, H.; Gaedigk, A.; Lee, S.B.; Claw, K.G.; Mehrotra, A.; Chaudhry, A.S.; Pearce, R.E.; Gaedigk, R.; et al. Hepatic Abundance and Activity of Androgen- and Drug-Metabolizing Enzyme UGT2B17 Are Associated with Genotype, Age, and Sex. Drug Metab. Dispos. 2018, 46, 888–896. [Google Scholar] [CrossRef]
- Richard, K.; Hume, R.; Kaptein, E.; Stanley, E.L.; Visser, T.J.; Coughtrie, M.W. Sulfation of thyroid hormone and dopamine during human development: Ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J. Clin. Endocrinol. Metab. 2001, 86, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.L.; Hume, R.; Coughtrie, M.W. Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol. Cell. Endocrinol. 2005, 240, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Her, C.; Kaur, G.P.; Athwal, R.S.; Weinshilboum, R.M. Human sulfotransferase SULT1C1: cDNA cloning, tissue-specific expression, and chromosomal localization. Genomics 1997, 41, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Strange, R.C.; Davis, B.A.; Faulder, C.G.; Cotton, W.; Bain, A.D.; Hopkinson, D.A.; Hume, R. The human glutathione S-transferases: Developmental aspects of the GST1, GST2, and GST3 loci. Biochem. Genet. 1985, 23, 1011–1028. [Google Scholar] [CrossRef] [PubMed]
- Raijmakers, M.T.; Steegers, E.A.; Peters, W.H. Glutathione S-transferases and thiol concentrations in embryonic and early fetal tissues. Hum. Reprod. 2001, 16, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Strange, R.C.; Howie, A.F.; Hume, R.; Matharoo, B.; Bell, J.; Hiley, C.; Jones, P.; Beckett, G.J. The development expression of alpha-, mu- and pi-class glutathione S-transferases in human liver. Biochim. Biophys. Acta 1989, 993, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Beckett, G.J.; Howie, A.F.; Hume, R.; Matharoo, B.; Hiley, C.; Jones, P.; Strange, R.C. Human glutathione S-transferases: Radioimmunoassay studies on the expression of alpha-, mu- and pi-class isoenzymes in developing lung and kidney. Biochim. Biophys. Acta 1990, 1036, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gu, Y.; James, M.O.; Hines, R.N.; Simpson, P.; Langaee, T.; Stacpoole, P.W. Prenatal and postnatal expression of glutathione transferase zeta 1 in human liver and the roles of haplotype and subject age in determining activity with dichloroacetate. Drug Metab. Dispos. 2012, 40, 232–239. [Google Scholar] [CrossRef]
- Frazier, K.S. Species Differences in Renal Development and Associated Developmental Nephrotoxicity. Birth Defects Res. 2017, 109, 1243–1256. [Google Scholar] [CrossRef]
- Kandasamy, Y.; Rudd, D.; Smith, R.; Lumbers, E.R.; Wright, I.M. Extra uterine development of preterm kidneys. Pediatr. Nephrol. 2018, 33, 1007–1012. [Google Scholar] [CrossRef]
- Solhaug, M.J.; Bolger, P.M.; Jose, P.A. The developing kidney and environmental toxins. Pediatrics 2004, 113 (Suppl. 4), 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Bueters, R.; Bael, A.; Gasthuys, E.; Chen, C.; Schreuder, M.F.; Frazier, K.S. Ontogeny and Cross-species Comparison of Pathways Involved in Drug Absorption, Distribution, Metabolism, and Excretion in Neonates (Review): Kidney. Drug Metab. Dispos. 2020, 48, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Prediction of total and renal clearance of renally secreted drugs in neonates and infants (≤3 months of age). J. Clin. Transl. Res. 2022, 8, 445–452. [Google Scholar] [PubMed]
- Filler, G.; Bhayana, V.; Schott, C.; Diaz-Gonzalez de Ferris, M.E. How should we assess renal function in neonates and infants? Acta Paediatr. 2021, 110, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Loebstein, R.; Koren, G. Clinical pharmacology and therapeutic drug monitoring in neonates and children. Pediatr. Rev. 1998, 19, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Rhodin, M.M.; Anderson, B.J.; Peters, A.M.; Coulthard, M.G.; Wilkins, B.; Cole, M.; Chatelut, E.; Grubb, A.; Veal, G.J.; Keir, M.J.; et al. Human renal function maturation: A quantitative description using weight and postmenstrual age. Pediatr. Nephrol. 2009, 24, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Iacobelli, S.; Loprieno, S.; Bonsante, F.; Latorre, G.; Esposito, L.; Gouyon, J.B. Renal function in early childhood in very low birthweight infants. Am. J. Perinatol. 2007, 24, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mehta, N.; Muhari-Stark, E.; Burckart, G.J.; van den Anker, J.; Wang, J. Pediatric Renal Ontogeny and Applications in Drug Development. J. Clin. Pharmacol. 2019, 59 (Suppl. 1), S9–S20. [Google Scholar] [CrossRef]
- van den Anker, J.; Allegaert, K. Considerations for Drug Dosing in Premature Infants. J. Clin. Pharmacol. 2021, 61 (Suppl. 1), S141–S151. [Google Scholar] [CrossRef]
- Thabit, A.K. Antibiotics in the Biliary Tract: A Review of the Pharmacokinetics and Clinical Outcomes of Antibiotics Penetrating the Bile and Gallbladder Wall. Pharmacotherapy 2020, 40, 672–691. [Google Scholar] [CrossRef]
- Balistreri, W.F. Immaturity of hepatic excretory function and the ontogeny of bile acid metabolism. J. Pediatr. Gastroenterol. Nutr. 1983, 2 (Suppl. 1), S207–S214. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, M.T.; Hassan, A.S. Development of bile acid biogenesis and its significance in cholesterol homeostasis. Adv. Lipid Res. 1982, 19, 137–161. [Google Scholar] [PubMed]
- Grijalva, J.; Vakili, K. Neonatal liver physiology. Semin. Pediatr. Surg. 2013, 22, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Heubi, J.E.; Balistreri, W.F.; Suchy, F.J. Bile salt metabolism in the first year of life. J. Lab. Clin. Med. 1982, 100, 127–136. [Google Scholar] [PubMed]
- Ho, R.H.; Kim, R.B. Transporters and drug therapy: Implications for drug disposition and disease. Clin. Pharmacol. Ther. 2005, 78, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Sato, H.; Sugiyama, Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Konieczna, A.; Erdosova, B.; Lichnovska, R.; Jandl, M.; Cizkova, K.; Ehrmann, J. Differential expression of ABC transporters (MDR1, MRP1, BCRP) in developing human embryos. J. Mol. Histol. 2011, 42, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Prasad, B.; Patilea, G.; Gupta, A.; Salphati, L.; Evers, R.; Hop, C.E.; Unadkat, J.D. Quantitative transporter proteomics by liquid chromatography with tandem mass spectrometry: Addressing methodologic issues of plasma membrane isolation and expression-activity relationship. Drug Metab. Dispos. 2015, 43, 284–288. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Aleksunes, L.M. Xenobiotic, bile acid, and cholesterol transporters: Function and regulation. Pharmacol. Rev. 2010, 62, 1–96. [Google Scholar] [CrossRef]
- Deo, A.K.; Prasad, B.; Balogh, L.; Lai, Y.; Unadkat, J.D. Interindividual variability in hepatic expression of the multidrug resistance-associated protein 2 (MRP2/ABCC2): Quantification by liquid chromatography/tandem mass spectrometry. Drug Metab. Dispos. 2012, 40, 852–855. [Google Scholar] [CrossRef]
- Prasad, B.; Gaedigk, A.; Vrana, M.; Gaedigk, R.; Leeder, J.S.; Salphati, L.; Chu, X.; Xiao, G.; Hop, C.; Evers, R.; et al. Ontogeny of Hepatic Drug Transporters as Quantified by LC-MS/MS Proteomics. Clin. Pharmacol. Ther. 2016, 100, 362–370. [Google Scholar] [CrossRef] [PubMed]
- van Groen, B.D.; van de Steeg, E.; Mooij, M.G.; van Lipzig, M.M.H.; de Koning, B.A.E.; Verdijk, R.M.; Wortelboer, H.M.; Gaedigk, R.; Bi, C.; Leeder, J.S.; et al. Proteomics of human liver membrane transporters: A focus on fetuses and newborn infants. Eur. J. Pharm. Sci. 2018, 124, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Abanda, N.N.; Riches, Z.; Collier, A.C. Lobular Distribution and Variability in Hepatic ATP Binding Cassette Protein B1 (ABCB1, P-gp): Ontogenetic Differences and Potential for Toxicity. Pharmaceutics 2017, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Mooij, M.G.; van de Steeg, E.; van Rosmalen, J.; Windster, J.D.; de Koning, B.A.; Vaes, W.H.; van Groen, B.D.; Tibboel, D.; Wortelboer, H.M.; de Wildt, S.N. Proteomic Analysis of the Developmental Trajectory of Human Hepatic Membrane Transporter Proteins in the First Three Months of Life. Drug Metab. Dispos. 2016, 44, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Burgess, K.S.; Philips, S.; Benson, E.A.; Desta, Z.; Gaedigk, A.; Gaedigk, R.; Segar, M.W.; Liu, Y.; Skaar, T.C. Age-Related Changes in MicroRNA Expression and Pharmacogenes in Human Liver. Clin. Pharmacol. Ther. 2015, 98, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ellis, E.C.; Gramignoli, R.; Dorko, K.; Tahan, V.; Hansel, M.; Mattison, D.R.; Caritis, S.N.; Hines, R.N.; Venkataramanan, R.; et al. Hepatobiliary disposition of 17-OHPC and taurocholate in fetal human hepatocytes: A comparison with adult human hepatocytes. Drug Metab. Dispos. 2013, 41, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Hahn, D.; Emoto, C.; Vinks, A.A.; Fukuda, T. Developmental Changes in Hepatic Organic Cation Transporter OCT1 Protein Expression from Neonates to Children. Drug Metab. Dispos. 2017, 45, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Chen, H.L.; Liu, Y.J.; Feng, C.H.; Wu, C.Y.; Shyu, M.K.; Yuan, R.H.; Chang, M.H. Developmental expression of canalicular transporter genes in human liver. J. Hepatol. 2005, 43, 472–477. [Google Scholar] [CrossRef]
- Cheung, K.W.K.; van Groen, B.D.; Spaans, E.; van Borselen, M.D.; de Bruijn, A.; Simons-Oosterhuis, Y.; Tibboel, D.; Samsom, J.N.; Verdijk, R.M.; Smeets, B.; et al. A Comprehensive Analysis of Ontogeny of Renal Drug Transporters: mRNA Analyses, Quantitative Proteomics, and Localization. Clin. Pharmacol. Ther. 2019, 106, 1083–1092. [Google Scholar] [CrossRef]
- Lam, J.; Baello, S.; Iqbal, M.; Kelly, L.E.; Shannon, P.T.; Chitayat, D.; Matthews, S.G.; Koren, G. The ontogeny of P-glycoprotein in the developing human blood-brain barrier: Implication for opioid toxicity in neonates. Pediatr. Res. 2015, 78, 417–421. [Google Scholar] [CrossRef]
- Miki, Y.; Suzuki, T.; Tazawa, C.; Blumberg, B.; Sasano, H. Steroid and xenobiotic receptor (SXR), cytochrome P450 3A4 and multidrug resistance gene 1 in human adult and fetal tissues. Mol. Cell. Endocrinol. 2005, 231, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Bodenham, A.; Shelly, M.P.; Park, G.R. The altered pharmacokinetics and pharmacodynamics of drugs commonly used in critically ill patients. Clin. Pharmacokinet. 1988, 14, 347–373. [Google Scholar] [CrossRef] [PubMed]
- Cristea, S.; Smits, A.; Kulo, A.; Knibbe, C.A.J.; van Weissenbruch, M.; Krekels, E.H.J.; Allegaert, K. Amikacin Pharmacokinetics To Optimize Dosing in Neonates with Perinatal Asphyxia Treated with Hypothermia. Antimicrob. Agents Chemother. 2017, 61, e01282-17. [Google Scholar] [CrossRef] [PubMed]
- Raffaeli, G.; Pokorna, P.; Allegaert, K.; Mosca, F.; Cavallaro, G.; Wildschut, E.D.; Tibboel, D. Drug Disposition and Pharmacotherapy in Neonatal ECMO: From Fragmented Data to Integrated Knowledge. Front. Pediatr. 2019, 7, 360. [Google Scholar] [CrossRef] [PubMed]
- Van Den Anker, J.N.; Van Der Heijden, B.J.; Hop, W.C.; Schoemaker, R.C.; Broerse, H.M.; Neijens, H.J.; De Groot, R. The effect of asphyxia on the pharmacokinetics of ceftazidime in the term newborn. Pediatr. Res. 1995, 38, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Frymoyer, A.; Meng, L.; Bonifacio, S.L.; Verotta, D.; Guglielmo, B.J. Gentamicin pharmacokinetics and dosing in neonates with hypoxic ischemic encephalopathy receiving hypothermia. Pharmacotherapy 2013, 33, 718–726. [Google Scholar] [CrossRef]
- Samardzic, J.; Allegaert, K.; Wilbaux, M.; Pfister, M.; van den Anker, J.N. Quantitative clinical pharmacology practice for optimal use of antibiotics during the neonatal period. Expert Opin. Drug Metab. Toxicol. 2016, 12, 367–375. [Google Scholar] [CrossRef]
- Vet, N.J.; de Hoog, M.; Tibboel, D.; de Wildt, S.N. The effect of inflammation on drug metabolism: A focus on pediatrics. Drug Discov. Today 2011, 16, 435–442. [Google Scholar] [CrossRef]
- Chytra, I.; Stepan, M.; Benes, J.; Pelnar, P.; Zidkova, A.; Bergerova, T.; Pradl, R.; Kasal, E. Clinical and microbiological efficacy of continuous versus intermittent application of meropenem in critically ill patients: A randomized open-label controlled trial. Crit. Care 2012, 16, R113. [Google Scholar] [CrossRef]
- Udy, A.A.; Varghese, J.M.; Altukroni, M.; Briscoe, S.; McWhinney, B.C.; Ungerer, J.P.; Lipman, J.; Roberts, J.A. Subtherapeutic initial beta-lactam concentrations in select critically ill patients: Association between augmented renal clearance and low trough drug concentrations. Chest 2012, 142, 30–39. [Google Scholar] [CrossRef]
- Prowle, J.R.; Molan, M.P.; Hornsey, E.; Bellomo, R. Measurement of renal blood flow by phase-contrast magnetic resonance imaging during septic acute kidney injury: A pilot investigation. Crit. Care Med. 2012, 40, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- D’Agate, S.; Musuamba, F.T.; Della Pasqua, O. Dose Rationale for Amoxicillin in Neonatal Sepsis When Referral Is Not Possible. Front. Pharmacol. 2020, 11, 521933. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.S.; Ransom, J.L.; Gal, P.; Carlos, R.Q.; Smith, M.; Schall, S.A. Gentamicin pharmacokinetics in neonates with patent ductus arteriosus. Crit. Care Med. 1997, 25, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Mueller, B.A. Antibiotic Dosing in Patients with Acute Kidney Injury: “Enough But Not Too Much”. J. Intensive Care Med. 2016, 31, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Bunglawala, F.; Rajoli, R.K.R.; Mirochnick, M.; Owen, A.; Siccardi, M. Prediction of dolutegravir pharmacokinetics and dose optimization in neonates via physiologically based pharmacokinetic (PBPK) modelling. J. Antimicrob. Chemother. 2020, 75, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Neeli, H.; Hanna, N.; Abduljalil, K.; Cusumano, J.; Taft, D.R. Application of Physiologically Based Pharmacokinetic-Pharmacodynamic Modeling in Preterm Neonates to Guide Gentamicin Dosing Decisions and Predict Antibacterial Effect. J. Clin. Pharmacol. 2021, 61, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Zazo, H.; Lagarejos, E.; Prado-Velasco, M.; Sanchez-Herrero, S.; Serna, J.; Rueda-Ferreiro, A.; Martin-Suarez, A.; Calvo, M.V.; Perez-Blanco, J.S.; Lanao, J.M. Physiologically-based pharmacokinetic modelling and dosing evaluation of gentamicin in neonates using PhysPK. Front. Pharmacol. 2022, 13, 977372. [Google Scholar] [CrossRef]
- Li, S.; Xie, F. Foetal and neonatal exposure prediction and dosing evaluation for ampicillin using a physiologically-based pharmacokinetic modelling approach. Br. J. Clin. Pharmacol. 2023, 89, 1402–1412. [Google Scholar] [CrossRef]
- Hornik, C.P.; Wu, H.; Edginton, A.N.; Watt, K.; Cohen-Wolkowiez, M.; Gonzalez, D. Development of a Pediatric Physiologically-Based Pharmacokinetic Model of Clindamycin Using Opportunistic Pharmacokinetic Data. Clin. Pharmacokinet. 2017, 56, 1343–1353. [Google Scholar] [CrossRef]
- Willmann, S.; Frei, M.; Sutter, G.; Coboeken, K.; Wendl, T.; Eissing, T.; Lippert, J.; Stass, H. Application of Physiologically-Based and Population Pharmacokinetic Modeling for Dose Finding and Confirmation During the Pediatric Development of Moxifloxacin. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 654–663. [Google Scholar] [CrossRef]
- Conner, T.M.; Nikolian, V.C.; Georgoff, P.E.; Pai, M.P.; Alam, H.B.; Sun, D.; Reed, R.C.; Zhang, T. Physiologically based pharmacokinetic modeling of disposition and drug-drug interactions for valproic acid and divalproex. Eur. J. Pharm. Sci. 2018, 111, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Ladumor, M.K.; Bhatt, D.K.; Gaedigk, A.; Sharma, S.; Thakur, A.; Pearce, R.E.; Leeder, J.S.; Bolger, M.B.; Singh, S.; Prasad, B. Ontogeny of Hepatic Sulfotransferases and Prediction of Age-Dependent Fractional Contribution of Sulfation in Acetaminophen Metabolism. Drug Metab. Dispos. 2019, 47, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, J.G.; Watt, K.M.; Edginton, A.; Wade, K.C.; Salerno, S.N.; Benjamin, D.K.; Smith, P.B.; Hornik, C.P.; Cohen-Wolkowiez, M.; Duara, S.; et al. Physiologically-Based Pharmacokinetic Modeling of Fluconazole Using Plasma and Cerebrospinal Fluid Samples From Preterm and Term Infants. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.M.; Cohen-Wolkowiez, M.; Barrett, J.S.; Sevestre, M.; Zhao, P.; Brouwer, K.L.R.; Edginton, A.N. Physiologically Based Pharmacokinetic Approach to Determine Dosing on Extracorporeal Life Support: Fluconazole in Children on ECMO. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Thai, H.T.; Mazuir, F.; Cartot-Cotton, S.; Veyrat-Follet, C. Optimizing pharmacokinetic bridging studies in paediatric oncology using physiologically-based pharmacokinetic modelling: Application to docetaxel. Br. J. Clin. Pharmacol. 2015, 80, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.D.; Mathias, A.; German, P.; Pikora, C.; Reddy, S.; Kirby, B.J. Physiologically-Based Pharmacokinetic Modeling of Remdesivir and Its Metabolites to Support Dose Selection for the Treatment of Pediatric Patients with COVID-19. Clin. Pharmacol. Ther. 2021, 109, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Rehmel, J.; Ferguson-Sells, L.; Morse, B.L.; Li, B.; Dickinson, G.L. Physiologically based pharmacokinetic modeling of tadalafil to inform pediatric dose selection in children with pulmonary arterial hypertension. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.P.; Balusu, R.; Thotakura, S.; Pasnoor, A.K.; Raju, A.P.; Lal, S.M.; Lewis, L.E.; Mallayasamy, S. Development of Physiologically Based Pharmacokinetic Model and Assessment of the Impact of Renal Underdevelopment in Preterm Infants on the Pharmacokinetics of Aminophylline. J. Pharmacol. Pharmacother. 2022, 13, 72–78. [Google Scholar] [CrossRef]
- Rashid, M.; Sarfraz, M.; Arafat, M.; Hussain, A.; Abbas, N.; Sadiq, M.W.; Rasool, M.F.; Bukhari, N.I. Prediction of lisinopril pediatric dose from the reference adult dose by employing a physiologically based pharmacokinetic model. BMC Pharmacol. Toxicol. 2020, 21, 56. [Google Scholar] [CrossRef]
- Cho, Y.S.; Shin, J.G. Physiologically-based pharmacokinetic modeling of nafamostat to support dose selection for treatment of pediatric patients with COVID-19. Transl. Clin. Pharmacol. 2022, 30, 24–36. [Google Scholar] [CrossRef]
- Wei, L.; Mansoor, N.; Khan, R.A.; Czejka, M.; Ahmad, T.; Ahmed, M.; Ali, M.; Yang, D.H. WB-PBPK approach in predicting zidovudine pharmacokinetics in preterm neonates. Biopharm. Drug Dispos. 2019, 40, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Mousa, Y.M.; Zhao, L.; Raines, K.; Seo, P.; Wu, F. Using a Physiologically Based Pharmacokinetic Absorption Model to Establish Dissolution Bioequivalence Safe Space for Oseltamivir in Adult and Pediatric Populations. AAPS J. 2020, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- Boberg, M.; Vrana, M.; Mehrotra, A.; Pearce, R.E.; Gaedigk, A.; Bhatt, D.K.; Leeder, J.S.; Prasad, B. Age-Dependent Absolute Abundance of Hepatic Carboxylesterases (CES1 and CES2) by LC-MS/MS Proteomics: Application to PBPK Modeling of Oseltamivir In Vivo Pharmacokinetics in Infants. Drug Metab. Dispos. 2017, 45, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Parrott, N.; Davies, B.; Hoffmann, G.; Koerner, A.; Lave, T.; Prinssen, E.; Theogaraj, E.; Singer, T. Development of a physiologically based model for oseltamivir and simulation of pharmacokinetics in neonates and infants. Clin. Pharmacokinet. 2011, 50, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Hahn, D.; Emoto, C.; Euteneuer, J.C.; Mizuno, T.; Vinks, A.A.; Fukuda, T. Influence of OCT1 Ontogeny and Genetic Variation on Morphine Disposition in Critically Ill Neonates: Lessons From PBPK Modeling and Clinical Study. Clin. Pharmacol. Ther. 2019, 105, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; Johnson, T.N.; Neuhoff, S.; Hahn, D.; Vinks, A.A.; Fukuda, T. PBPK Model of Morphine Incorporating Developmental Changes in Hepatic OCT1 and UGT2B7 Proteins to Explain the Variability in Clearances in Neonates and Small Infants. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Verscheijden, L.F.M.; Litjens, C.H.C.; Koenderink, J.B.; Mathijssen, R.H.J.; Verbeek, M.M.; de Wildt, S.N.; Russel, F.G.M. Physiologically based pharmacokinetic/pharmacodynamic model for the prediction of morphine brain disposition and analgesia in adults and children. PLoS Comput. Biol. 2021, 17, e1008786. [Google Scholar] [CrossRef] [PubMed]
- McPhail, B.T.; Emoto, C.; Fukuda, T.; Butler, D.; Wiles, J.R.; Akinbi, H.; Vinks, A.A. Utilizing Pediatric Physiologically Based Pharmacokinetic Models to Examine Factors That Contribute to Methadone Pharmacokinetic Variability in Neonatal Abstinence Syndrome Patients. J. Clin. Pharmacol. 2020, 60, 453–465. [Google Scholar] [CrossRef]
- Michelet, R.; Van Bocxlaer, J.; Allegaert, K.; Vermeulen, A. The use of PBPK modeling across the pediatric age range using propofol as a case. J. Pharmacokinet. Pharmacodyn. 2018, 45, 765–785. [Google Scholar] [CrossRef]
- Kovar, L.; Schrapel, C.; Selzer, D.; Kohl, Y.; Bals, R.; Schwab, M.; Lehr, T. Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates. Pharmaceutics 2020, 12, 578. [Google Scholar] [CrossRef]
- van Hoogdalem, M.W.; Johnson, T.N.; McPhail, B.T.; Kamatkar, S.; Wexelblatt, S.L.; Ward, L.P.; Christians, U.; Akinbi, H.T.; Vinks, A.A.; Mizuno, T. Physiologically-Based Pharmacokinetic Modeling to Investigate the Effect of Maturation on Buprenorphine Pharmacokinetics in Newborns with Neonatal Opioid withdrawal Syndrome. Clin. Pharmacol. Ther. 2022, 111, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Shimizu, M.; Kamiya, Y.; Emoto, C.; Fukuda, T.; Yamazaki, H. Adult and infant pharmacokinetic profiling of dihydrocodeine using physiologically based pharmacokinetic modeling. Biopharm. Drug Dispos. 2019, 40, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Kovar, L.; Weber, A.; Zemlin, M.; Kohl, Y.; Bals, R.; Meibohm, B.; Selzer, D.; Lehr, T. Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients. Pharmaceutics 2020, 12, 908. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; Fukuda, T.; Johnson, T.N.; Adams, D.M.; Vinks, A.A. Development of a Pediatric Physiologically Based Pharmacokinetic Model for Sirolimus: Applying Principles of Growth and Maturation in Neonates and Infants. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, e17. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.; Bonner, J.J.; Johnson, T.N.; Neuhoff, S.; Ghazaly, E.A.; Gribben, J.G.; Boddy, A.V.; Veal, G.J. Development of a physiologically based pharmacokinetic model of actinomycin D in children with cancer. Br. J. Clin. Pharmacol. 2016, 81, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Idkaidek, N.; Hamadi, S.; Bani-Domi, R.; Al-Adham, I.; Alsmadi, M.; Awaysheh, F.; Aqrabawi, H.; Al-Ghazawi, A.; Rabayah, A. Saliva versus Plasma Therapeutic Drug Monitoring of Gentamicin in Jordanian Preterm Infants. Development of a Physiologically-Based Pharmacokinetic (PBPK) Model and Validation of Class II Drugs of Salivary Excretion Classification System. Drug Res. 2020, 70, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, N.; Ahmed, M.; Czejka, M.; Sharib, S.; Hassan, S.; Hassan, A. Pharmacokinetics of Midazolam in preterm neonates with an insight in brain Tissue: A PBPK approach. Pak. J. Pharm. Sci. 2022, 35, 1459–1465. [Google Scholar]
- Jiang, X.L.; Zhao, P.; Barrett, J.S.; Lesko, L.J.; Schmidt, S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e80. [Google Scholar] [CrossRef]
- Zhao, W.; Le Guellec, C.; Benjamin, D.K.; Hope, W.W.; Bourgeois, T.; Watt, K.M.; van den Anker, J.N.; Matrot, B.; Saxen, H.; Hoppu, K.; et al. First Dose in Neonates: Are Juvenile Mice, Adults and In Vitro-In Silico Data Predictive of Neonatal Pharmacokinetics of Fluconazole. Clin. Pharmacokinet. 2014, 53, 1005–1018. [Google Scholar] [CrossRef]
- Xu, R.; Tang, H.; Chen, L.; Ge, W.; Yang, J. Developing a physiologically based pharmacokinetic model of apixaban to predict scenarios of drug-drug interactions, renal impairment and paediatric populations. Br. J. Clin. Pharmacol. 2021, 87, 3244–3254. [Google Scholar] [CrossRef]
- Donovan, M.D.; Abduljalil, K.; Cryan, J.F.; Boylan, G.B.; Griffin, B.T. Application of a physiologically-based pharmacokinetic model for the prediction of bumetanide plasma and brain concentrations in the neonate. Biopharm. Drug Dispos. 2018, 39, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Olafuyi, O.; Abbasi, M.Y.; Allegaert, K. Physiologically based pharmacokinetic modelling of acetaminophen in preterm neonates-The impact of metabolising enzyme ontogeny and reduced cardiac output. Biopharm. Drug Dispos. 2021, 42, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yu, Z.; Liu, H.; Wang, X.; Li, H.; Yao, X.; Liu, D. Model-informed drug development: The mechanistic HSK3486 physiologically based pharmacokinetic model informing dose decisions in clinical trials of specific populations. Biopharm. Drug Dispos. 2023, 44, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.J.; Burt, H.; Johnson, T.N.; Whitaker, M.J.; Porter, J.; Ross, R.J. Development and verification of an endogenous PBPK model to inform hydrocortisone replacement dosing in children and adults with cortisol deficiency. Eur. J. Pharm. Sci. 2021, 165, 105913. [Google Scholar] [CrossRef] [PubMed]
- McGavin, J.K.; Goa, K.L. Ganciclovir: An update of its use in the prevention of cytomegalovirus infection and disease in transplant recipients. Drugs 2001, 61, 1153–1183. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Wu, F.; Moore, J.N.; Fisher, J.; Crentsil, V.; Gonzalez, D.; Zhang, L.; Burckart, G.J.; Wang, J. Assessing CYP2C19 Ontogeny in Neonates and Infants Using Physiologically Based Pharmacokinetic Models: Impact of Enzyme Maturation Versus Inhibition. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 158–166. [Google Scholar] [CrossRef]
- Abduljalil, K.; Jamei, M.; Rostami-Hodjegan, A.; Johnson, T.N. Changes in individual drug-independent system parameters during virtual paediatric pharmacokinetic trials: Introducing time-varying physiology into a paediatric PBPK model. AAPS J. 2014, 16, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Salem, F.; Small, B.G.; Johnson, T.N. Development and application of a pediatric mechanistic kidney model. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 854–866. [Google Scholar] [CrossRef]
- Pan, X.; Stader, F.; Abduljalil, K.; Gill, K.L.; Johnson, T.N.; Gardner, I.; Jamei, M. Development and Application of a Physiologically-Based Pharmacokinetic Model to Predict the Pharmacokinetics of Therapeutic Proteins from Full-term Neonates to Adolescents. AAPS J. 2020, 22, 76. [Google Scholar] [CrossRef]
- Edginton, A.N.; Schmitt, W.; Willmann, S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin. Pharmacokinet. 2006, 45, 1013–1034. [Google Scholar] [CrossRef]
- van der Heijden, J.E.M.; Freriksen, J.J.M.; de Hoop-Sommen, M.A.; van Bussel, L.P.M.; Driessen, S.H.P.; Orlebeke, A.E.M.; Verscheijden, L.F.M.; Greupink, R.; de Wildt, S.N. Feasibility of a Pragmatic PBPK Modeling Approach: Towards Model-Informed Dosing in Pediatric Clinical Care. Clin. Pharmacokinet. 2022, 61, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Fisher, J.W.; Yoshida, K.; Zhang, L.; Burckart, G.J.; Wang, J. Physiologically Based Pharmacokinetic Prediction of Linezolid and Emtricitabine in Neonates and Infants. Clin. Pharmacokinet. 2017, 56, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Abduljalil, K.; Pan, X.; Pansari, A.; Jamei, M.; Johnson, T.N. Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population. Clin. Pharmacokinet. 2020, 59, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Ince, I.; Dallmann, A.; Frechen, S.; Coboeken, K.; Niederalt, C.; Wendl, T.; Block, M.; Meyer, M.; Eissing, T.; Burghaus, R.; et al. Predictive Performance of Physiology-Based Pharmacokinetic Dose Estimates for Pediatric Trials: Evaluation with 10 Bayer Small-Molecule Compounds in Children. J. Clin. Pharmacol. 2021, 61 (Suppl. 1), S70–S82. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.J.; Aldibani, H.K.A.; Rostami-Hodjegan, A. In-depth analysis of patterns in selection of different physiologically based pharmacokinetic modeling tools: Part I—Applications and rationale behind the use of open source-code software. Biopharm. Drug Dispos. 2023, 44, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Aldibani, H.K.A.; Rajput, A.J.; Rostami-Hodjegan, A. In-depth analysis of patterns in selection of different physiologically-based pharmacokinetic modeling tools: Part II—Assessment of model reusability and comparison between open and non-open source-code software. Biopharm. Drug Dispos. 2023, 44, 292–300. [Google Scholar] [CrossRef] [PubMed]
- El-Khateeb, E.; Burkhill, S.; Murby, S.; Amirat, H.; Rostami-Hodjegan, A.; Ahmad, A. Physiological-based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20-years; in-depth analysis of applications, organizations, and platforms. Biopharm. Drug Dispos. 2021, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- T’Jollyn, H.; Vermeulen, A.; Van Bocxlaer, J. PBPK and its Virtual Populations: The Impact of Physiology on Pediatric Pharmacokinetic Predictions of Tramadol. AAPS J. 2019, 21, 8. [Google Scholar] [CrossRef]
- Dinh, T.H.; Delaney, K.P.; Goga, A.; Jackson, D.; Lombard, C.; Woldesenbet, S.; Mogashoa, M.; Pillay, Y.; Shaffer, N. Impact of Maternal HIV Seroconversion during Pregnancy on Early Mother to Child Transmission of HIV (MTCT) Measured at 4–8 Weeks Postpartum in South Africa 2011–2012: A National Population-Based Evaluation. PLoS ONE 2015, 10, e0125525. [Google Scholar]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T.; et al. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar] [CrossRef]
- Zhuravel, S.V.; Khmelnitskiy, O.K.; Burlaka, O.O.; Gritsan, A.I.; Goloshchekin, B.M.; Kim, S.; Hong, K.Y. Nafamostat in hospitalized patients with moderate to severe COVID-19 pneumonia: A randomised Phase II clinical trial. EClinicalMedicine 2021, 41, 101169. [Google Scholar] [CrossRef] [PubMed]
- van Donge, T.; Pfister, M.; Bielicki, J.; Csajka, C.; Rodieux, F.; van den Anker, J.; Fuchs, A. Quantitative Analysis of Gentamicin Exposure in Neonates and Infants Calls into Question Its Current Dosing Recommendations. Antimicrob. Agents Chemother. 2018, 62, e02004-17. [Google Scholar] [CrossRef] [PubMed]
- Dallos, P.; Wang, C.Y. Bioelectric correlates of kanamycin intoxication. Audiology 1974, 13, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.; McGee, T.J. Development of hearing loss in kanamycin treated chinchillas. Ann. Otol. Rhinol. Laryngol. 1977, 86, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Huth, M.E.; Ricci, A.J.; Cheng, A.G. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011, 2011, 937861. [Google Scholar] [CrossRef] [PubMed]
- Contrepois, A.; Brion, N.; Garaud, J.J.; Faurisson, F.; Delatour, F.; Levy, J.C.; Deybach, J.C.; Carbon, C. Renal disposition of gentamicin, dibekacin, tobramycin, netilmicin, and amikacin in humans. Antimicrob. Agents Chemother. 1985, 27, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.B.; Cohen-Wolkowiez, M.; Castro, L.M.; Poindexter, B.; Bidegain, M.; Weitkamp, J.H.; Schelonka, R.L.; Ward, R.M.; Wade, K.; Valencia, G.; et al. Population pharmacokinetics of meropenem in plasma and cerebrospinal fluid of infants with suspected or complicated intra-abdominal infections. Pediatr. Infect. Dis. J. 2011, 30, 844–849. [Google Scholar] [CrossRef]
- Lim, S.Y.; Miller, J.L. Ampicillin Dose for Early and Late-Onset Group B Streptococcal Disease in Neonates. Am. J. Perinatol. 2022, 39, 717–725. [Google Scholar] [CrossRef]
- Ecker, K.L.; Donohue, P.K.; Kim, K.S.; Shepard, J.A.; Aucott, S.W. The impact of group B Streptococcus prophylaxis on early onset neonatal infections. J. Neonatal Perinat. Med. 2013, 6, 37–44. [Google Scholar] [CrossRef]
- Castagnola, E.; Jacqz-Aigrain, E.; Kaguelidou, F.; Maragliano, R.; Stronati, M.; Rizzollo, S.; Farina, D.; Manzoni, P. Fluconazole use and safety in the nursery. Early Hum. Dev. 2012, 88 (Suppl. 2), S11–S15. [Google Scholar] [CrossRef]
- Tamimi, J.J.; Salem, I.I.; Alam, S.M.; Zaman, Q.; Dham, R. Bioequivalence evaluation of two brands of lisinopril tablets (Lisotec and Zestril) in healthy human volunteers. Biopharm. Drug Dispos. 2005, 26, 335–339. [Google Scholar] [CrossRef]
- Hirota, K.; Yoshioka, H.; Kabara, S.; Kudo, T.; Ishihara, H.; Matsuki, A. A comparison of the relaxant effects of olprinone and aminophylline on methacholine-induced bronchoconstriction in dogs. Anesth. Analg. 2001, 93, 230–233. [Google Scholar] [CrossRef]
- Armanian, A.M.; Badiee, Z.; Afghari, R.; Salehimehr, N.; Hassanzade, A.; Sheikhzadeh, S.; Sharif Tehrani, M.; Rezvan, G. Prophylactic aminophylline for prevention of apnea at higher-risk preterm neonates. Iran. Red Crescent Med. J. 2014, 16, e12559. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Edginton, A.N.; Fotaki, N. Assessment of Age-Related Changes in Pediatric Gastrointestinal Solubility. Pharm. Res. 2016, 33, 52–71. [Google Scholar] [CrossRef] [PubMed]
| Parameters | Population | Main Performance |
|---|---|---|
| Gastric pH | Preterm neonates | 1. Relatively higher than term infants [13]. |
| Neonates | 1. Drops from approximately 7 to approximately 2 after birth, then rises to above 4 [42]. | |
| Infants | 1. Declines back to approximately 2 in two years [42]. | |
| Adults | 1. Approximately 1–2. | |
| Gastric volume | Neonates | 1. Decreased compared with older children and adults. |
| Gastric motility | Highly preterm neonates | 1. Lower than full-term neonates and infants [43]. |
| Term neonates | 1. Slower than that in older children and adults, and matures rapidly after birth [43]. | |
| Adults | 1. Biphasic emptying [51]. | |
| Intestinal pH | Neonates/infants | 1. 6.6 ± 0.4 for duodenal pH; 6.6 ± 0.4 and 6.8 ± 0.7 for the pH of jejunum and ileum, respectively [52]. |
| Adults | 1. Slightly lower than neonates [53]. | |
| Intestinal transit time | Preterm newborns | 1. Approximately four-fold that of term infants [48]. |
| Term neonates | 1. Approximately four hours proven by an in vitro model [48]. | |
| Adults | 1. Approximately four hours. | |
| Intestinal surface area | Neonates/infants/children | 1. Reduced surface-to-volume ratio compared with adults [54]. |
| Intestinal permeability | Preterm neonates | 1. The intestinal permeability in preterm neonates (GW 26–36) was higher than full-term newborns [55]. |
| Intestinal microbiome | Preterm neonates | 1. Reduced microbial diversity and increased pathogenic organism colonization vs. term neonates [56]. |
| Term neonates | 1. Immature and matures rapidly during the first year of life [56]. | |
| Intestinal fluid composition | Neonates/infants | 1. Lower bile acid and salt concentration, no secondary bile salts, and higher total protein and lipid concentrations compared to adults [57]. |
| Digestive enzyme secretion | Preterm neonates | 1. Enterokinase secretion at GW 24 is approximately 25% of the values of older infants [41]. 2. Lactase activity at GW 34 is only 30% of the levels in term neonates [58]. |
| Term neonates | 1. Trypsin secretion reaches approximately 90% of childhood levels at term [59]. 2. Pepsin expression is not completed in neonates and matures with age [48]. 3. Pancreatic triglyceride lipase is lower in neonates than in adults [60]. | |
| Intestinal P-gp | Preterm neonates | 1. Expression is lower than term infants, children, and adults [49]. |
| Term neonates | 1. Lower than adults [49]. | |
| Intestinal CYP3A4 | Neonates | 1. Low expression after birth and increases from neonates to adults [46,50]. |
| Parameters | Population | Main Performance |
|---|---|---|
| Total body water (%) | Preterm neonates | As high as 90% of body weight [61,62,63]. |
| Term neonates | Higher than adults (80–85%) [61,62,63]. | |
| Adults | Approximately 60–65% of body weight [61,62,63]. | |
| Total body fat (%) | Preterm neonates | 3% in a premature neonate with deficient birth weight [63,64,65]. |
| Term neonates | 12% in full-term babies [63,64,65]. | |
| Weight loss | Preterm neonates | A 10–15% weight loss at the end of first week [61]. |
| Term neonates | A 5–7% weight loss at the end of first week [61]. | |
| HAS levels | Preterm neonates | The mean albumin level in preterm infants (GW 23–34) is 2.36 g/dL [68]. |
| Term neonates | The mean albumin level in full-term neonates is 3.43 g/dL [68]. | |
| Adults | The mean albumin level in adults is 4.0 g/dL [74]. | |
| AAG levels | Preterm newborns | Stay stable at a low level until 260 days of GA and significantly increase afterward [69]. |
| Term neonates | About 50% of adult level [75]. |
| Transporters | Measure Methods | Expression Related to Age |
|---|---|---|
| Hepatic Transporters | ||
| MDR1 | Quantitative proteomics | 1. P-gp expression was significantly lower in neonatal or infant livers and increased with age [161]. 2. Lower expression in neonates than in adults, whereas no difference between preterm and term newborns [162]. |
| Western blot | 1. Lower expression in S9 liver fractions from children (seven days to 18 years old) than that from adults [163]. | |
| Gene expression | 1. Increase in the first years [164,165]. | |
| BCRP | Quantitative proteomics | 1. Stable from neonate to adults [161,162]. |
| Western blot | 1. Stable expression in neonates and adults [72]. | |
| Immunohistochemistry | 1. Detected in fetuses at GW 5.5 [157]. | |
| Gene expression | 1. Gene expressed in fetuses is 3-fold lower than that in adults [166]. 2. Expression in neonates is lower than children > 7 years [159]. | |
| OATP1B1 | Quantitative proteomics | 1. High expression in fetuses and low expression in term neonates [162]. 2. No age-dependent changes [161]. |
| Gene expression | 1. Expression in adults > fetuses > neonates [50]. | |
| OATP1B3 | Quantitative proteomics | 1. Stable from fetuses to adults [162]. 2. Expression is lower in neonates than in adults and increased with age [161]. |
| Immunohistochemistry | 1. Expressed in early childhood; increases with age [157]. | |
| Gene expression | 1. Higher in adults than in fetuses and neonates [50]. | |
| OATP2B1 | Quantitative proteomics | 1. Stable from neonates to adults [161,162]. |
| Immunohistochemistry | 1. Expressed in early childhood, overexpression in neonates and young infants [157]. | |
| Gene expression | 1. Gene expression is much higher in adults than in fetuses and neonates [50]. | |
| NTCP | Quantitative proteomics | 1. Stable from neonates to adults [161]. 2. Lower expression in preterm neonates than that in adults [162]. |
| Western blot | 1. Similar expression in neonates and adults [72]. | |
| Gene expression | 1. Lower gene expression in neonates than in adults [50]. | |
| OCT1 | Quantitative proteomics | 1. Lower expression in term neonates than in adults [162]. 2. Lower expression in neonates than in young infants and increases to adult age [161]. |
| Western blot | 1. Low expression in newborns, increases from birth up to 8–12 years old [167]. | |
| MRP2 | Quantitative proteomics | 1. Lower expression in term neonates than in adults [162]. 2. No age-dependent changes in expression [161]. |
| Gene expression | Lower gene expression in fetuses and neonates than that in adults [50]. | |
| MRP3 | Quantitative proteomics | 1. Lower expression in term neonates than in adults [162]. 2. Lower expression in infants and adolescents than that in adults [161]. |
| Gene expression | 1. Lower gene expression in fetuses than that in adults [166]. | |
| MRP1 | Quantitative proteomics | 2. Lower expression in term neonates than in adults [162]. |
| Immunohistochemistry | 1. Detectable in fetal livers [157]. | |
| MRP4 | Gene expression | 1. No age-dependent changes in expression [166]. |
| MRP6 | Gene expression | 1. Expression increases in an age-dependent manner from neonates to adults [159]. |
| BSEP | Quantitative proteomics | 1. Lower expression in fetuses and term neonates than in adults [162]. 2. No age-dependent changes in expression [161]. |
| Immunohistochemistry | 1. Detectable in second-trimester fetuses [168]. | |
| Gene expression | 1. Lower expression in fetuses and neonates than that in adults [159,166]. | |
| MATE1 | Immunohistochemistry | 1. No age-dependent changes in protein abundance [157]. |
| Gene expression | 1. Expression increase in an age-dependent behavior [159]. | |
| GLUT1 | Quantitative proteomics | 1. Higher in fetal livers than in term neonates, children, and adults [162]. |
| MCT1 | Quantitative proteomics | 1. Stable expression from fetuses to adults [162]. |
| ATP1A1 | Quantitative proteomics | 1. Stable expression from fetuses to adults [162]. 2. Lower expression in neonates and increases with age [161]. |
| Renal Transporters | ||
| MDR1 | Quantitative proteomics | 1. Lowest protein abundance in neonates and reaches adult level during childhood (2–12 yr) [169]. |
| Immunohistochemistry | 1. Detectable in the kidney by GW 5.5 [157]. | |
| Gene expression | 1. Expression in premature and/or term newborns was significantly lower than in the older age groups, no difference between preterm and term newborns [169]. | |
| BCRP | Quantitative proteomics | 1. Protein abundance is similar between neonates and adults [169]. |
| Immunohistochemistry | 1. High in newborns and reduces with age [157]. | |
| Gene expression | 1. mRNA expression is higher in term newborns than in children and adolescents [169]. | |
| MRP1 | Immunohistochemistry | 1. Detectable in the kidney by GW 5.5 [157]. |
| MRP2 | Gene expression | 1. Expression is similar between all age groups (preterm newborn, term newborn, infants, children, adolescents, and adults [169]. |
| MRP4 | Immunohistochemistry | 1. Detectable in the kidney by GW 27 [169]. |
| Gene expression | 1. Expression is similar between all age groups (preterm newborn, term newborn, infants, children, adolescents, and adults [169]. | |
| OCT2 | Quantitative proteomics | 1. Protein abundance was lower in term neonates and infants than in older populations [169]. |
| Gene expression | 1. Expression in premature and/or term newborns was significantly lower than in older age groups [169]. | |
| OAT1 | Quantitative proteomics | 1. Protein abundance is lowest in term newborns and infants, approaching adult levels in children or adolescents [169]. |
| Gene expression | 1. Expression in premature and/or term newborns was significantly lower than in older age groups [169]. | |
| OAT3 | Quantitative proteomics | 1. Protein abundance is lowest in term newborns and infants, reaching adult levels in adolescence [169]. |
| Gene expression | 1. Expression in premature and/or term newborns was significantly lower than in older age groups [169]. | |
| URAT1 | Quantitative proteomics | 1. Protein abundance is lower in term newborns and infants, reaching adult levels during childhood [169]. |
| Gene expression | 1. mRNA expression in infants and children is higher in term neonates and adults [169]. | |
| MATE1 | Quantitative proteomics | 1. No age-dependent changes in expression [169]. |
| Gene expression | 1. No age-dependent changes in expression [169]. | |
| Transporters in the blood–brain barrier | ||
| MDR1 | Immunohistochemistry | 1. Low expression at birth (approximately 30% to 50% of adults), increases with postnatal maturation, reaching adult levels at around 3–6 months [170]. |
| Intestinal transporters | ||
| MDR1 | Quantitative proteomics | 1. Similar levels in preterm newborns, full-term neonates, and adults [161,162]. |
| Immunohistochemistry | 1. Similar between children of different ages and adults [157]. | |
| Gene expression | 1. Detectable at GW 14-22 [171]. 2. Expression in duodenal and jejunal was stable in children from 1 month to adulthood [49]. 3. Expression in neonatal and infant intestines is similar to that in adult intestines [50]. | |
| MRP2 | Immunohistochemistry | 1. Similar between children of different ages and adults [157]. |
| Gene expression | 1. Stable in expression from neonates to adults [50]. | |
| OATP2B1 | Quantitative proteomics | 1. Similar levels in preterm newborns, full-term neonates, and adults [162]. |
| Immunohistochemistry | 1. Higher expression in neonates and young infants than in adults [157]. | |
| Gene expression | 2. Higher expression in neonates than in adults [50]. | |
| Study Types/Ref. | Drugs | Platform | Main Outcomes |
|---|---|---|---|
| Dose Optimization/Regimens/Selection | |||
| [185] | Dolutegravir | MATLAB SimBiology | 1. Recommended regimen for neonates: Day 1–20 = 5 mg every 48 h (q48h); Day 21–28 = 5 mg every 24 h (q24h). 2. If the mother has taken dolutegravir 2–24 h before delivery, the first dose for the newborn may be delayed until 24–48 h after birth. |
| [186] | Gentamicin | Simcyp® | 1. This model suggested that a higher dose with an extended-dosing interval (5 mg/kg q36h) was beneficial for neonates with PMA 30–34 weeks PNA 8–28 days and PMA ≥ 35 weeks PNA 0–7 days. |
| [187] | Gentamicin | PhysPK® | 1. Extended-interval dosing regimens (6 mg/kg q36h and 6 mg/kg q48h for term and preterm neonates) were recommended because of higher efficacy and lower toxicity. |
| [188] | Ampicillin | Simcyp® | 1. Recommended dosing regimens: 50 mg/kg q8h for neonates; 1 g ampicillin for a duration ≤ 4 h before delivery for intrapartum prophylaxis. |
| [189] | Clindamycin | PK-Sim® | 1. The optimal dosing supported by this PBPK model was 9 mg/kg/dose for children ≤ 5 months of age, 12 mg/kg/dose for children > 5 months–6 years of age, and 10 mg/kg/dose for children 6–18 years of age, all administered every 8 h. |
| [190] | Moxifloxacin | PK-Sim® | 1. Initial dosing regimen based on PBPK model: 5 mg/kg for school children (6–14 years), 7 mg/kg for pre-school children (2–6 years), and 9 mg/kg for infants and toddlers (3 months–2 years). 2. The popPK model suggested that an alternative dosing regimen (200 mg q12h i.v.) can be developed for children aged 12–18 years. |
| [191] | Valproic acid | Simcyp® | 1. Dosage recommendation (bid): 31.25 mg for neonate, 62.5 mg for infants; 125 mg for toddler and pre-schooler; 250 mg for school age; 375–500 mg for adolescent. |
| [192] | Acetaminophen | GastroPlusTM | 1. A proteomics-informed PBPK supported the FDA label dose of acetaminophen injection in neonates and infants. |
| [193] | Fluconazole | PK-sim® | 1. Target drug concentration in plasma and CSF was reached more quickly after using a 25 mg/kg loading dose in neonates. |
| [194] | Fluconazole | PK-sim® | 1. Loading dose recommendations for children on ECMO in the first 24 h of therapy: 30 mg/kg for neonates, 35 mg/kg for infants and children (2 years to <12 years), and 30 mg/kg for adolescents. |
| [195] | Docetaxel | Simcyp® | 1. The PBPK simulation suggested that the revised dose of docetaxel for a child > 1.5 years old was higher than the adult dose, while this was the same for children aged 1 to 1.5 years. |
| [7] | Propofol | Simcyp® | 1. The change in propofol clearance following CO reductions increased with age. 2. If CO was reduced by 40–50%, the dose of propofol should be reduced by 15% for newborns, infants, and children, and 25% for adolescents and adults. |
| [196] | Remdesivir | Simcyp® | 1. Dose recommendation: adult dosage regimen for pediatric patients ≥ 40 kg; a weight-based regimen for pediatric patients weighing 2.5 to <40 kg (5 mg/kg loading dose on Day 1 then 2.5 mg/kg daily maintenance dose starting on Day 2). |
| [197] | Tadalafil | Simcyp® | 1. A PBPK model was developed to explore tadalafil doses for children less than 2 years old: children aged birth to <1 month (2 mg), children aged 1 to <6 months (3 mg), children aged 6 months to <1 year (4 mg), and children aged 1 to <2 years (6 mg). |
| [198] | Aminophylline | PK-sim® | 1. PNA had a significant influence on clearance of the drug. 2. Dosing for neonates with renal underdevelopment should be conservative. |
| [33] | Meropenem | PK-Sim® | 1. This PBPK model supported the meropenem dosing regimens recommended in the product label for infants < 3 months of age with complicated intraabdominal. |
| [199] | Lisinopril | PK-Sim® | 1. 1.0 and 1.5 to 2.5 mg for neonates to infants and infants to toddlers; 5 and 10 mg for adolescents; 20 mg for adults. |
| [200] | Nafamostat | Simcyp® | 1. A whole-body PBPK model of nafamostat in adults was built and scaled to children including newborns, providing evidence for using a weight-based dosing regimen in pediatric COVID-19 patients. |
| [201] | Zidovudine | PK-sim® | 1. The preterm newborns of lesser GA have less capacity for drug clearance. 2. Zidovudine dosages in preterm infants may need to be adjusted for GA. |
| [9] | Carbamazepine | GastroPlusTM | 1. Dose reduction was recommended: 1.0 and 1.5 to 2.5 mg for neonates and infants to toddlers. |
| Special application of neonatal PBPK modeling | |||
| [202] | Oseltamivir | GastroPlusTM | 1. The generic drugs with 10% slower dissolution profile than the reference drugs could maintain BE in adults, while the dissolution boundary for pediatrics is restricted (6% slower for adolescents, 4% slower for 0–2-month neonates) to maintain BE. |
| Neonatal PBPK model establishment and simulation | |||
| [203] | Oseltamivir | Simcyp® | 1. The protein abundance data of hepatic CESs were imported into a pediatric PBPK model and the concentration of oseltamivir in infants (0–1 year of age) was successfully predicted. |
| [204] | Oseltamivir | GastroPlusTM | 1. The exposure of intravenous oseltamivir in neonates was 3-fold higher than those observed with the same oral doses. |
| [205] | Morphine | Simcyp® | 1. The clearance of morphine increased from preterm to term to post-term neonates. 2. OCT1 genotype influences morphine clearance in term and post-term neonates. |
| [206] | Morphine | Simcyp® | 1. Morphine clearance in neonates was more sensitive to developmental changes in UGT2B7 activity. |
| [207] | Morphine | Simcyp® | 1. The variation in BBB expression of P-gp transporter was not responsible for differences in brain exposure of the drug. 2. The PBPK-PD modeling suggested that neonates were more sensitive to morphine than adults and older children. |
| [208] | Methadone | Simcyp® | 1. Changes in CYP2B6 and CYP3A4 activity, AGP, and MPPGL affected the drug clearance in neonates. 2. The effect of cardiac output on drug disposition was negligible. |
| [209] | Propofol | Simcyp® | 1. A PBPK model combining in vitro and in vivo data made a good prediction on clearance and concentration–time profiles of propofol across a broad age span from preterm to adults. |
| [210] | Buprenorphine | PK-Sim® | 1. A whole-body parent-metabolite PBPK model of buprenorphine for an adult and two pediatric populations (children aged 4.6–7.5 years and preterm neonates with PMA 27–34 weeks) was successfully developed and displayed an excellent predictive model performance. |
| [211] | Buprenorphine | Simcyp® | 1. The PK variability of buprenorphine in neonates was influenced by the extent of biliary clearance, oral mucosa absorption, and CYP3A4 abundance. |
| [212] | Dihydrocodeine | Simcyp® | 1. A full PBPK model of dihydrocodeine for healthy adults was established and scaled to pediatric patients with varied ages (from 1 month old to 14 years old). |
| [213] | Fentanyl | PK-Sim® | 1. A whole-body parent-metabolite PBPK model of fentanyl for adults was built and successfully scaled to several pediatric subpopulations (from preterm neonates to up to 3-year-old children). |
| [214] | Sirolimus | Simcyp® | 1. The maturation pattern of sirolimus clearance in pediatric patients was successfully investigated through a novel PBPK model. |
| [215] | Actinomycin D | Simcyp® | 1. A PBPK model of actinomycin D in infants and younger children was established with perfect predictive performance. |
| [216] | Gentamicin | PK-Sim® | 1. The simulation found a good correlation between plasma and saliva exposures, supporting saliva concentration as an alternative for TDM of gentamicin in premature infants. |
| [217] | Midazolam | PK-sim® | 1. The predicted midazolam disposition in preterm neonates was comparable to the observed data. 2. The MRT in neonatal brain was higher than the MRT in plasma. |
| [218] | Acetaminophen | Simcyp® | 1. A pediatric PBPK model integrating physiological changes and enzyme ontogeny successfully described the PK profiles from birth to adulthood (0–17 years). |
| [219] | Fluconazole | Simcyp® | 1. A model-based bridging approach (from juvenile mice/adults/in vitro–in silico data to neonates) successfully predicted fluconazole PK profiles in neonates. |
| [220] | Apixaban | Simcyp® | 1. Apixaban levels reduced with weight-normalized clearance in infants younger than 1 year old. |
| [221] | Bumetanide | Simcyp® | 1. This model suggested that the brain concentration of bumetanide in neonates enrolled in NEMO clinical trial was lower than therapeutic concentration. |
| [222] | Acetaminophen | Simcyp® | 1. The validated model found that a reduced CO by up to 30% did not affect drug disposition in preterm neonates. |
| [223] | HSK3486 | Simcyp® | 1. No change in the systemic exposure of HSK386 in neonates compared to that in adults. |
| [224] | Hydrocortisone | Simcyp® | 1. This hydrocortisone PBPK model successfully predicted PK parameters of both immediate- and modified-release hydrocortisone formulations in adults and pediatrics. |
| [101] | Theophylline and midazolam | MATLAB SimBiology | 1. Total clearance of these two drugs was very low in neonates and increased by 5 years old, then decreased in adults. |
| [225] | Ganciclovir and valganciclovir | GastroPlusTM | 1. The drug exposure in neonates was slightly under-predicted through this modeling. |
| [226] | Pantoprazole and esomeprazole | Simcyp® | 1. The interplay of CYP maturation and inhibition in neonates might be age-dependent. |
| [227] | Sildenafil and phenytoin | Simcyp® | 1. The PK of both sildenafil and phenytoin were predicted better at the end of a prolonged study using the time-varying compared to fixed PBPK models. |
| [228] | Cimetidine, ciprofloxacin, metformin, tenofovir, and zidovudine | Simcyp® | 1. Predictions in neonates and early infants (up to 14 weeks PNA) were reasonable. |
| [229] | Recombinant human erythropoietin, infliximab, etanercept, basiliximab, anakinra, and enfuvirtide | Simcyp® | 1. A PBPK model incorporating age-dependent physiology changes was developed to predict PK profiles of different therapeutic proteins in the pediatric population, including full-term neonates. |
| [8] | Theophylline, paracetamol, and ketoconazole | Simcyp® | 1. A pediatric absorption model integrating the available information on pediatric gastrointestinal physiology and its ontogeny was developed to predict oral drug absorption. |
| [230] | Paracetamol, alfentanil, morphine, theophylline, and levofloxacin | PK-Sim® | 1. The plasma concentration in neonates was greater than that in the adults, and the concentration in older children was less than that in the adults. 2. No age-dependent bias for term neonates to 18 years of age when examining volumes of distribution and t1/2. |
| [231] | Meropenem, ceftazidime, azithromycin, propofol, midazolam, lorazepam, and caffeine | Simcyp® | 1. The predictive performance of this PBPK model appeared to decrease in the (pre)term neonatal population. |
| [21] | Midazolam, caffeine, carbamazepine, cisapride, theophylline, diclofenac, omeprazole, S-warfarin, phenytoin, gentamicin, and vancomycin | Simcyp® | 1. The PBPK modeling using Simcyp® software was more accurate than that of simple allometry scaling, especially in small children (<2 years old) |
| [232] | Linezolid and emtricitabine | Simcyp® | 1. A PBPK model incorporating renal maturation ontogeny successfully predicted the PK of linezolid and emtricitabine in the pediatric population, including neonates. |
| [233] | Alfentanil, midazolam, caffeine, ibuprofen, gentamicin, and vancomycin | Simcyp® | 1. All PK parameter predictions of these six drugs for preterm neonates were within twofold error criteria. |
| [234] | Amikacin, ciprofloxacin, copanlisib, gadovist and magnevist, levonorgestrel, moxifloxacin, regorafenib, riociguat, and rivaroxaban | PK-Sim® | 1. The pediatric PBPK models successfully predict the PK profiles of 10 small-molecule compounds in children with different age. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Zhang, Q.; Cao, Z.; Zheng, L.; Hu, W. Physiologically Based Pharmacokinetic Modeling in Neonates: Current Status and Future Perspectives. Pharmaceutics 2023, 15, 2765. https://doi.org/10.3390/pharmaceutics15122765
Zhang W, Zhang Q, Cao Z, Zheng L, Hu W. Physiologically Based Pharmacokinetic Modeling in Neonates: Current Status and Future Perspectives. Pharmaceutics. 2023; 15(12):2765. https://doi.org/10.3390/pharmaceutics15122765
Chicago/Turabian StyleZhang, Wei, Qian Zhang, Zhihai Cao, Liang Zheng, and Wei Hu. 2023. "Physiologically Based Pharmacokinetic Modeling in Neonates: Current Status and Future Perspectives" Pharmaceutics 15, no. 12: 2765. https://doi.org/10.3390/pharmaceutics15122765
APA StyleZhang, W., Zhang, Q., Cao, Z., Zheng, L., & Hu, W. (2023). Physiologically Based Pharmacokinetic Modeling in Neonates: Current Status and Future Perspectives. Pharmaceutics, 15(12), 2765. https://doi.org/10.3390/pharmaceutics15122765