
Biomimetic Inorganic Nanovectors as Tumor-Targeting Theranostic Platform against Triple-Negative Breast Cancer

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Isolation of Cancer Cell Membranes
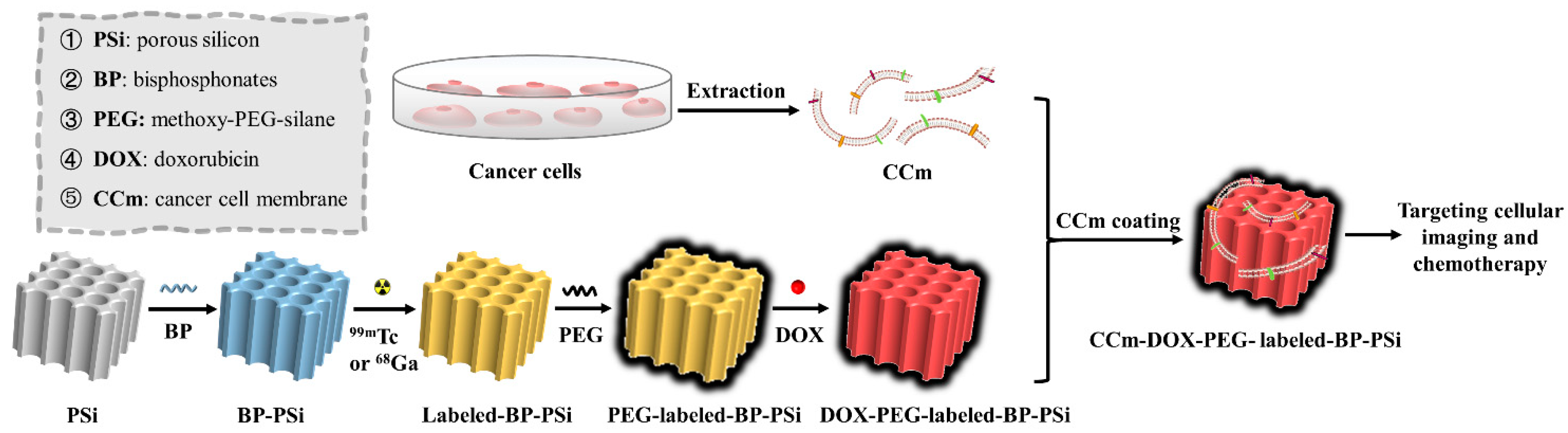
2.3. Preparation of Nanoparticles
2.4. Physicochemical Characterization
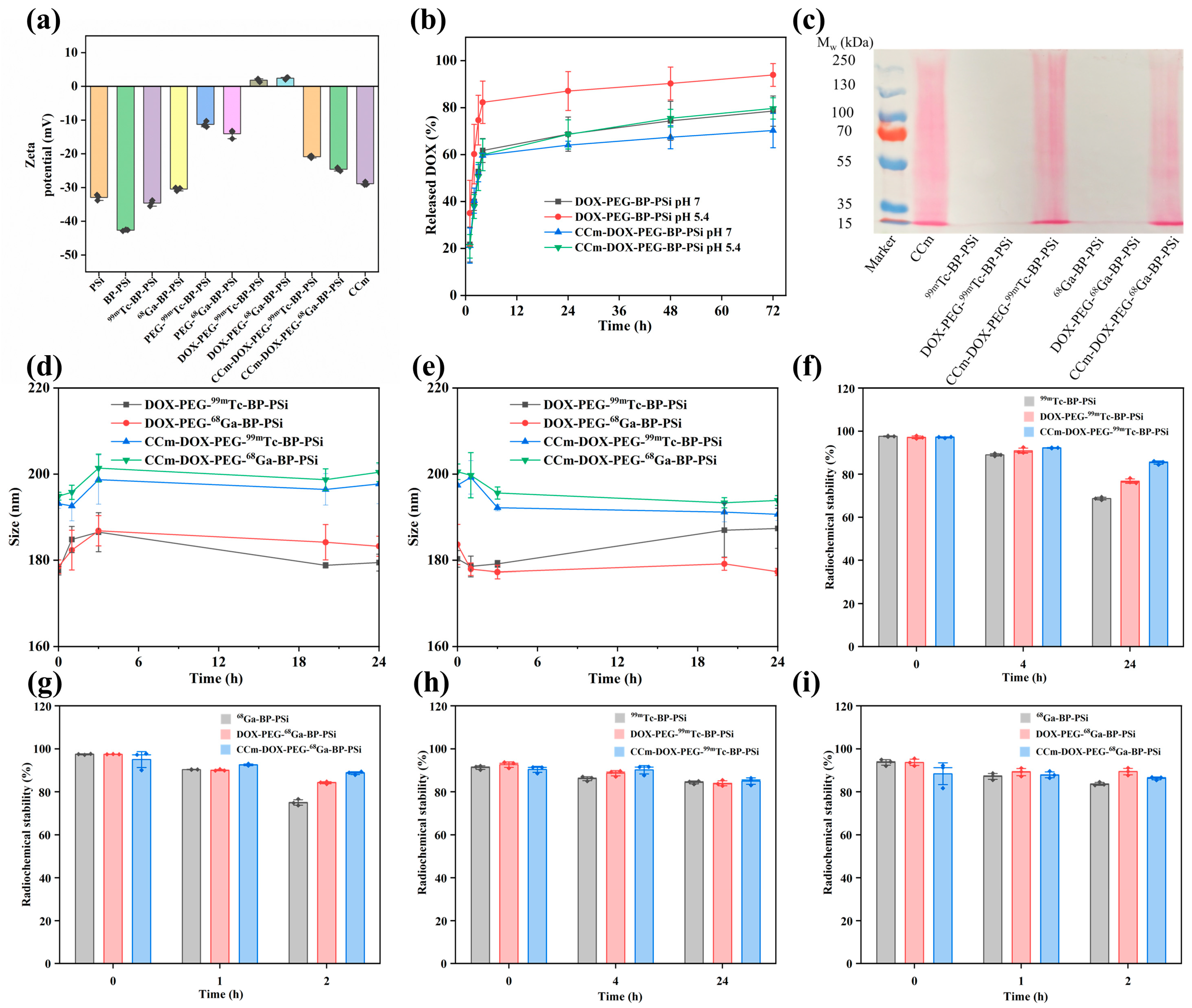
2.5. Drug Release
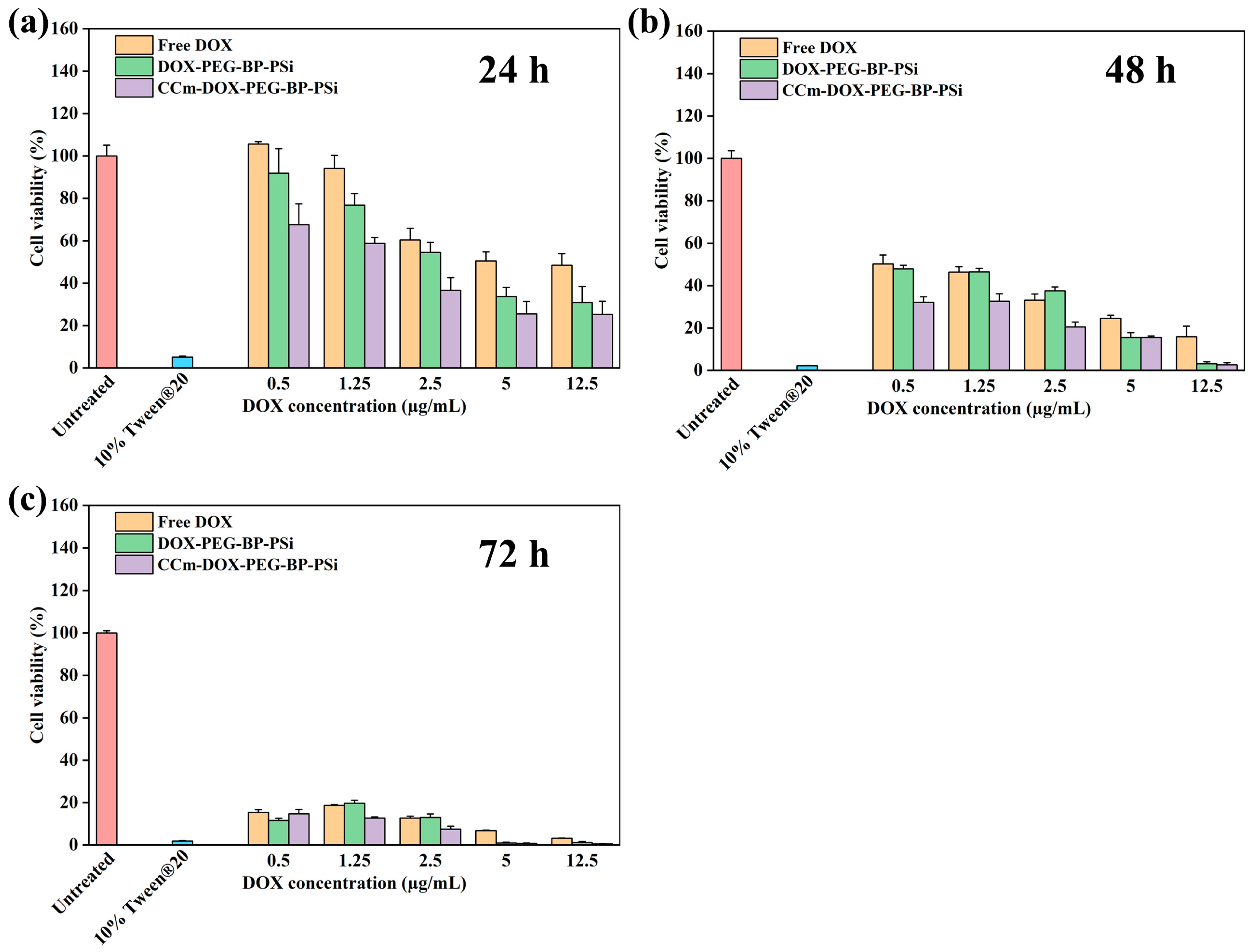
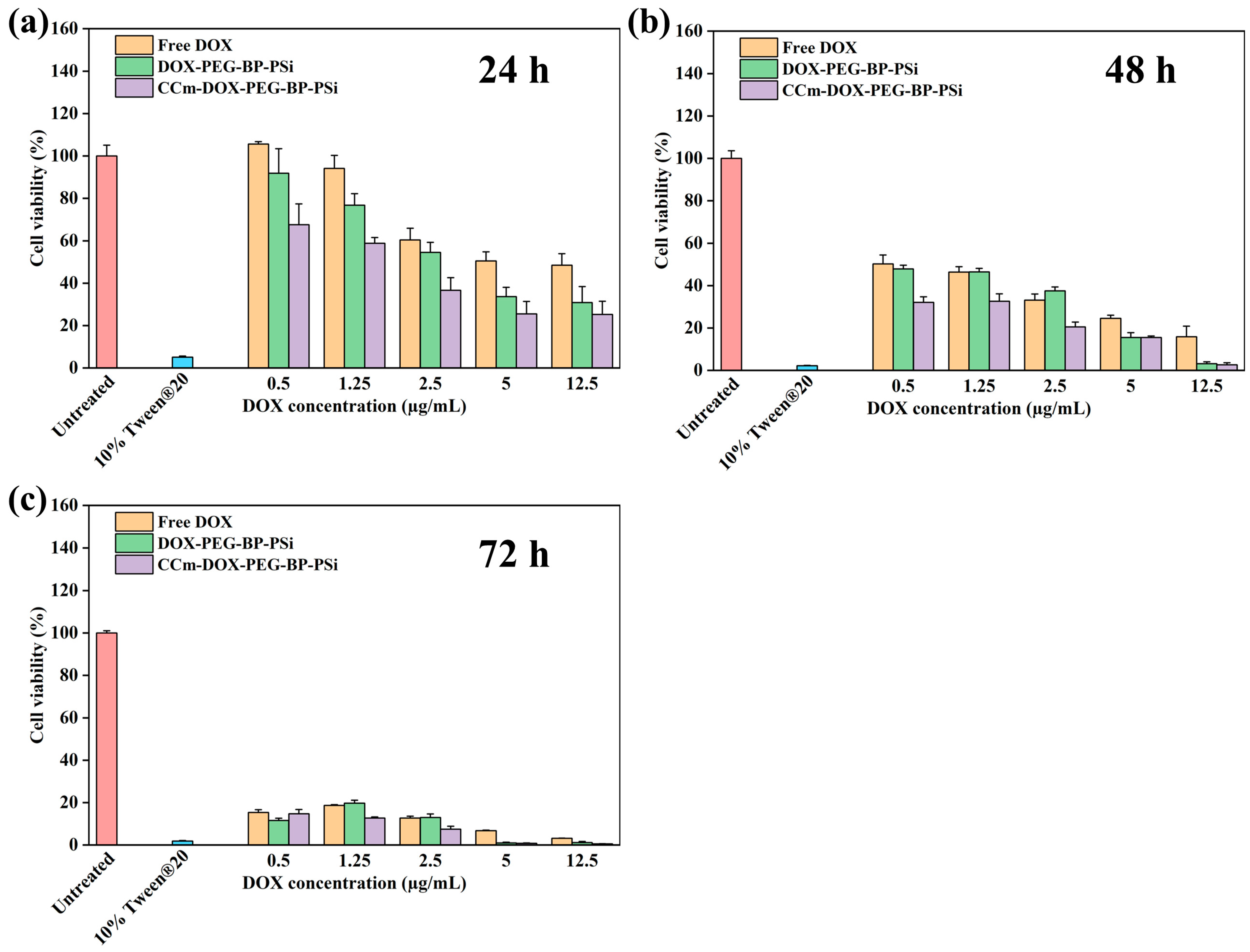
2.6. Cell Cytotoxicity
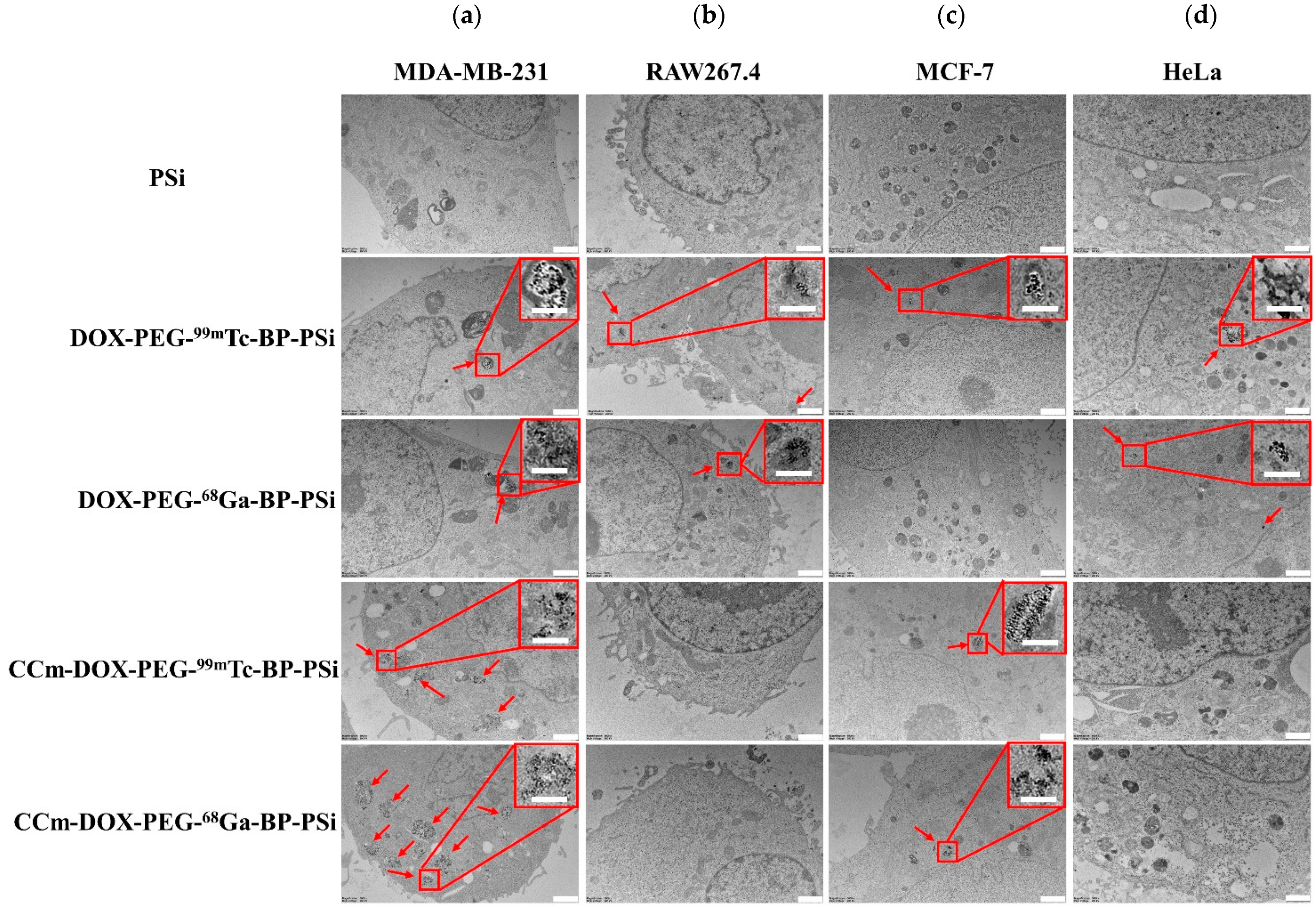
2.7. Homotypic Targeting Studies
3. Result and Discussion
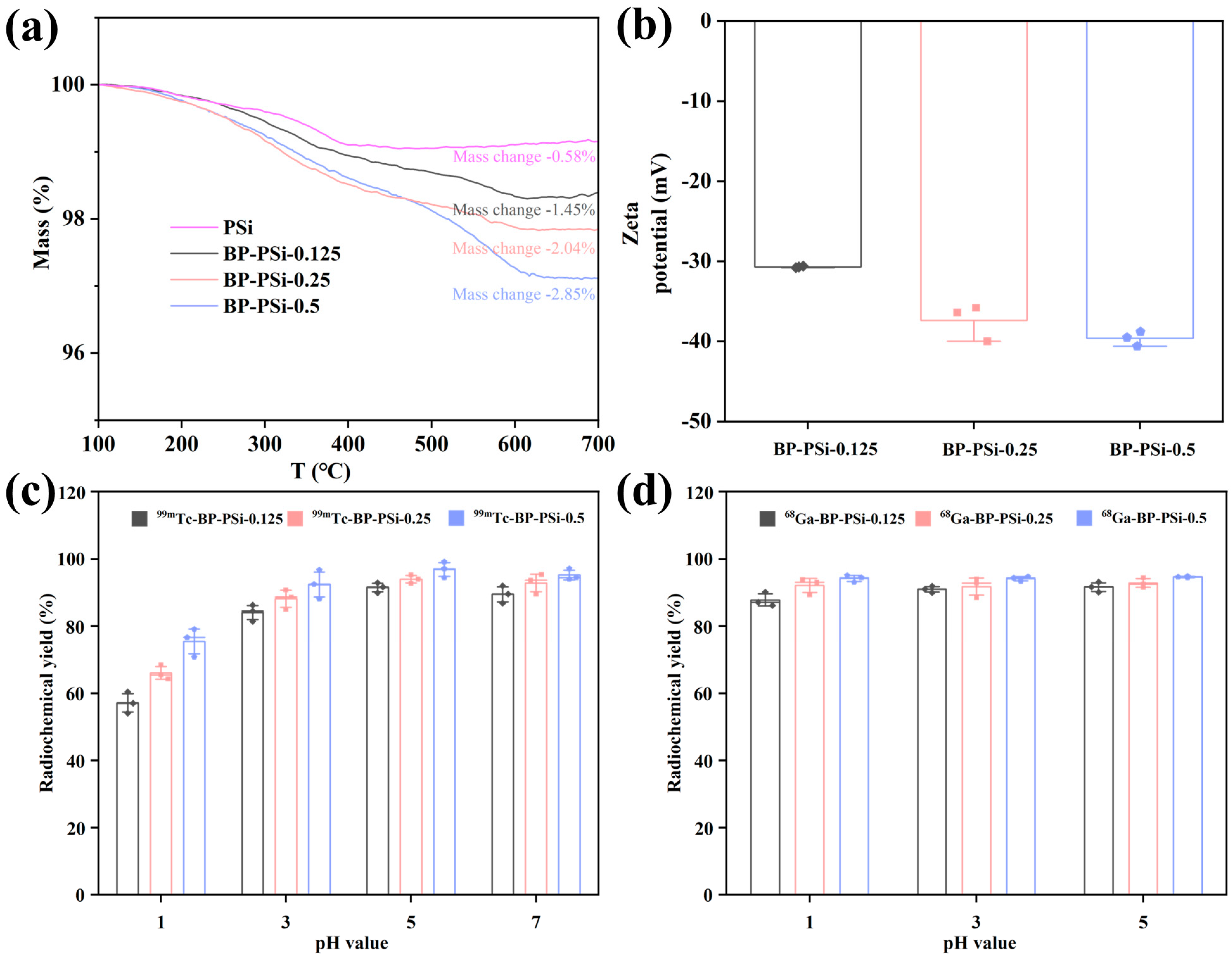
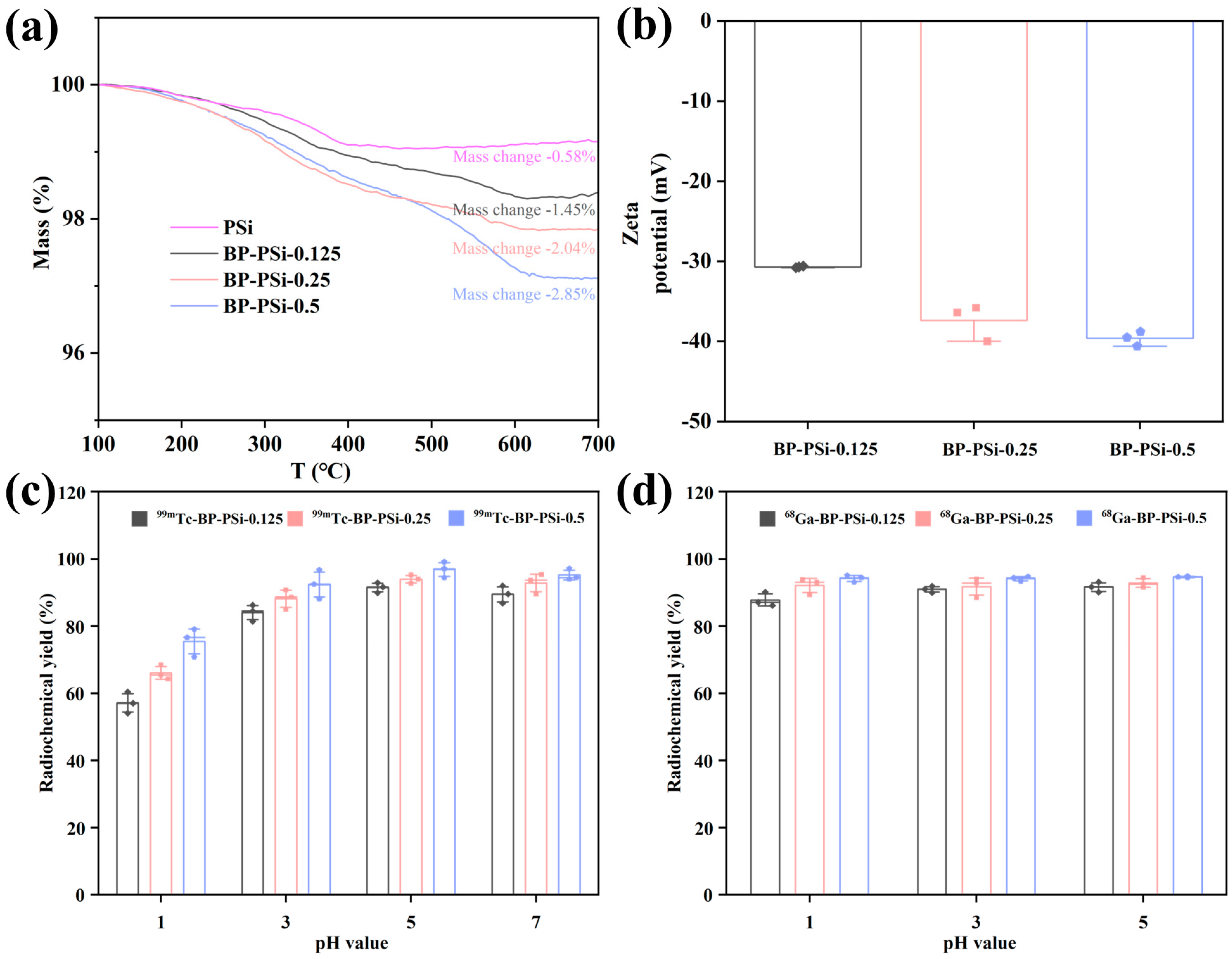
3.1. The Effects of BP Amount and pH Value on Radiolabeling
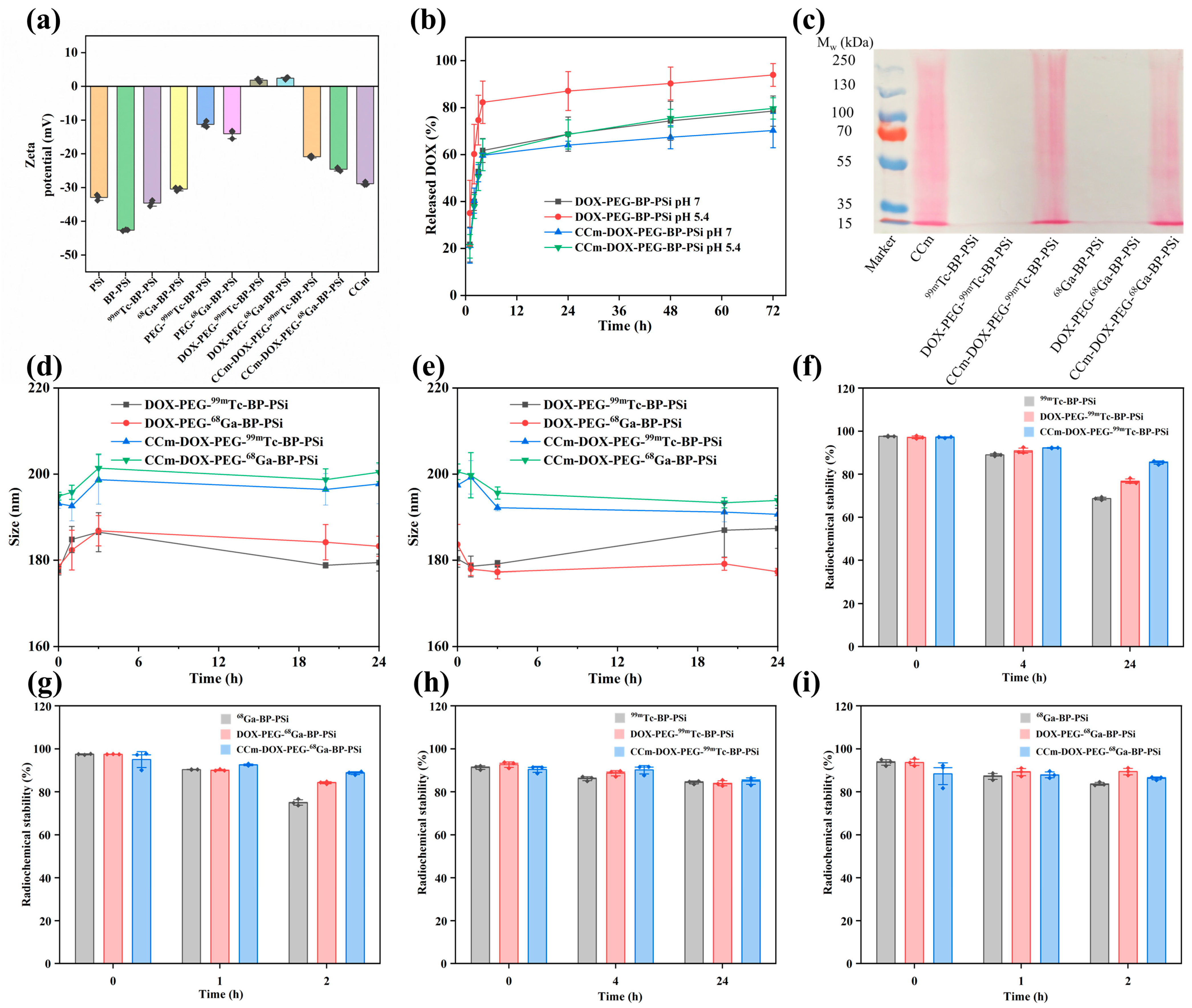
3.2. Physiochemical Characterizations of PSi-Based Platform
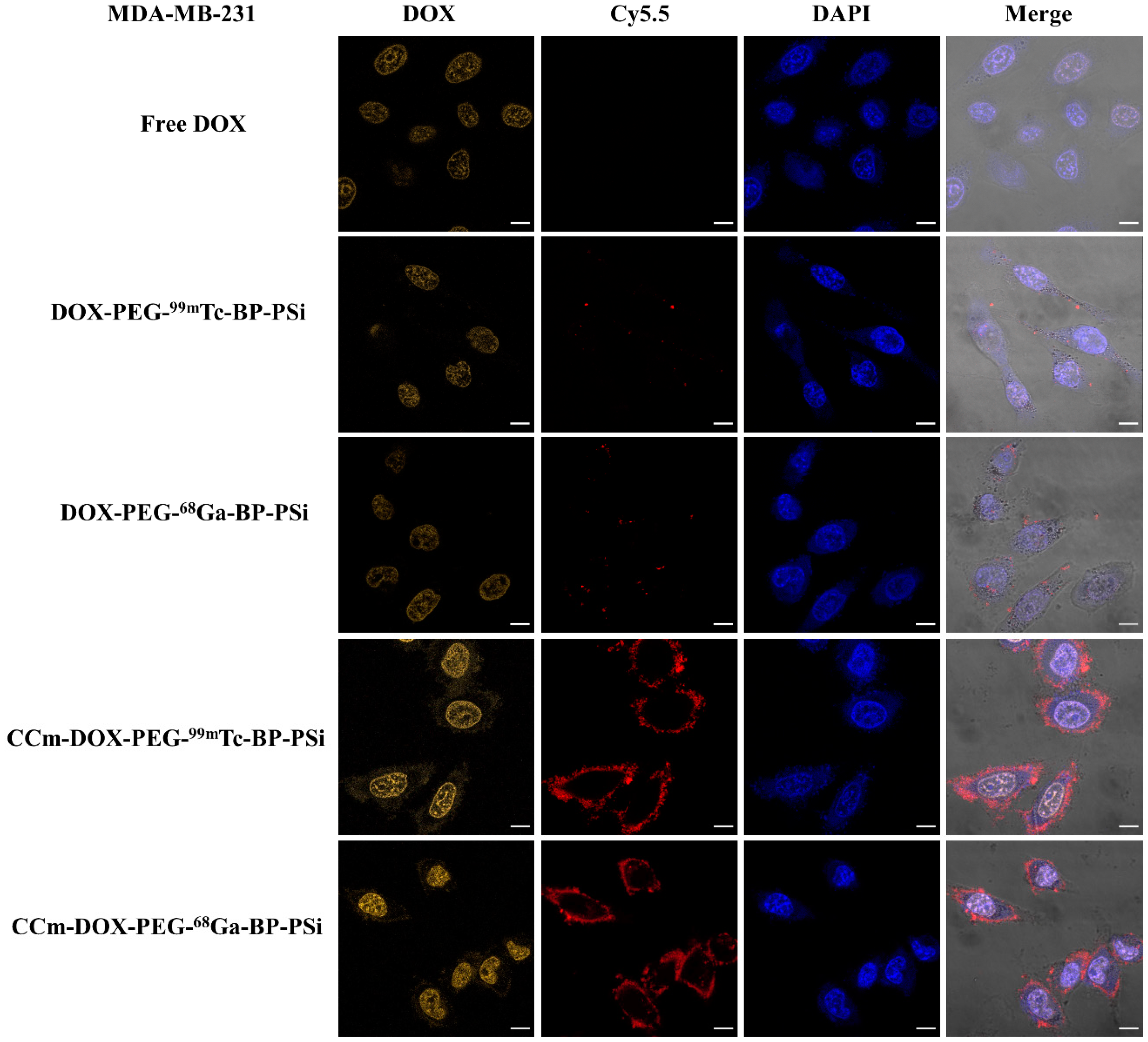
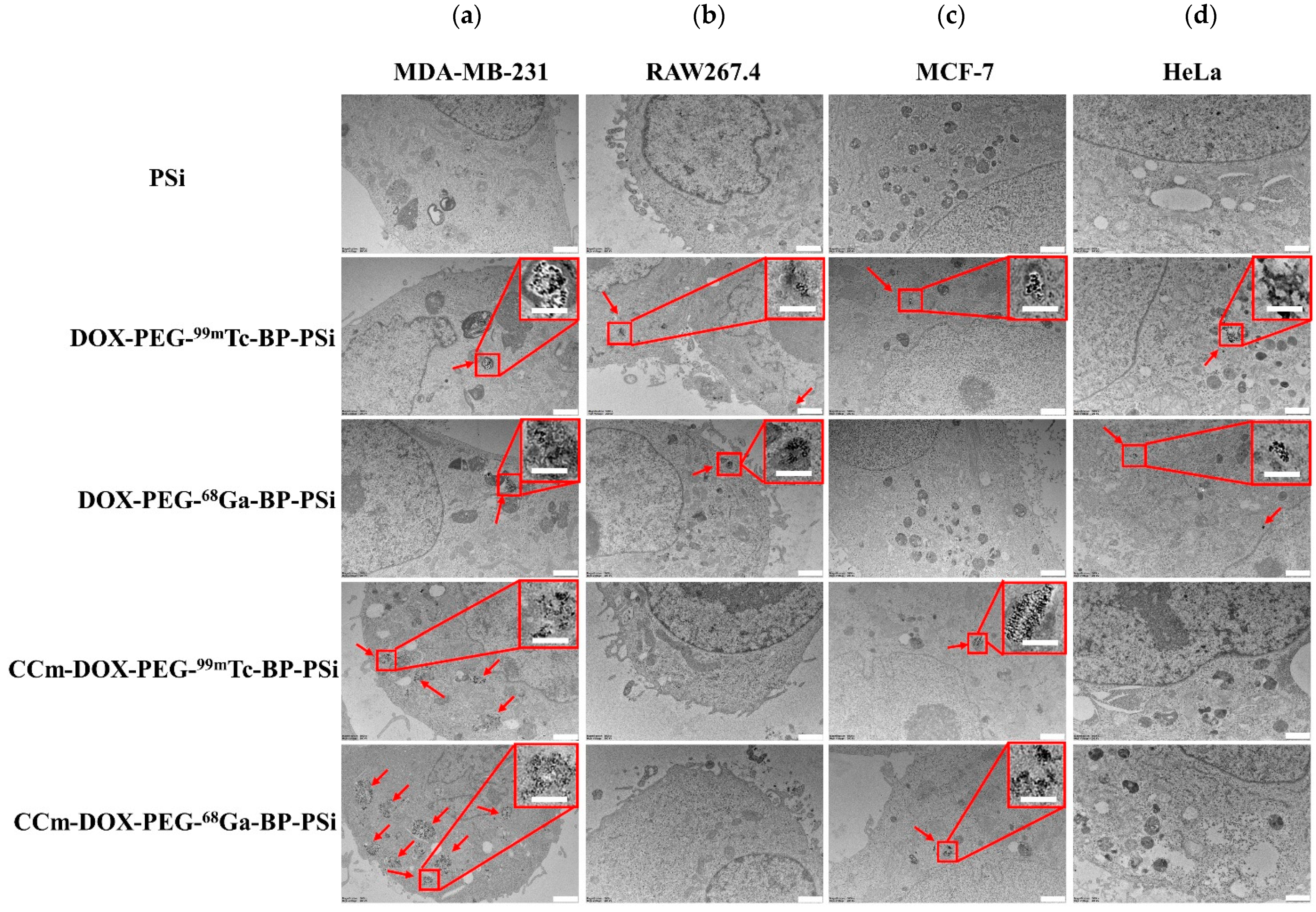
3.3. In Vitro Homotypic Targeting Experiments and Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, J.; Richard, S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol. Cell 2021, 81, 4357–4368. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-c. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Dang, Y.; Guan, J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater. Med. 2020, 1, 10–19. [Google Scholar] [CrossRef]
- Liu, L.; Bai, X.; Martikainen, M.-V.; Kårlund, A.; Roponen, M.; Xu, W.; Hu, G.; Tasciotti, E.; Lehto, V.-P. Cell membrane coating integrity affects the internalization mechanism of biomimetic nanoparticles. Nat. Commun. 2021, 12, 5726. [Google Scholar] [CrossRef]
- Liu, L.; Pan, D.; Chen, S.; Martikainen, M.-V.; Kårlund, A.; Ke, J.; Pulkkinen, H.; Ruhanen, H.; Roponen, M.; Käkelä, R. Systematic design of cell membrane coating to improve tumor targeting of nanoparticles. Nat. Commun. 2022, 13, 6181. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Dai, Z.; Li, J.; Yang, Y.; Xi, Z.; Wang, J.; Zhang, W.; Qian, K.; Guo, S.; Zhu, C. Cancer-cell-membrane-coated nanoparticles with a yolk–shell structure augment cancer chemotherapy. Nano Lett. 2019, 20, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, Z.; Pan, Q.; Bai, Y.; Yu, M.; Zhou, Y. Targeted Therapy of Oral Squamous Cell Carcinoma with Cancer Cell Membrane Coated Co-Fc Nanoparticles Via Autophagy Inhibition. Adv. Funct. Mater. 2023, 33, 2300235. [Google Scholar] [CrossRef]
- Pan, W.-L.; Tan, Y.; Meng, W.; Huang, N.-H.; Zhao, Y.-B.; Yu, Z.-Q.; Huang, Z.; Zhang, W.-H.; Sun, B.; Chen, J.-X. Microenvironment-driven sequential ferroptosis, photodynamic therapy, and chemotherapy for targeted breast cancer therapy by a cancer-cell-membrane-coated nanoscale metal-organic framework. Biomaterials 2022, 283, 121449. [Google Scholar] [CrossRef]
- Jia, L.; Li, X.; Liu, H.; Xia, J.; Shi, X.; Shen, M. Ultrasound-enhanced precision tumor theranostics using cell membrane-coated and pH-responsive nanoclusters assembled from ultrasmall iron oxide nanoparticles. Nano Today 2021, 36, 101022. [Google Scholar] [CrossRef]
- Fathy Abd-Ellatef, G.-E.; Gazzano, E.; Chirio, D.; Ragab Hamed, A.; Belisario, D.C.; Zuddas, C.; Peira, E.; Rolando, B.; Kopecka, J.; Assem Said Marie, M. Curcumin-loaded solid lipid nanoparticles bypass p-glycoprotein mediated doxorubicin resistance in triple negative breast cancer cells. Pharmaceutics 2020, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Pullan, J.; Dailey, K.; Bhallamudi, S.; Feng, L.; Alhalhooly, L.; Froberg, J.; Osborn, J.; Sarkar, K.; Molden, T.; Sathish, V. Modified bovine milk exosomes for doxorubicin delivery to triple-negative breast cancer cells. ACS Appl. Bio Mater. 2022, 5, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Zhang, Y.; Xu, Y.; Li, Y.; Xie, Z.; Wang, H.; Lin, Y.; Lin, Q.; Gong, T. Live macrophage-delivered doxorubicin-loaded liposomes effectively treat triple-negative breast cancer. ACS Nano 2022, 16, 9799–9809. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Näkki, S.; Wang, J.T.-W.; Wu, J.; Fan, L.; Rantanen, J.; Nissinen, T.; Kettunen, M.I.; Backholm, M.; Ras, R.H.; Al-Jamal, K.T. Designed inorganic porous nanovector with controlled release and MRI features for safe administration of doxorubicin. Int. J. Pharm. 2019, 554, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Helmi, O.; Elshishiny, F.; Mamdouh, W. Targeted doxorubicin delivery and release within breast cancer environment using PEGylated chitosan nanoparticles labeled with monoclonal antibodies. Int. J. Biol. Macromol. 2021, 184, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Williams, G.R.; Wu, J.; Wu, J.; Zhang, X.; Chen, X.; Li, S.; Jiao, J.; Zhu, L.-M. A chitosan-based cascade-responsive drug delivery system for triple-negative breast cancer therapy. J. Nanobiotechnol. 2019, 17, 95. [Google Scholar] [CrossRef]
- Schneible, J.D.; Young, A.T.; Daniele, M.; Menegatti, S. Chitosan hydrogels for synergistic delivery of chemotherapeutics to triple negative breast cancer cells and spheroids. Pharm. Res. 2020, 37, 142. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, T.; Sun, S.; Ren, W.; Wu, A.; Xu, H. Ultrasound-mediated cavitation enhances EGFR-targeting PLGA-PEG nano-micelle delivery for triple-negative breast cancer treatment. Cancers 2021, 13, 3383. [Google Scholar] [CrossRef]
- Mekseriwattana, W.; Phungsom, A.; Sawasdee, K.; Wongwienkham, P.; Kuhakarn, C.; Chaiyen, P.; Katewongsa, K.P. Dual Functions of Riboflavin-functionalized Poly (lactic-co-glycolic acid) Nanoparticles for Enhanced Drug Delivery Efficiency and Photodynamic Therapy in Triple-negative Breast Cancer Cells. Photochem. Photobiol. 2021, 97, 1548–1557. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, X.; Zhou, W.; Wu, Q.; Yan, J.; Xu, X.; Ghimire, B.; Rosenholm, J.M.; Feng, J.; Wang, D. Combination of photothermal, prodrug and tumor cell camouflage technologies for triple-negative breast cancer treatment. Mater. Today Adv. 2022, 13, 100199. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, H.; Li, L.; Guo, Z.; Song, J.; Yang, X.; Wan, G.; Li, R.; Wang, Y. Leukocyte/platelet hybrid membrane-camouflaged dendritic large pore mesoporous silica nanoparticles co-loaded with photo/chemotherapeutic agents for triple negative breast cancer combination treatment. Bioact. Mater. 2021, 6, 3865–3878. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Närvänen, A.; Jokivarsi, K.; Poutiainen, P.; Xu, W.; Lehto, V.-P. A robust approach to make inorganic nanovectors biotraceable. Int. J. Pharm. 2022, 624, 122040. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Tan, B.; Venkatakrishnan, K. Cell selective apoptosis induced by polymorphic alteration of self-assembled silica nanowebs. ACS Appl. Mater. Interfaces 2017, 9, 6292–6305. [Google Scholar] [CrossRef]
- Polyak, A.; Ross, T.L. Nanoparticles for SPECT and PET imaging: Towards personalized medicine and theranostics. Curr. Med. Chem. 2018, 25, 4328–4353. [Google Scholar] [CrossRef]
- Van der Geest, T.; Laverman, P.; Metselaar, J.M.; Storm, G.; Boerman, O.C. Radionuclide imaging of liposomal drug delivery. Expert Opin. Drug Deliv. 2016, 13, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, J.; Nissinen, T.; Alanne, A.; Thapa, R.; Fioux, P.; Bonne, M.; Rigolet, S.; Morlet-Savary, F.; Aussenac, F.; Marichal, C.; et al. Stable surface functionalization of carbonized mesoporous silicon. Inorg. Chem. Front. 2020, 7, 631–641. [Google Scholar] [CrossRef]
- Green, J.M.; Donohoe, M.E.; Foster, M.E.; Glajch, J.L. Thin-layer chromatographic procedures for the characterization of technetium-99m bicisate. J. Nucl. Med. Technol. 1994, 22, 21–26. [Google Scholar]
- Thapa, R. Recovery of Scandium and Uranium with Bisphosphonate Modified Mesoporous Silicon; Itä-Suomen yliopisto: Kuopio, Finland, 2021. [Google Scholar]
- Thapa, R.; Nissinen, T.; Turhanen, P.; Määttä, J.; Vepsäläinen, J.; Lehto, V.-P.; Riikonen, J. Bisphosphonate modified mesoporous silicon for scandium adsorption. Microporous Mesoporous Mater. 2020, 296, 109980. [Google Scholar] [CrossRef]
- Tamarov, K.; Wang, J.T.-W.; Kari, J.; Happonen, E.; Vesavaara, I.; Niemela, M.; Peramaki, P.; Al-Jamal, K.T.; Xu, W.; Lehto, V.-P. Comparison between fluorescence imaging and elemental analysis to determine biodistribution of inorganic nanoparticles with strong light absorption. ACS Appl. Mater. Interfaces 2021, 13, 40392–40400. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Chen, W.; Wang, Z.; Lu, W.; Cheng, R.; Deng, C.; Meng, F.; Liu, H.; Zhong, Z. Acid-Activatable Prodrug Nanogels for Efficient Intracellular Doxorubicin Release. Biomacromolecules 2011, 12, 3612–3620. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, H.; Poutiainen, P.; Batnasan, E.; Latonen, L.; Lehto, V.-P.; Xu, W. Biomimetic Inorganic Nanovectors as Tumor-Targeting Theranostic Platform against Triple-Negative Breast Cancer. Pharmaceutics 2023, 15, 2507. https://doi.org/10.3390/pharmaceutics15102507
Wen H, Poutiainen P, Batnasan E, Latonen L, Lehto V-P, Xu W. Biomimetic Inorganic Nanovectors as Tumor-Targeting Theranostic Platform against Triple-Negative Breast Cancer. Pharmaceutics. 2023; 15(10):2507. https://doi.org/10.3390/pharmaceutics15102507
Chicago/Turabian StyleWen, Huang, Pekka Poutiainen, Enkhzaya Batnasan, Leena Latonen, Vesa-Pekka Lehto, and Wujun Xu. 2023. "Biomimetic Inorganic Nanovectors as Tumor-Targeting Theranostic Platform against Triple-Negative Breast Cancer" Pharmaceutics 15, no. 10: 2507. https://doi.org/10.3390/pharmaceutics15102507
APA StyleWen, H., Poutiainen, P., Batnasan, E., Latonen, L., Lehto, V.-P., & Xu, W. (2023). Biomimetic Inorganic Nanovectors as Tumor-Targeting Theranostic Platform against Triple-Negative Breast Cancer. Pharmaceutics, 15(10), 2507. https://doi.org/10.3390/pharmaceutics15102507