Anticancer Peptides Derived from Aldolase A and Induced Tumor-Suppressing Cells Inhibit Pancreatic Ductal Adenocarcinoma Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Assay and EdU Assay
2.3. Scratch Assay
2.4. Transwell Invasion Assay
2.5. Western Blot Analysis
2.6. Human Peripheral Blood Mononuclear Cells (PBMCs)
2.7. Ex Vivo Tissue Assay
2.8. Molecular Docking Analysis
2.9. Vibration Assay
2.10. Statistical Analyses
3. Results
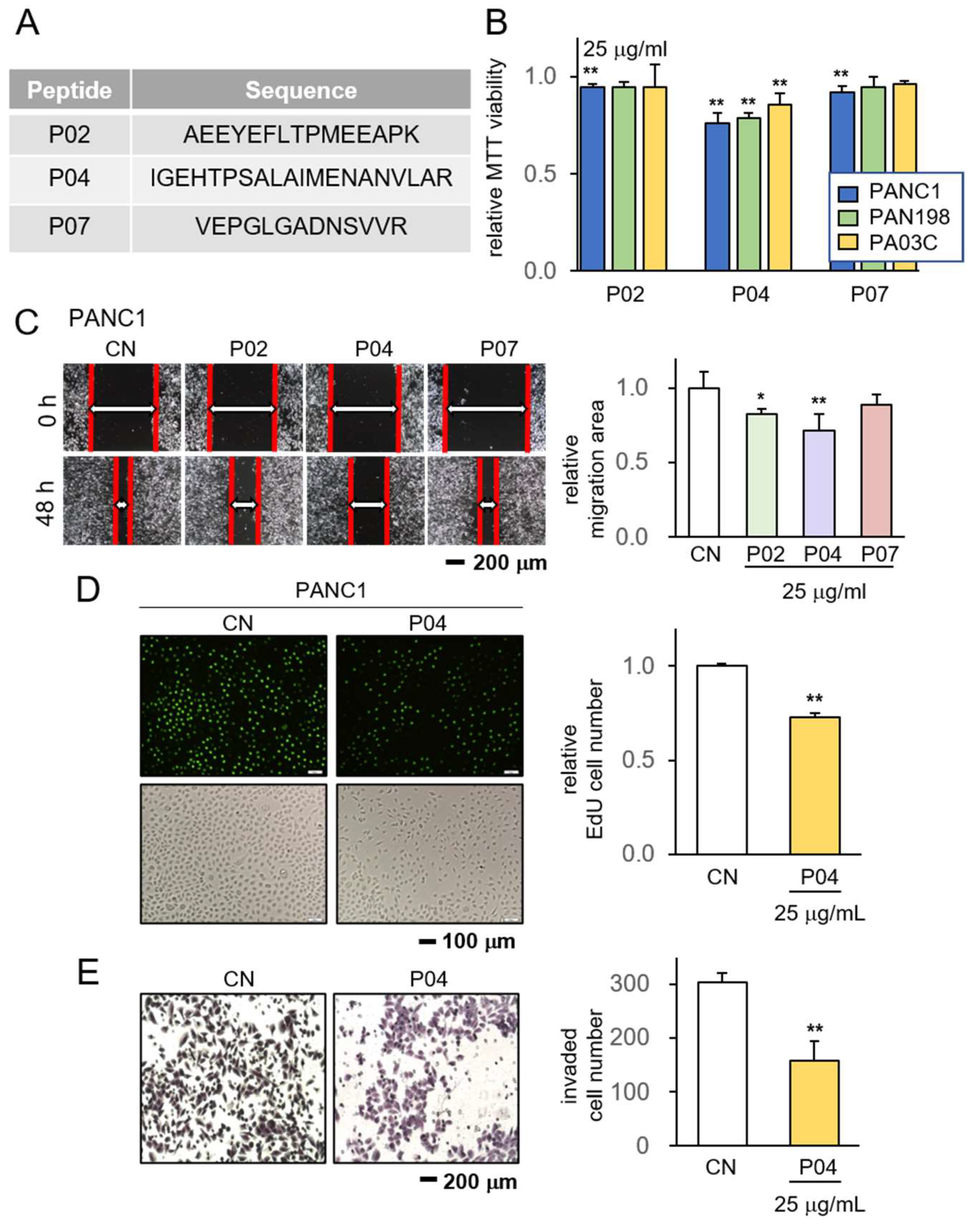
3.1. Anti-Tumor Effect of P02, P04, and P07 on PDAC Cells
3.2. Anti-Tumor Effect of P04 on Human PDAC Tissue Fragments
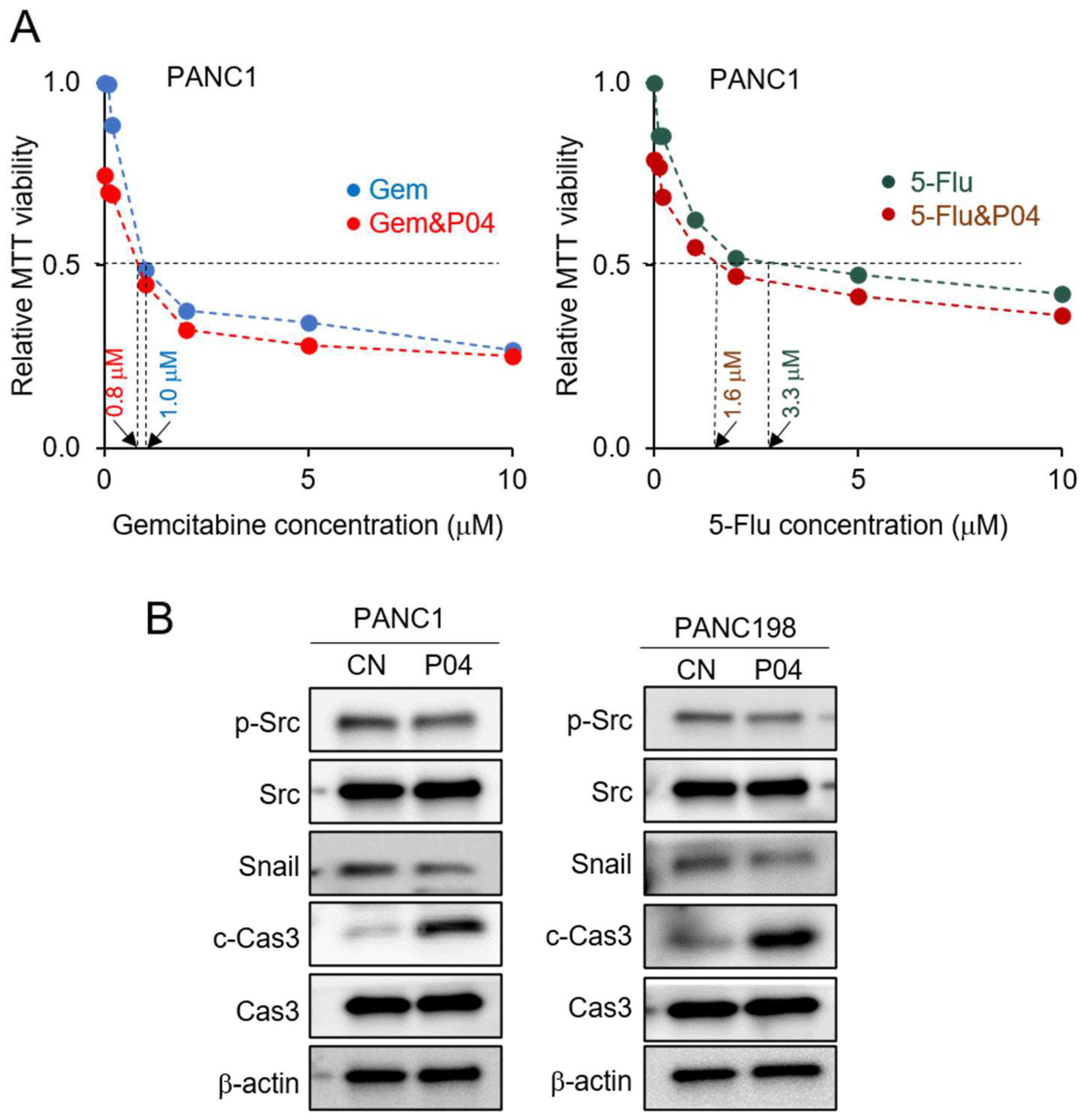
3.3. Enhanced Anti-Tumor Effects in Combination with Chemotherapeutic Agents
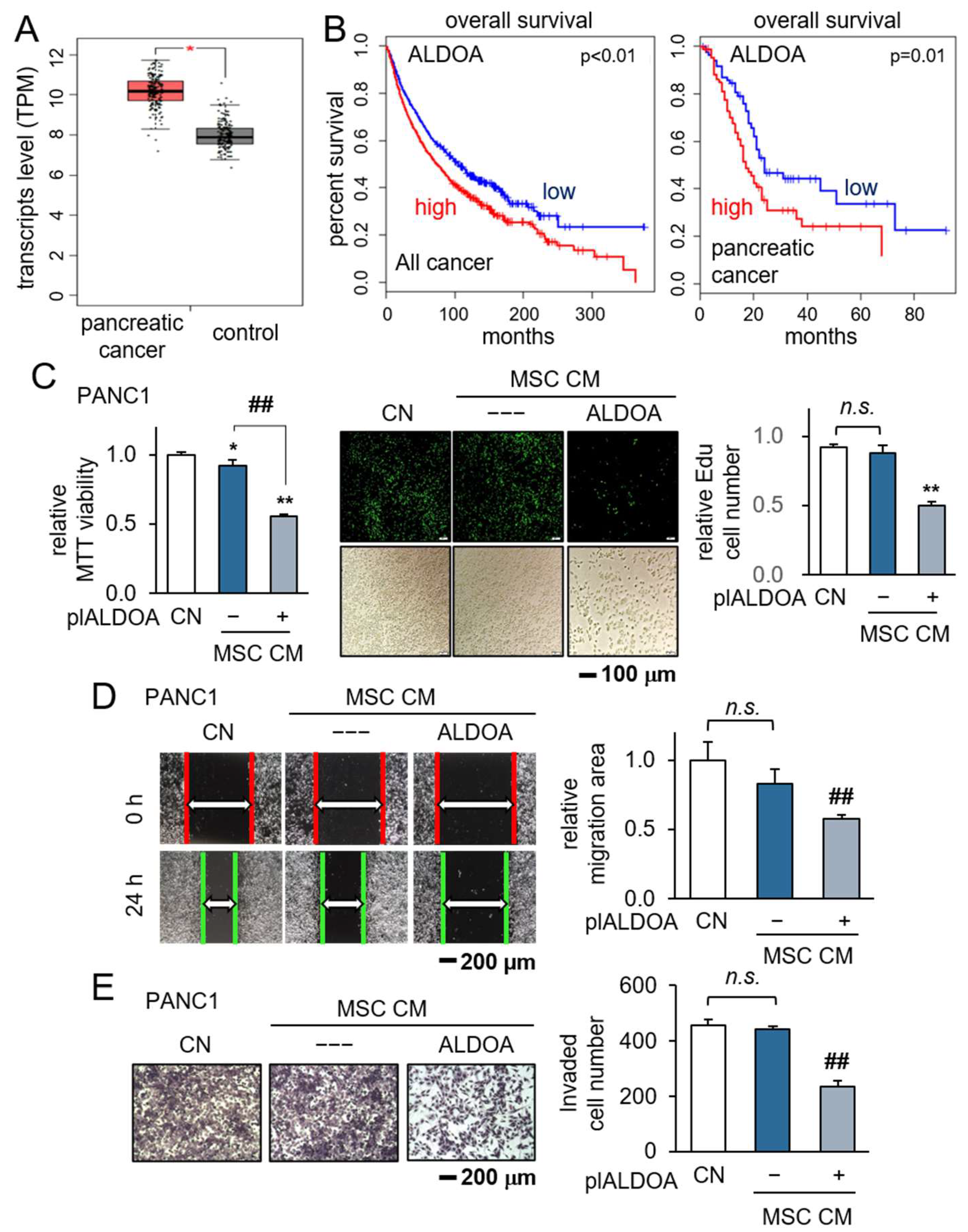
3.4. Generation of Antitumor CM by the Overexpression of ALDOA in MSCs
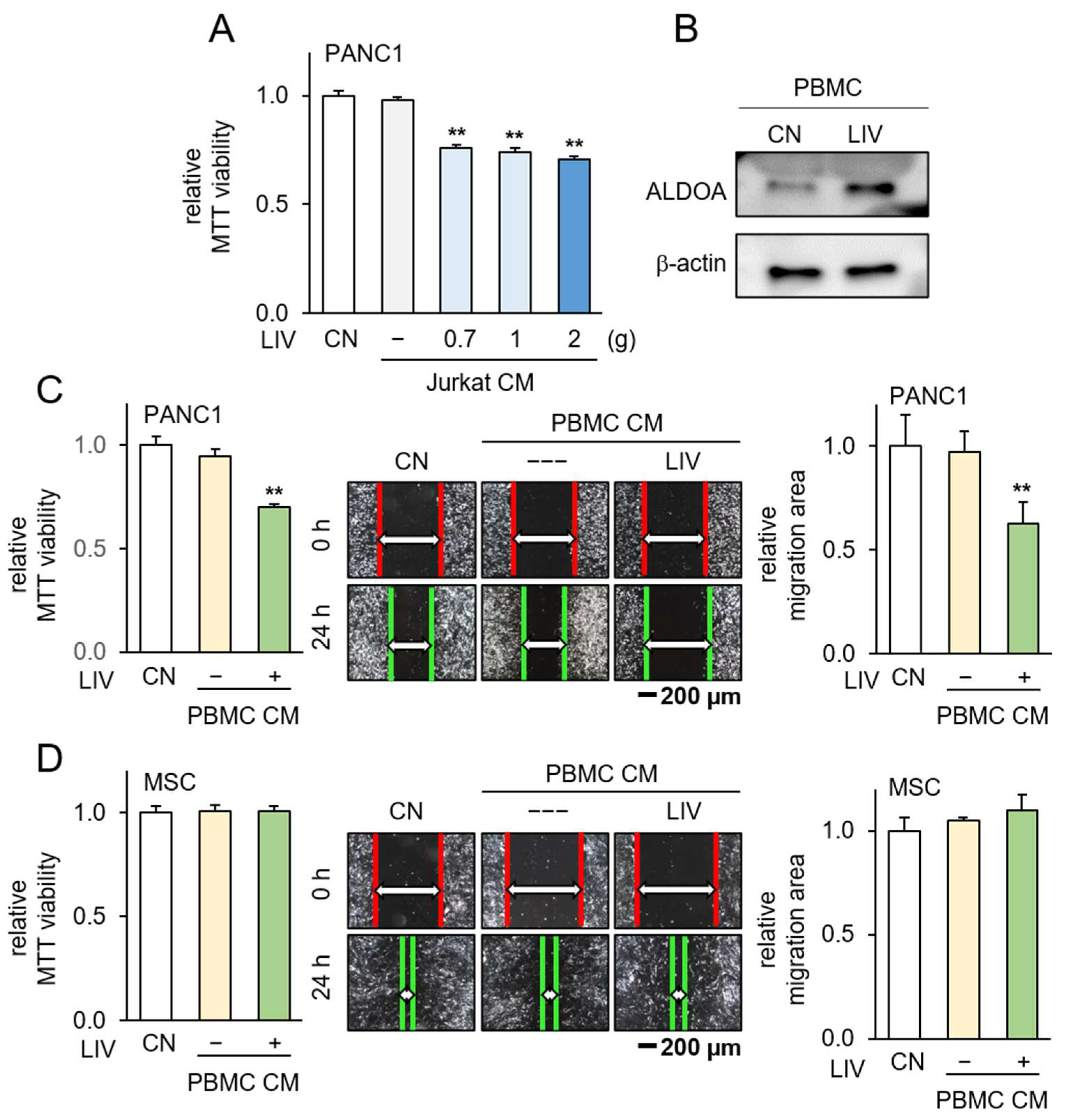
3.5. Generation of Tumor-Suppressive CM from PBMCs by Overexpression ALDOA
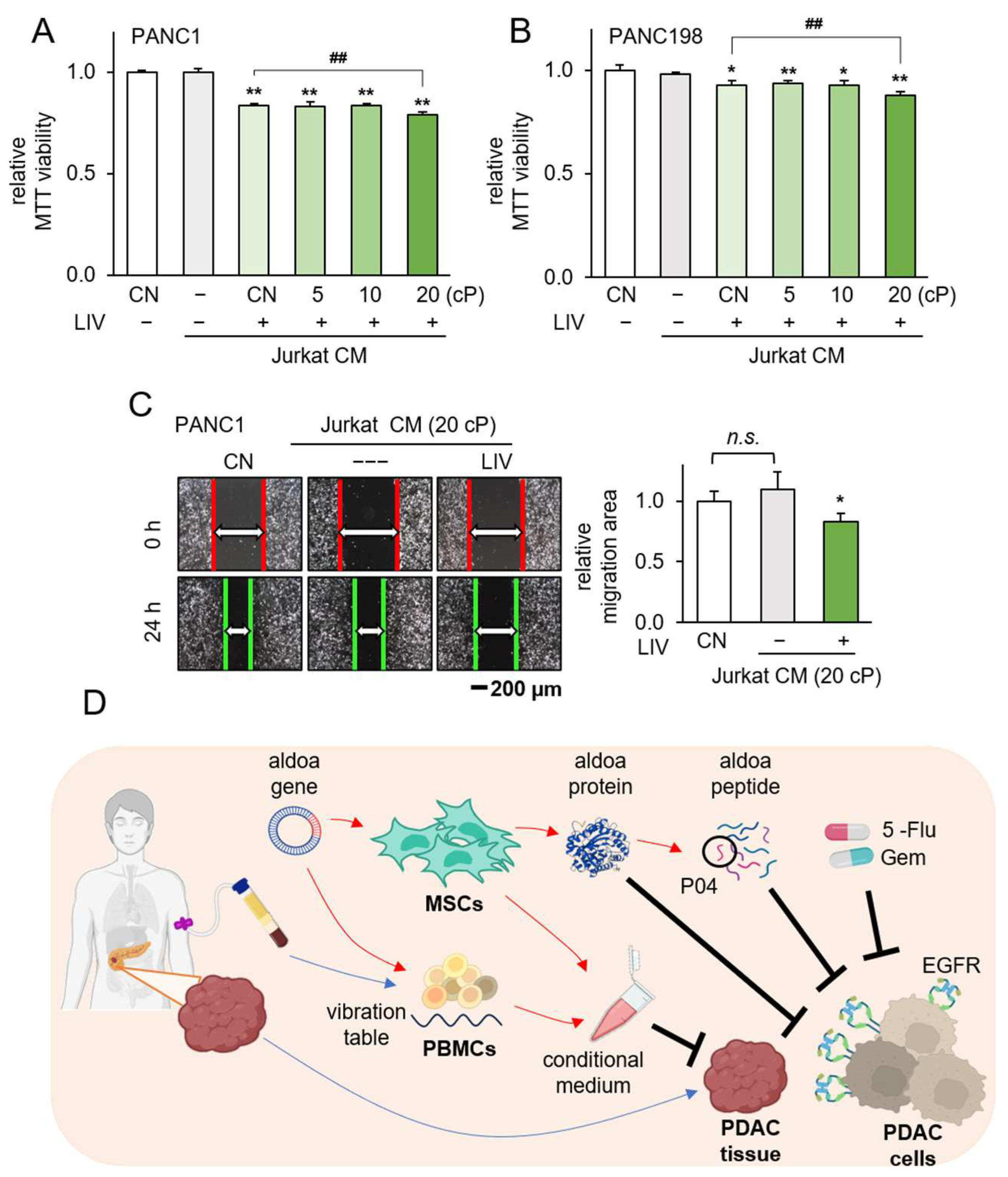
3.6. Generation of Tumor-Suppressive CM Using Low-Intensity Vibration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moffat, G.T.; Epstein, A.S.; O’Reilly, E.M. Pancreatic cancer-A disease in need: Optimizing and integrating supportive care. Cancer 2019, 125, 3927–3935. [Google Scholar] [CrossRef] [PubMed]
- Sturm, N.; Ettrich, T.J.; Perkhofer, L. The Impact of Biomarkers in Pancreatic Ductal Adenocarcinoma on Diagnosis, Surveillance and Therapy. Cancers 2022, 14, 217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hayashi, H.; Matsumura, K.; Uemura, N.; Shiraishi, Y.; Sato, H.; Baba, H. Biological and Clinical Impacts of Glucose Metabolism in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 498. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Sho, M.; Akahori, T.; Nakagawa, K.; Nakamura, K. Application of liquid biopsy for surgical management of pancreatic cancer. Ann. Gastroenterol. Surg. 2020, 4, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.; Vernerey, D.; Bachet, J.B.; Tuech, J.J.; Portales, F.; Michel, P.; Cunha, A.S. Resectable pancreatic adenocarcinoma neo-adjuvant FOLF(IRIN)OX-based chemotherapy—A multicenter, non-comparative, randomized, phase II trial (PANACHE01-PRODIGE48 study). BMC Cancer 2018, 18, 762. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Parikh, A.R.; Szabolcs, A.; Allen, J.N.; Clark, J.W.; Wo, J.Y.; Raabe, M.; Thel, H.; Hoyos, D.; Mehta, A.; Arshad, S.; et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat. Cancer 2021, 2, 1124–1135. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef]
- Mader, J.S.; Hoskin, D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [PubMed]
- León-Letelier, R.A.; Katayama, H.; Hanash, S. Mining the Immunopeptidome for Antigenic Peptides in Cancer. Cancers 2022, 14, 4968. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Stopfer, L.E.; Ahn, R.; Sanders, E.A.; Sandel, D.A.; Freed-Pastor, W.A.; Rideout, W.M., 3rd; Naranjo, S.; Fessenden, T.; Nguyen, K.B.; et al. Deciphering the immunopeptidome in vivo reveals new tumour antigens. Nature 2022, 607, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.R.; Biswas, S.; Ali, M.A.; Halim, M.A.; Ullah, M.O. In silico peptide-based therapeutics against human colorectal cancer by the activation of TLR5 signaling pathways. J. Mol. Model. 2023, 29, 35. [Google Scholar] [CrossRef]
- Akiyama, Y.; Komiyama, M.; Nakamura, Y.; Iizuka, A.; Oshita, C.; Kume, A.; Nogami, M.; Miyata, H.; Ashizawa, T.; Yoshikawa, S.; et al. Identification of novel MAGE-A6- and MAGE-A12-derived HLA-A24-restricted cytotoxic T lymphocyte epitopes using an in silico peptide-docking assay. Cancer Immunol. Immunother. CII 2012, 61, 2311–2319. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Hase, M.; Zha, R.; Feng, Y.; Li, B.Y.; Yokota, H. Suppression of breast cancer-associated bone loss with osteoblast proteomes via Hsp90ab1/moesin-mediated inhibition of TGFβ/FN1/CD44 signaling. Theranostics 2022, 12, 929–943. [Google Scholar] [CrossRef]
- Li, K.; Sun, X.; Zha, R.; Liu, S.; Feng, Y.; Sano, T.; Aryal, U.K.; Sudo, A.; Li, B.Y.; Yokota, H. Counterintuitive production of tumor-suppressive secretomes from Oct4- and c-Myc-overexpressing tumor cells and MSCs. Theranostics 2022, 12, 3084–3103. [Google Scholar] [CrossRef]
- Li, K.; Huo, Q.; Dimmitt, N.H.; Qu, G.; Bao, J.; Pandya, P.H.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Kacena, M.A.; Pollok, K.E.; et al. Osteosarcoma-enriched transcripts paradoxically generate osteosarcoma-suppressing extracellular proteins. eLife 2023, 12, e83768. [Google Scholar] [CrossRef]
- Li, K.X.; Sun, X.; Li, B.Y.; Yokota, H. Conversion of Osteoclasts into Bone-Protective, Tumor-Suppressing Cells. Cancers 2021, 13, 5593. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.X.; Figueiredo, M.L.; Lin, C.C.; Li, B.Y.; Yokota, H. Generation of the Chondroprotective Proteomes by Activating PI3K and TNFα Signaling. Cancers 2022, 14, 3039. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Zha, R.; Liu, S.; Fan, Y.; Wu, D.; Hase, M.; Aryal, U.K.; Lin, C.C.; Li, B.Y.; et al. Preventing tumor progression to the bone by induced tumor-suppressing MSCs. Theranostics 2021, 11, 5143–5159. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Sun, X.; Minami, K.; Tamari, K.; Ogawa, K.; Li, H.; Ma, H.; Zhou, M.; Na, S.; Li, B.Y.; et al. Proteomes from AMPK-inhibited peripheral blood mononuclear cells suppress the progression of breast cancer and bone metastasis. Theranostics 2023, 13, 1247–1263. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, X.; Li, K.; Zha, R.; Feng, Y.; Sano, T.; Dong, C.; Liu, Y.; Aryal, U.K.; Sudo, A.; et al. Generation of the tumor-suppressive secretome from tumor cells. Theranostics 2021, 11, 8517–8534. [Google Scholar] [CrossRef]
- Manea, M.; Mezo, G.; Hudecz, F.; Przybylski, M. Mass spectrometric identification of the trypsin cleavage pathway in lysyl-proline containing oligotuftsin peptides. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2007, 13, 227–236. [Google Scholar] [CrossRef]
- Tian, W.; Zhou, J.; Chen, M.; Qiu, L.; Li, Y.; Zhang, W.; Guo, R.; Lei, N.; Chang, L. Bioinformatics analysis of the role of aldolase A in tumor prognosis and immunity. Sci. Rep. 2022, 12, 11632. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Shi, Y.; Lv, Y.; Yuan, S.; Ramirez, C.F.A.; Lieftink, C.; Wang, L.; Wang, S.; Wang, C.; Dias, M.H.; et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021, 595, 730–734. [Google Scholar] [CrossRef]
- Aissa, A.F.; Islam, A.; Ariss, M.M.; Go, C.C.; Rader, A.E.; Conrardy, R.D.; Gajda, A.M.; Rubio-Perez, C.; Valyi-Nagy, K.; Pasquinelli, M.; et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat. Commun. 2021, 12, 1628. [Google Scholar] [CrossRef]
- Ahsan, A.; Ramanand, S.G.; Bergin, I.L.; Zhao, L.; Whitehead, C.E.; Rehemtulla, A.; Ray, D.; Pratt, W.B.; Lawrence, T.S.; Nyati, M.K. Efficacy of an EGFR-specific peptide against EGFR-dependent cancer cell lines and tumor xenografts. Neoplasia 2014, 16, 105–114. [Google Scholar] [CrossRef]
- Cassel, D.L.; Hoxie, J.A.; Cooper, R.A. Phorbol ester modulation of T-cell antigens in the Jurkat lymphoblastic leukemia cell line. Cancer Res. 1983, 43, 4582–4586. [Google Scholar]
- Zuo, Q.; Liu, J.; Zhang, J.; Wu, M.; Guo, L.; Liao, W. Development of trastuzumab-resistant human gastric carcinoma cell lines and mechanisms of drug resistance. Sci. Rep. 2015, 5, 11634. [Google Scholar] [CrossRef]
- Kaiser, C.L.; Kamien, A.J.; Shah, P.A.; Chapman, B.J.; Cotanche, D.A. 5-Ethynyl-2′-deoxyuridine labeling detects proliferating cells in the regenerating avian cochlea. Laryngoscope 2009, 119, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Radstake, W.E.; Gautam, K.; Van Rompay, C.; Vermeesen, R.; Tabury, K.; Verslegers, M.; Baatout, S.; Baselet, B. Comparison of in vitro scratch wound assay experimental procedures. Biochem. Biophys. Rep. 2023, 33, 101423. [Google Scholar] [CrossRef] [PubMed]
- Pijuan, J.; Barceló, C.; Moreno, D.F.; Maiques, O.; Sisó, P.; Marti, R.M.; Macià, A.; Panosa, A. In vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front. Cell Dev. Biol. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Miura, K. Imaging technologies for the detection of multiple stains in proteomics. Proteomics 2003, 3, 1097–1108. [Google Scholar] [CrossRef]
- Aramyan, S.; McGregor, K.; Sandeep, S.; Haczku, A. SP-A binding to the SARS-CoV-2 spike protein using hybrid quantum and classical in silico modeling and molecular pruning by Quantum Approximate Optimization Algorithm (QAOA) Based MaxCut with ZDOCK. Front. Immunol. 2022, 13, 945317. [Google Scholar] [CrossRef]
- Barradas-Bautista, D.; Cao, Z.; Vangone, A.; Oliva, R.; Cavallo, L. A random forest classifier for protein-protein docking models. Bioinform. Adv. 2022, 2, vbab042. [Google Scholar] [CrossRef]
- Tang, A.H.; Hoefer, R.A.; Guye, M.L.; Bear, H.D. Persistent EGFR/K-RAS/SIAH pathway activation drives chemo-resistance and early tumor relapse in triple-negative breast cancer. Cancer Drug Resist. 2022, 5, 691–702. [Google Scholar] [CrossRef]
- Ghergurovich, J.M.; Esposito, M.; Chen, Z.; Wang, J.Z.; Bhatt, V.; Lan, T.; White, E.; Kang, Y.; Guo, J.Y.; Rabinowitz, J.D. Glucose-6-Phosphate Dehydrogenase Is Not Essential for K-Ras-Driven Tumor Growth or Metastasis. Cancer Res. 2020, 80, 3820–3829. [Google Scholar] [CrossRef]
- Ishizaki, H.; Tsunoda, T.; Wada, S.; Yamauchi, M.; Shibuya, M.; Tahara, H. Inhibition of tumor growth with antiangiogenic cancer vaccine using epitope peptides derived from human vascular endothelial growth factor receptor 1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 5841–5849. [Google Scholar] [CrossRef]
- Wada, S.; Tsunoda, T.; Baba, T.; Primus, F.J.; Kuwano, H.; Shibuya, M.; Tahara, H. Rationale for antiangiogenic cancer therapy with vaccination using epitope peptides derived from human vascular endothelial growth factor receptor 2. Cancer Res. 2005, 65, 4939–4946. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chiou, J.; Yang, Y.F.; Su, C.Y.; Lin, Y.F.; Yang, C.N.; Lu, P.J.; Huang, M.S.; Yang, C.J.; Hsiao, M. Therapeutic Targeting of Aldolase A Interactions Inhibits Lung Cancer Metastasis and Prolongs Survival. Cancer Res. 2019, 79, 4754–4766. [Google Scholar] [CrossRef]
- Pagnotti, G.M.; Thompson, W.R.; Guise, T.A.; Rubin, C.T. Suppression of cancer-associated bone loss through dynamic mechanical loading. Bone 2021, 150, 115998. [Google Scholar] [CrossRef] [PubMed]
- Wohl, I.; Sherman, E. Spectral Analysis of ATP-Dependent Mechanical Vibrations in T Cells. Front. Cell Dev. Biol. 2021, 9, 590655. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Lin, T.C.; Wang, L.; Chen, S.; Chen, X.; Yiu, P.M.; Tsui, O.K.C.; Chu, J.; Kiang, C.H.; Park, H. Mechanical Responses of Breast Cancer Cells to Substrates of Varying Stiffness Revealed by Single-Cell Measurements. J. Phys. Chem. Lett. 2020, 11, 7643–7649. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J. Continuous and simultaneous measurement of the biophysical properties of blood in a microfluidic environment. Anal. 2016, 141, 6583–6597. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Erickson, G.R.; Ting-Beall, H.P. The effects of osmotic stress on the viscoelastic and physical properties of articular chondrocytes. Biophys. J. 2002, 82, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Stirbat, T.V.; Mgharbel, A.; Bodennec, S.; Ferri, K.; Mertani, H.C.; Rieu, J.P.; Delanoë-Ayari, H. Fine tuning of tissues’ viscosity and surface tension through contractility suggests a new role for α-catenin. PLoS ONE 2013, 8, e52554. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, C.; Huo, Q.; Xiong, X.; Li, K.; Fishel, M.L.; Li, B.; Yokota, H. Anticancer Peptides Derived from Aldolase A and Induced Tumor-Suppressing Cells Inhibit Pancreatic Ductal Adenocarcinoma Cells. Pharmaceutics 2023, 15, 2447. https://doi.org/10.3390/pharmaceutics15102447
Cui C, Huo Q, Xiong X, Li K, Fishel ML, Li B, Yokota H. Anticancer Peptides Derived from Aldolase A and Induced Tumor-Suppressing Cells Inhibit Pancreatic Ductal Adenocarcinoma Cells. Pharmaceutics. 2023; 15(10):2447. https://doi.org/10.3390/pharmaceutics15102447
Chicago/Turabian StyleCui, Changpeng, Qingji Huo, Xue Xiong, Kexin Li, Melissa L. Fishel, Baiyan Li, and Hiroki Yokota. 2023. "Anticancer Peptides Derived from Aldolase A and Induced Tumor-Suppressing Cells Inhibit Pancreatic Ductal Adenocarcinoma Cells" Pharmaceutics 15, no. 10: 2447. https://doi.org/10.3390/pharmaceutics15102447
APA StyleCui, C., Huo, Q., Xiong, X., Li, K., Fishel, M. L., Li, B., & Yokota, H. (2023). Anticancer Peptides Derived from Aldolase A and Induced Tumor-Suppressing Cells Inhibit Pancreatic Ductal Adenocarcinoma Cells. Pharmaceutics, 15(10), 2447. https://doi.org/10.3390/pharmaceutics15102447