
TACkling Cancer by Targeting Selective Protein Degradation

 ,
,  , , and
, , and
Abstract
:1. Introduction
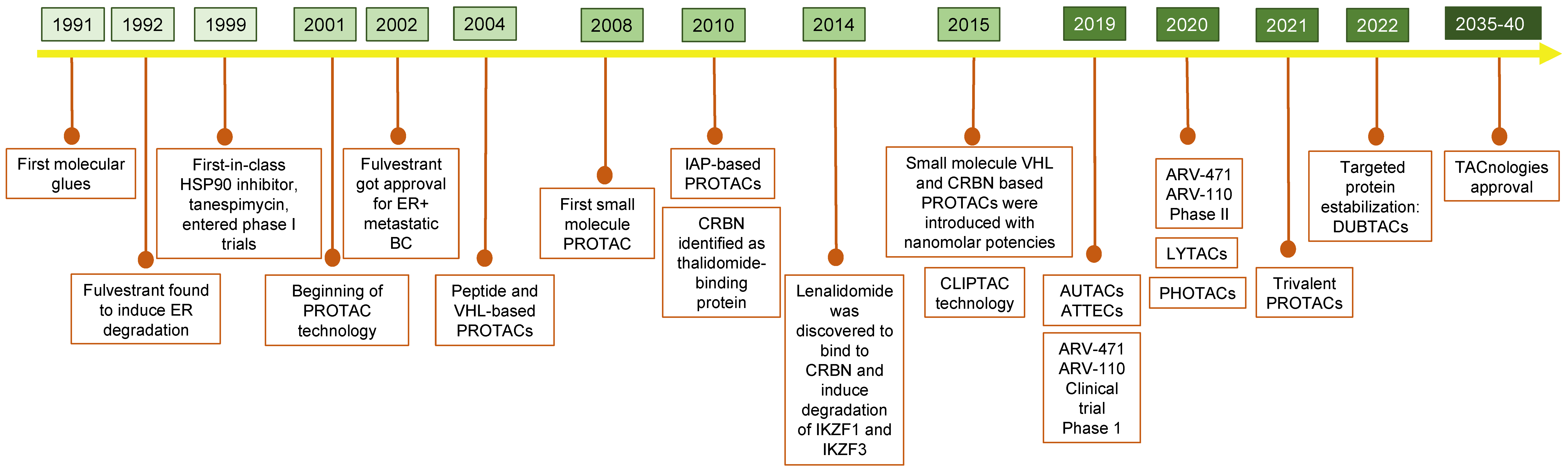
A Brief History of TPD
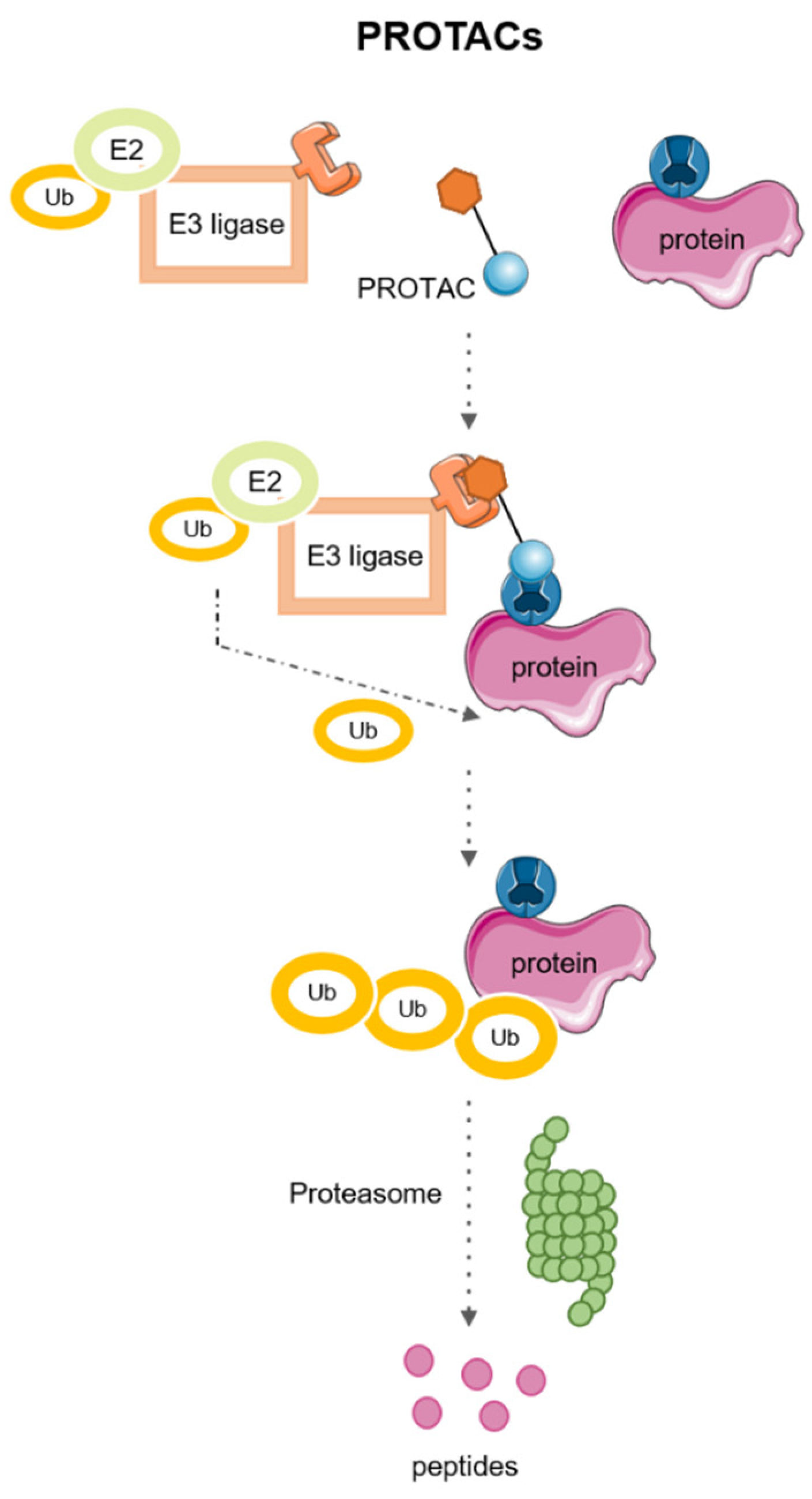
2. PROTACs: PRoteolysis TArgeting Chimeras
2.1. PROTACs: Examples of Applications
2.1.1. BCL-2 Protein Family
2.1.2. Cycling Dependent Kinases
2.1.3. Mitogen-Activated Protein Kinases
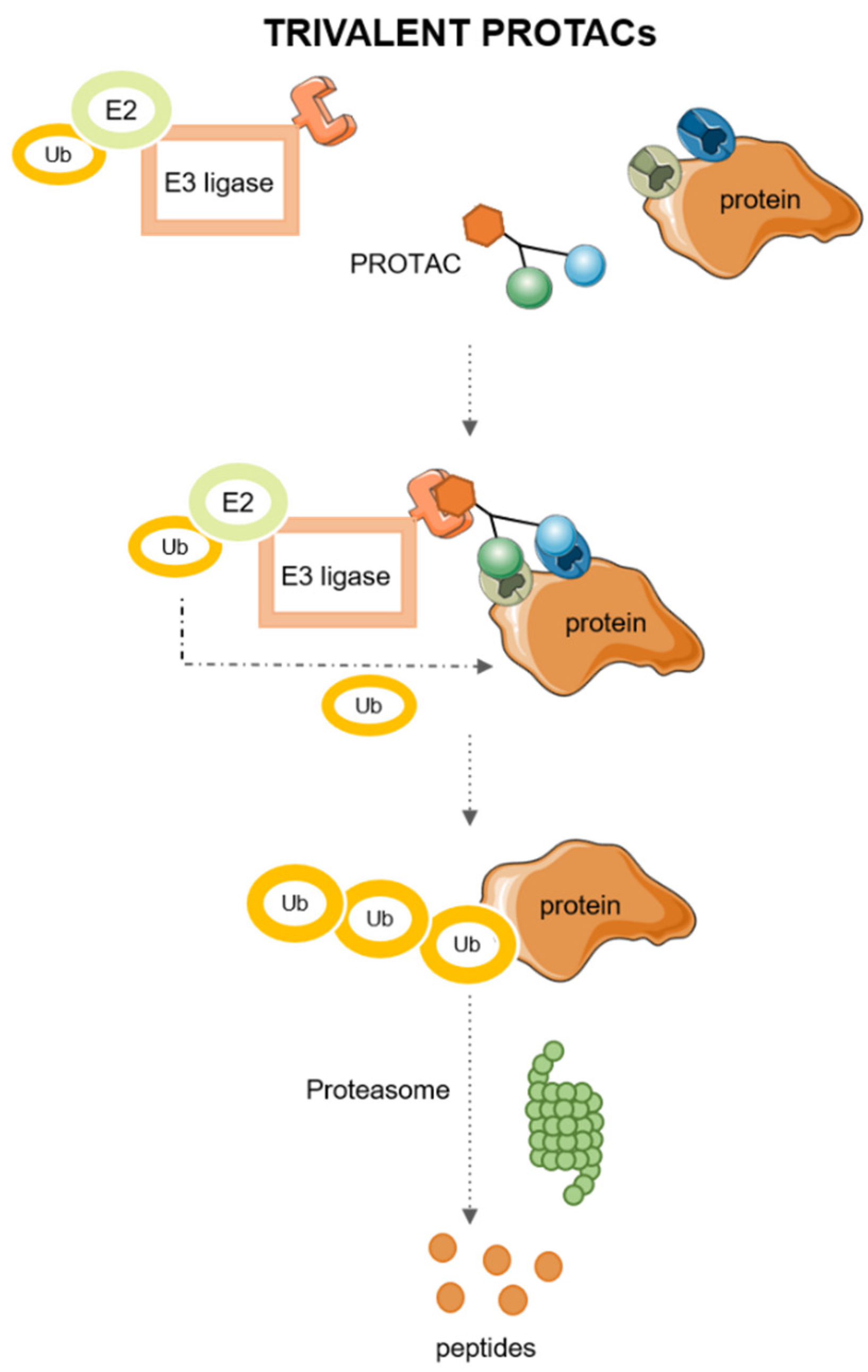
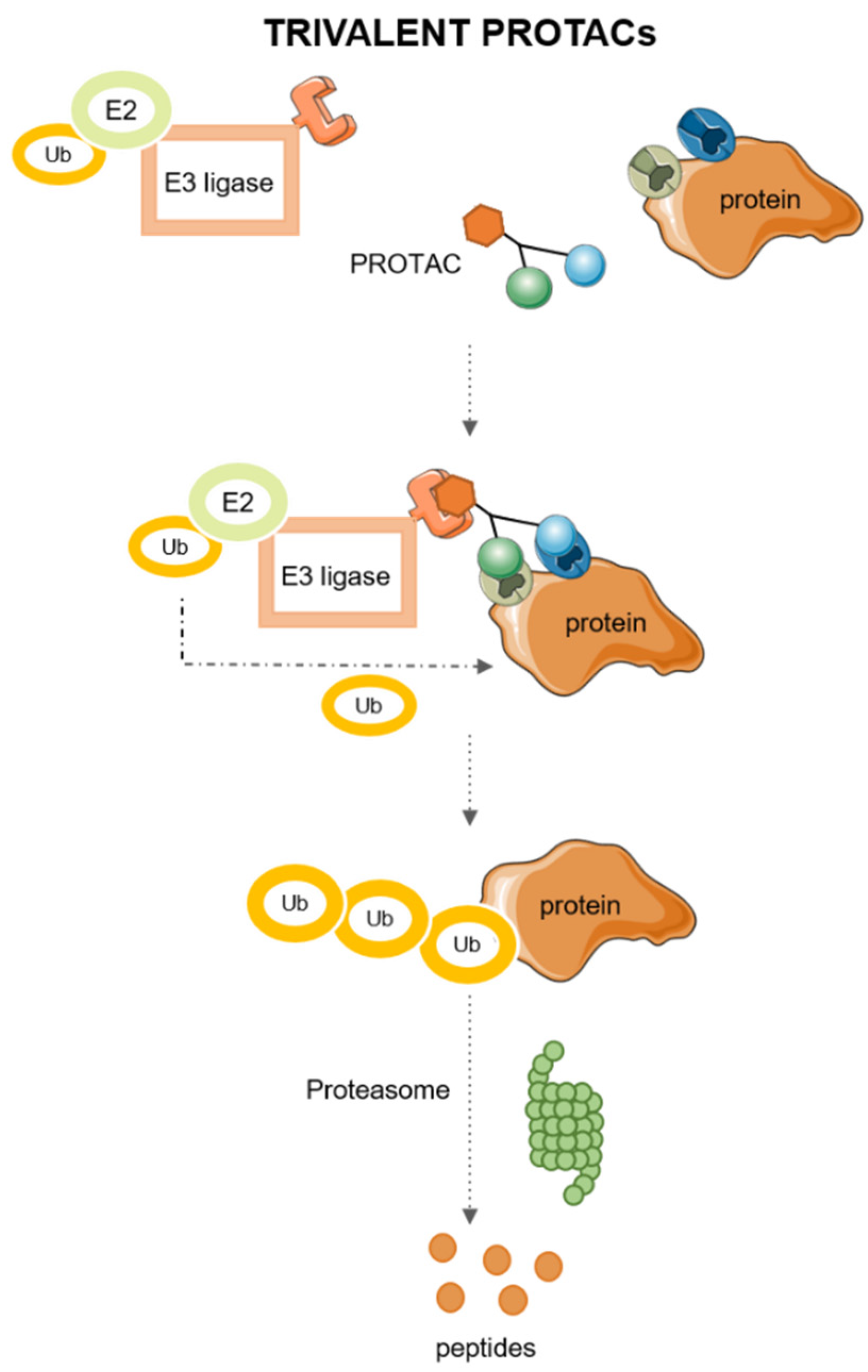
2.2. Trivalent PROTACs
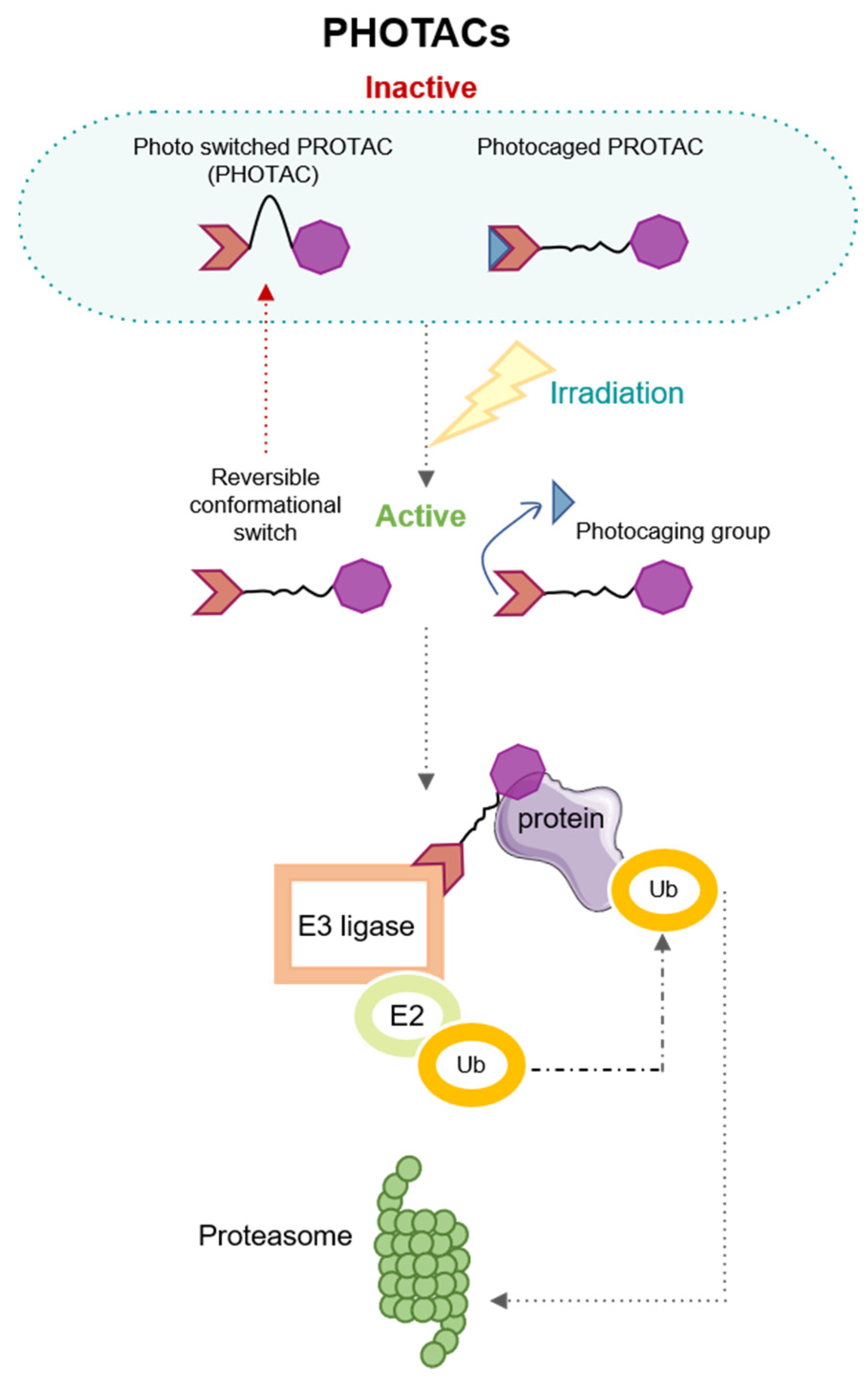
3. Modulating the Activation by Light: PHOtochemically TArgeting Chimeras (PHOTACs)
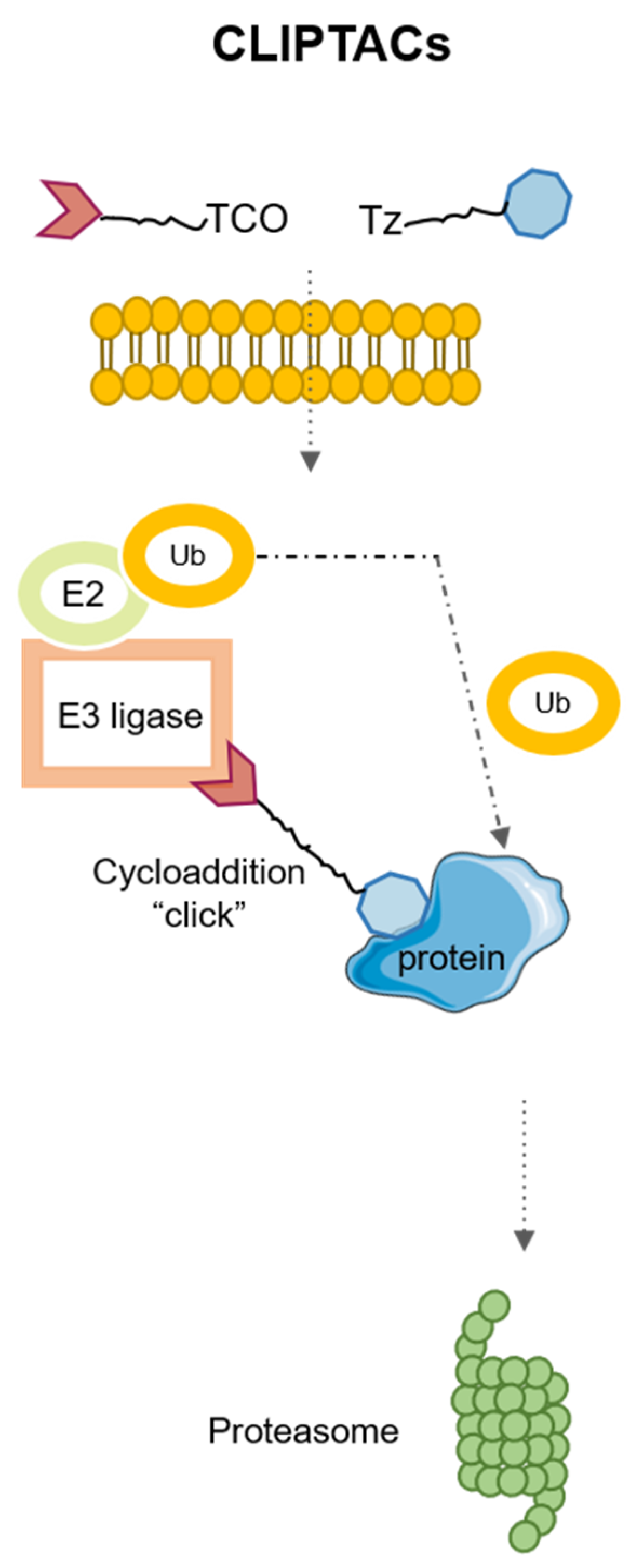
4. Application of Click Chemistry for PROTACs: CLIPTACs
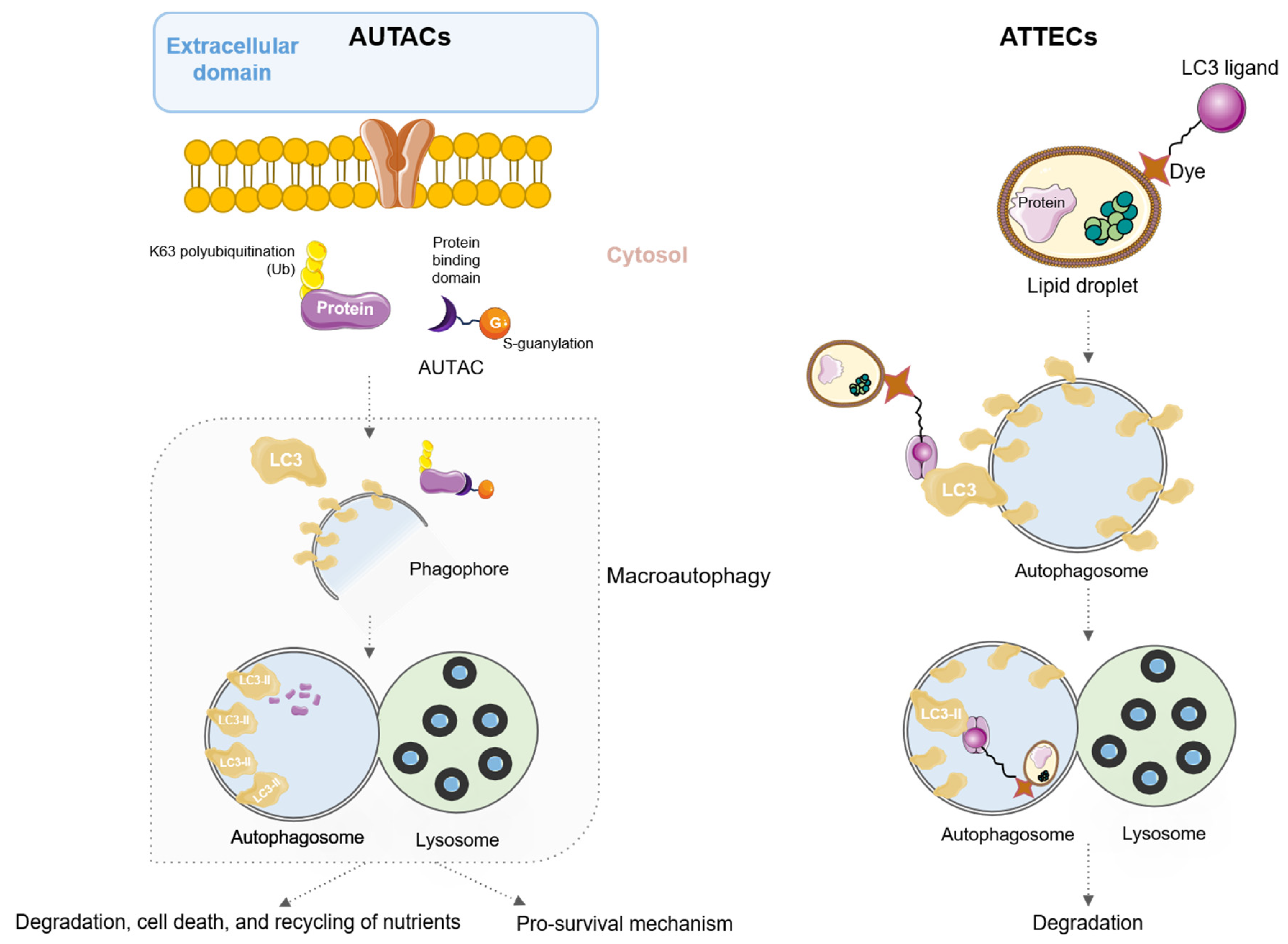
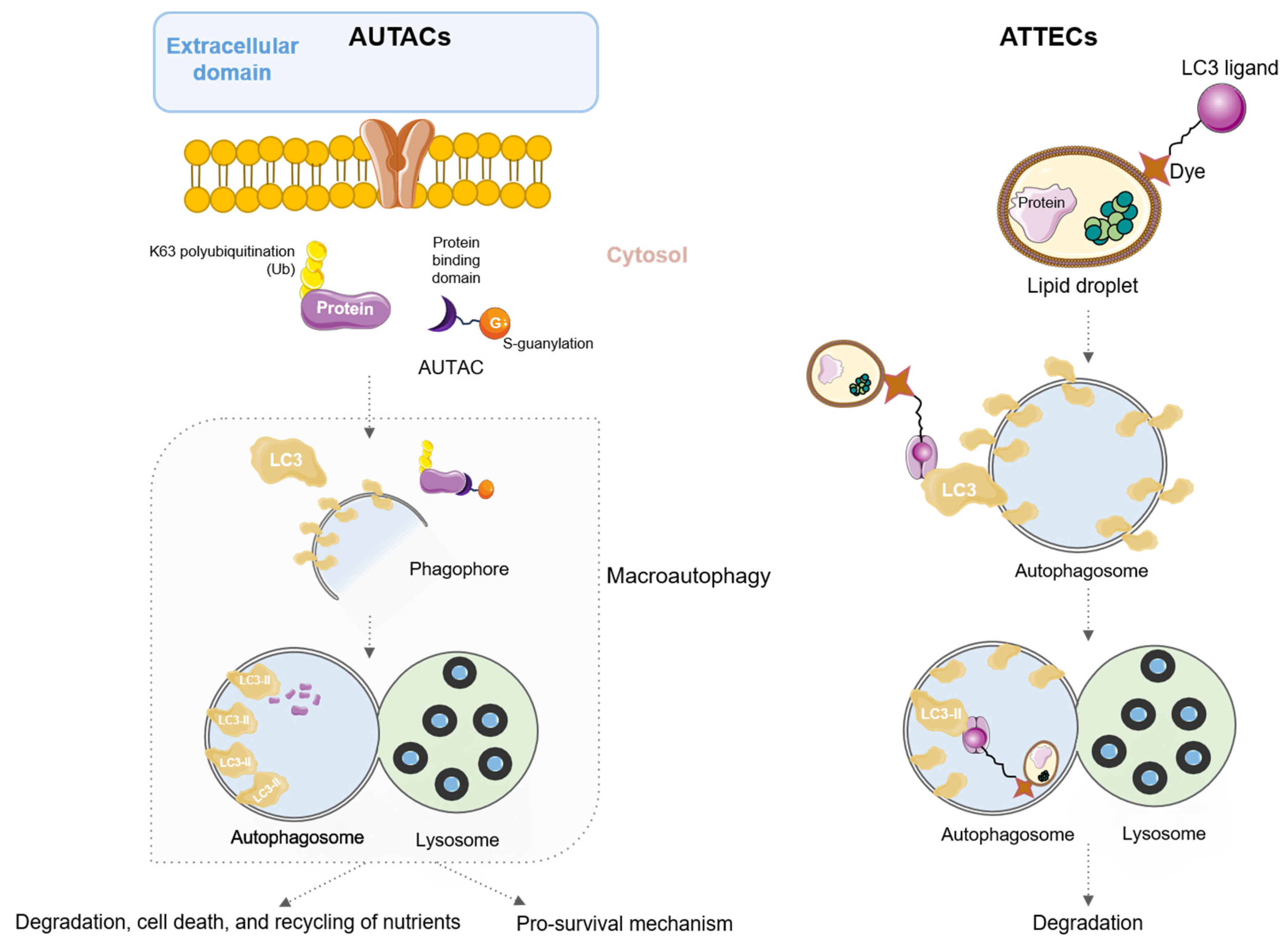
5. AUtophagy TArgeting Chimeras (AUTACs) and AuTophagosome TEthering Compounds (ATTECs)
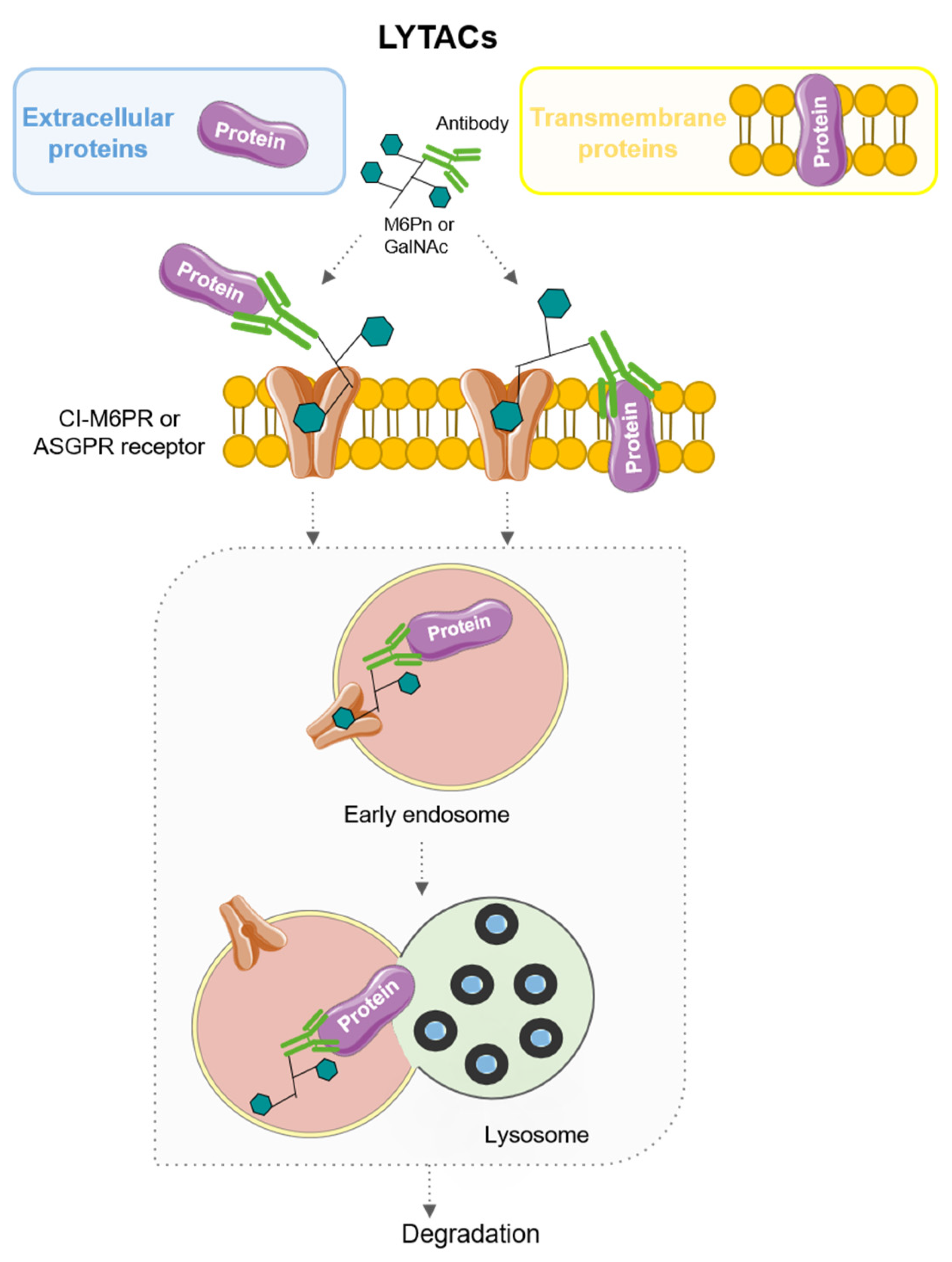
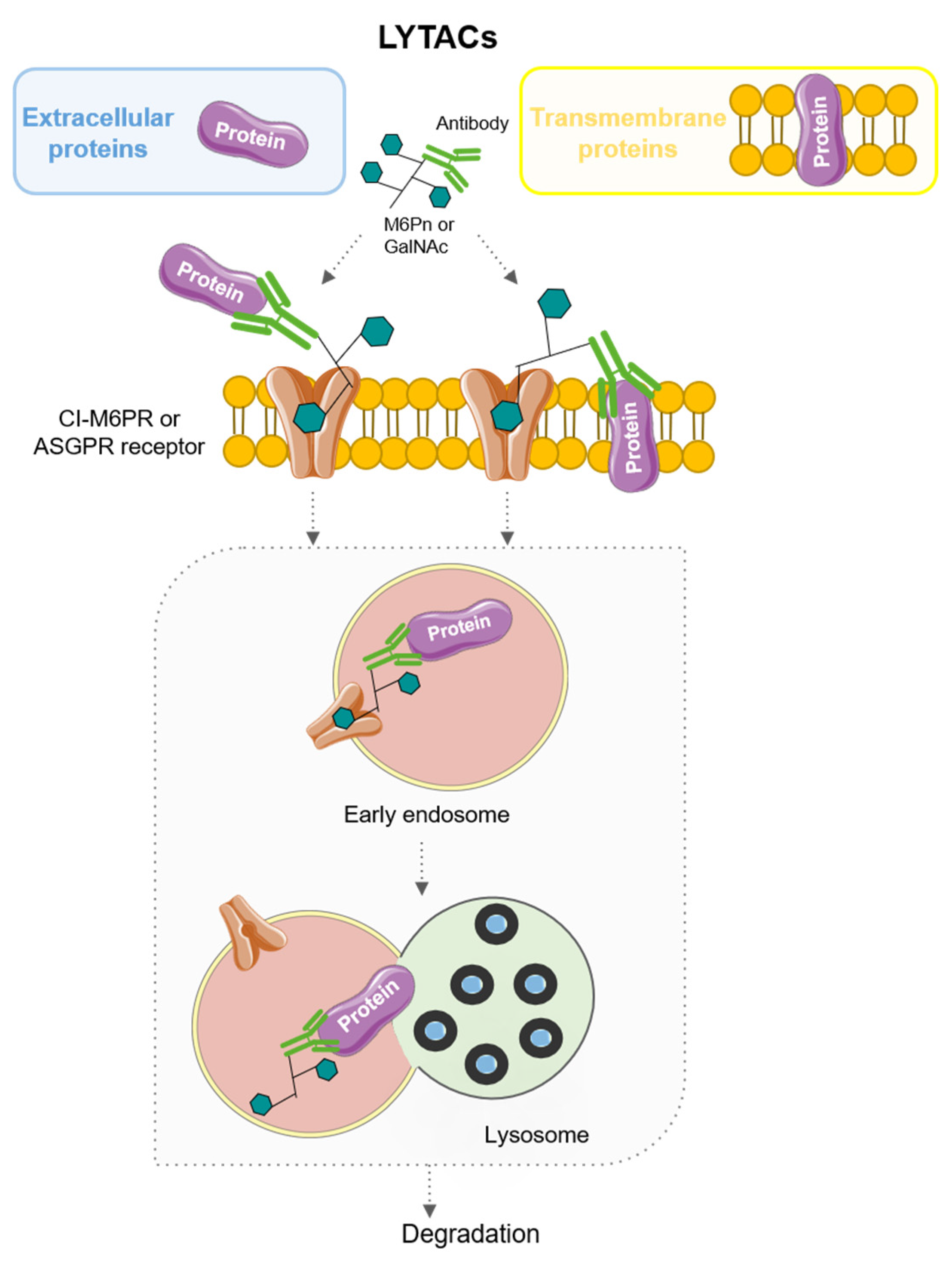
6. LYTACs: LYsosome-TArgeting Chimeras
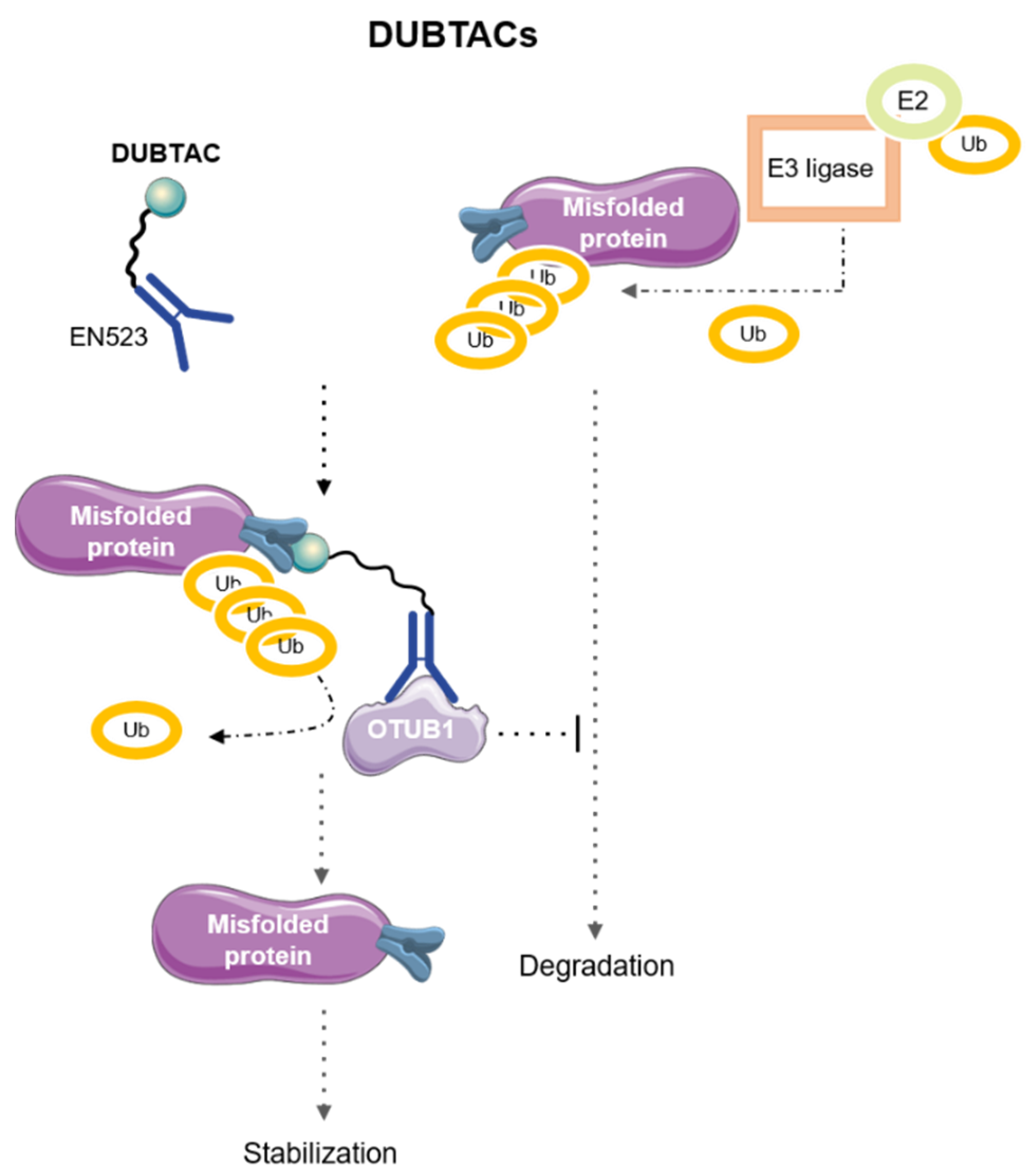
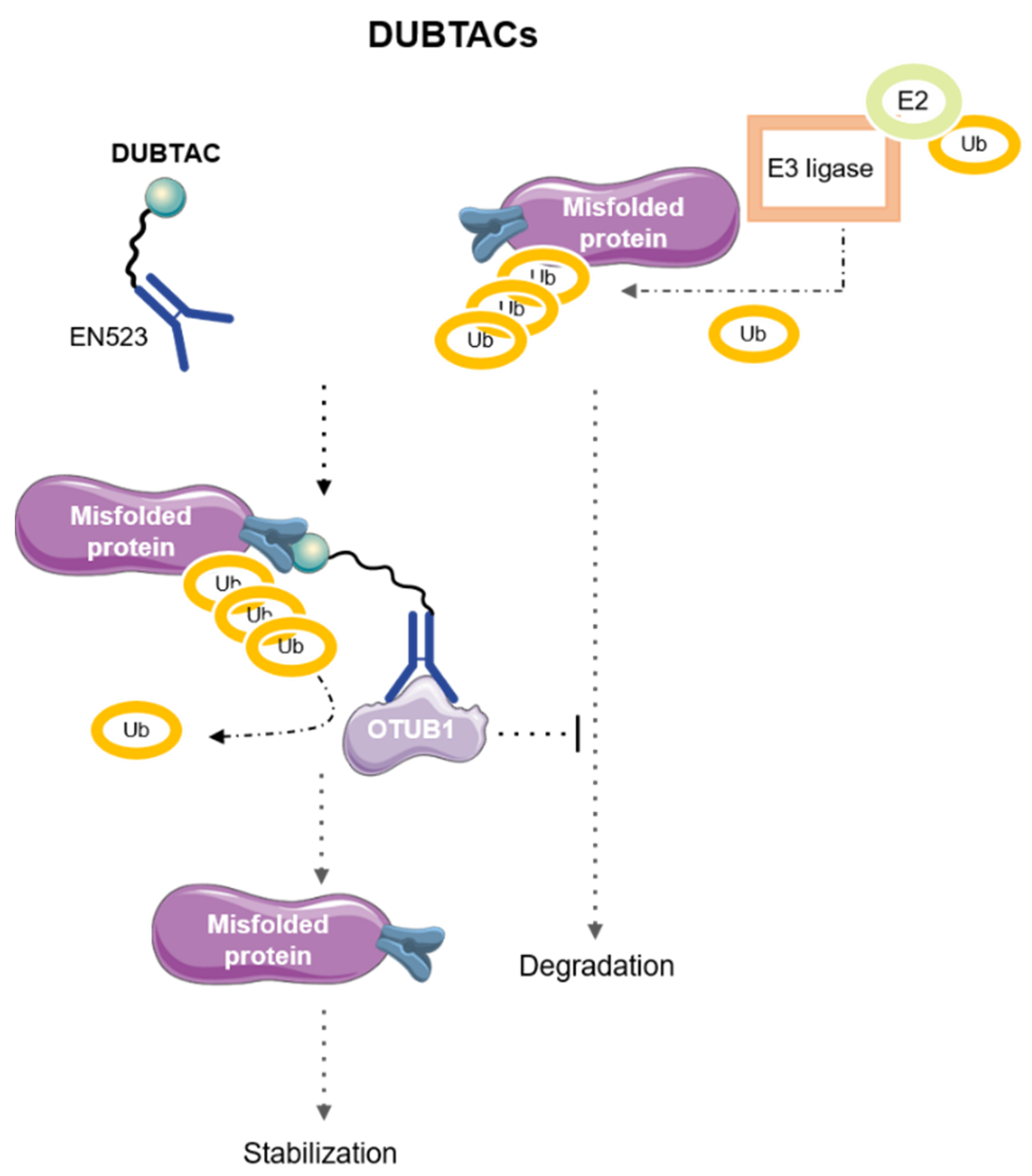
7. DeUBiquitinase-TArgeting Chimeras (DUBTACs)
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, M. Targeted Therapies for Cancer. BMC Med. 2022, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Zocchi, M.R. Natural Killer Cells and Immune-Checkpoint Inhibitor Therapy: Current Knowledge and New Challenges. Mol. Ther.-Oncolytics 2022, 24, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-L.; Chen, J.-S.; Chen, S.-C.; Chu, P.-H. Cardiovascular Toxicity of Molecular Targeted Therapy in Cancer Patients: A Double-Edged Sword. Acta Cardiol. Sin. 2013, 29, 295–303. [Google Scholar]
- Du, R.; Wang, X.; Ma, L.; Larcher, L.M.; Tang, H.; Zhou, H.; Chen, C.; Wang, T. Adverse Reactions of Targeted Therapy in Cancer Patients: A Retrospective Study of Hospital Medical Data in China. BMC Cancer 2021, 21, 206. [Google Scholar] [CrossRef]
- Assoun, S.; Lemiale, V.; Azoulay, E. Molecular Targeted Therapy-Related Life-Threatening Toxicity in Patients with Malignancies. A Systematic Review of Published Cases. Intensive Care Med. 2019, 45, 988. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/Proteasome Pathway Impairment in Neurodegeneration: Therapeutic Implications. Apoptosis 2010, 15, 1292–1311. [Google Scholar] [CrossRef]
- Mohibi, S.; Chen, X.; Zhang, J. Cancer the‘RBP’Eutics–RNA-Binding Proteins as Therapeutic Targets for Cancer. Pharmacol. Ther. 2019, 203, 107390. [Google Scholar] [CrossRef]
- Kenney, D.L.; Benarroch, E.E. The Autophagy-Lysosomal Pathway: General Concepts and Clinical Implications. Neurology 2015, 85, 634–645. [Google Scholar] [CrossRef]
- Schreiber, S.L. The Rise of Molecular Glues. Cell 2021, 184, 3–9. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin Is a Common Target of Cyclophilin-Cyclosporin A and FKBP-FK506 Complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Agrawal, A.; Robertson, J.F.R.; Cheung, K.L.; Gutteridge, E.; Ellis, I.O.; Nicholson, R.I.; Gee, J.M.W. Biological Effects of Fulvestrant on Estrogen Receptor Positive Human Breast Cancer: Short, Medium and Long-term Effects Based on Sequential Biopsies. Int. J. Cancer 2016, 138, 146–159. [Google Scholar] [CrossRef]
- Bross, P.F.; Cohen, M.H.; Williams, G.A.; Pazdur, R. FDA Drug Approval Summaries: Fulvestrant. Oncologist 2002, 7, 477–480. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Fu, L. Small-Molecule PROTACs: An Emerging and Promising Approach for the Development of Targeted Therapy Drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Sharma, C.; Choi, M.A.; Song, Y.; Seo, Y.H. Rational Design and Synthesis of HSF1-PROTACs for Anticancer Drug Development. Molecules 2022, 27, 1655. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Dai, H.; Fernandez, E.; Tinworth, C.P.; Churcher, I.; Cryan, J.; Denyer, J.; Harling, J.D.; Konopacka, A.; Queisser, M.A.; et al. Structural Insights into PROTAC-Mediated Degradation of Bcl-xL. ACS Chem. Biol. 2020, 15, 2316–2323. [Google Scholar] [CrossRef]
- Aublette, M.C.; Harrison, T.A.; Thorpe, E.J.; Gadd, M.S. Selective Wee1 Degradation by PROTAC Degraders Recruiting VHL and CRBN E3 Ubiquitin Ligases. Bioorg. Med. Chem. Lett. 2022, 64, 128636. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Fan, Z.; Zhou, Z.; Li, Y.; Cui, R.; Liu, C.; Zhou, G.; Diao, X.; Jiang, H.; Zheng, M.; et al. Discovery of the First-in-Class Agonist-Based SOS1 PROTACs Effective in Human Cancer Cells Harboring Various KRAS Mutations. J. Med. Chem. 2022, 65, 3923–3942. [Google Scholar] [CrossRef]
- Juan, A.; del Mar Noblejas-López, M.; Arenas-Moreira, M.; Alonso-Moreno, C.; Ocaña, A. Options to Improve the Action of PROTACs in Cancer: Development of Controlled Delivery Nanoparticles. Front. Cell Dev. Biol. 2022, 9, 805336. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.Ü.; et al. Light-Induced Control of Protein Destruction by Opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, S.; Yang, X.; Zhang, L.; Liang, D.; Dong, G.; Du, L.; Lv, Z.; Li, M. Development of Photocontrolled BRD4 PROTACs for Tongue Squamous Cell Carcinoma (TSCC). Eur. J. Med. Chem. 2021, 222, 113608. [Google Scholar] [CrossRef]
- Ito, T.; Yamaguchi, Y.; Handa, H. Exploiting Ubiquitin Ligase Cereblon as a Target for Small-Molecule Compounds in Medicine and Chemical Biology. Cell Chem. Biol. 2021, 28, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs Enable Optical Control of Protein Degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar] [CrossRef] [PubMed]
- Imaide, S.; Riching, K.M.; Makukhin, N.; Vetma, V.; Whitworth, C.; Hughes, S.J.; Trainor, N.; Mahan, S.D.; Murphy, N.; Cowan, A.D.; et al. Trivalent PROTACs Enhance Protein Degradation via Combined Avidity and Cooperativity. Nat. Chem. Biol. 2021, 17, 1157–1167. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef]
- Sun, X.; Rao, Y. PROTACs as Potential Therapeutic Agents for Cancer Drug Resistance. Biochemistry 2020, 59, 240–249. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Weng, G.; Shen, C.; Cao, D.; Gao, J.; Dong, X.; He, Q.; Yang, B.; Li, D.; Wu, J.; Hou, T. PROTAC-DB: An Online Database of PROTACs. Nucleic Acids Res. 2021, 49, D1381–D1387. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, Present and Future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.-T.; Kim, Y.A.; Kim, J.; Oh, H.J.; Park, J.H.; Choi, I.S.; Park, J.H.; Oh, S.; Chu, A.; Lee, J.Y.; et al. Prognostic Influences of BCL1 and BCL2 Expression on Disease-Free Survival in Breast Cancer. Sci. Rep. 2021, 11, 11942. [Google Scholar] [CrossRef] [PubMed]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Kaefer, A.; Yang, J.; Noertersheuser, P.; Mensing, S.; Humerickhouse, R.; Awni, W.; Xiong, H. Mechanism-Based Pharmacokinetic/Pharmacodynamic Meta-Analysis of Navitoclax (ABT-263) Induced Thrombocytopenia. Cancer Chemother. Pharmacol. 2014, 74, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-xL and BCL-2 Dual Degrader with Improved Anti-Leukemic Activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, Q.; Zhang, W.; Andreeff, M.; Jain, N.; Zhang, P.; Zheng, G.; Zhou, D.; Konopleva, M. Targeting BCL-XL and BCL-2 By Protac 753B Effectively Eliminates AML Cells and Enhances Efficacy of Chemotherapy By Targeting Senescent Cells. Blood 2021, 138, 2230. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Roskoski, R. Cyclin-Dependent Protein Serine/Threonine Kinase Inhibitors as Anticancer Drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Yu, B.; Du, Z.; Zhang, Y.; Li, Z.; Bian, J. Small-Molecule Degraders of Cyclin-Dependent Kinase Protein: A Review. Future Med. Chem. 2022, 14, 167–185. [Google Scholar] [CrossRef]
- Lu, M.; Wang, Y.; Zhan, X. The MAPK Pathway-Based Drug Therapeutic Targets in Pituitary Adenomas. Front. Endocrinol. 2019, 10, 330. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Z.; Pei, Y.; Song, N.; Xu, L.; Feng, B.; Wang, H.; Luo, X.; Hu, X.; Qiu, X.; et al. Discovery of Thalidomide-Based PROTAC Small Molecules as the Highly Efficient SHP2 Degraders. Eur. J. Med. Chem. 2021, 218, 113341. [Google Scholar] [CrossRef]
- Posternak, G.; Tang, X.; Maisonneuve, P.; Jin, T.; Lavoie, H.; Daou, S.; Orlicky, S.; Goullet de Rugy, T.; Caldwell, L.; Chan, K.; et al. Functional Characterization of a PROTAC Directed against BRAF Mutant V600E. Nat. Chem. Biol. 2020, 16, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wen, Y.; Wang, C.; Zhou, Y.; Zhou, Y.; Zhang, Z.-M.; Liu, T.; Lu, X. Efficient Targeted Oncogenic KRASG12C Degradation via First Reversible-Covalent PROTAC. Eur. J. Med. Chem. 2022, 230, 114088. [Google Scholar] [CrossRef] [PubMed]
- Farnaby, W.; Koegl, M.; McConnell, D.B.; Ciulli, A. Transforming Targeted Cancer Therapy with PROTACs: A Forward-Looking Perspective. Curr. Opin. Pharmacol. 2021, 57, 175–183. [Google Scholar] [CrossRef]
- Lacham-Hartman, S.; Shmidov, Y.; Radisky, E.S.; Bitton, R.; Lukatsky, D.B.; Papo, N. Avidity Observed between a Bivalent Inhibitor and an Enzyme Monomer with a Single Active Site. PLoS ONE 2021, 16, e0249616. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.F. Phototherapy: From Ancient Egypt to the New Millennium. J. Perinatol. 2001, 21, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Amendoeira, A.; García, L.R.; Fernandes, A.R.; Baptista, P.V. Light Irradiation of Gold Nanoparticles Toward Advanced Cancer Therapeutics. Adv. Ther. 2020, 3, 1900153. [Google Scholar] [CrossRef]
- Mendes, R.; Pedrosa, P.; Lima, J.C.; Fernandes, A.R.; Baptista, P.V. Photothermal Enhancement of Chemotherapy in Breast Cancer by Visible Irradiation of Gold Nanoparticles. Sci. Rep. 2017, 7, 10872. [Google Scholar] [CrossRef]
- Reynders, M.; Trauner, D. Optical Control of Targeted Protein Degradation. Cell Chem. Biol. 2021, 28, 969–986. [Google Scholar] [CrossRef]
- Delacour, Q.; Li, C.; Plamont, M.-A.; Billon-Denis, E.; Aujard, I.; Le Saux, T.; Jullien, L.; Gautier, A. Light-Activated Proteolysis for the Spatiotemporal Control of Proteins. ACS Chem. Biol. 2015, 10, 1643–1647. [Google Scholar] [CrossRef]
- Cecchini, C.; Pannilunghi, S.; Tardy, S.; Scapozza, L. From Conception to Development: Investigating PROTACs Features for Improved Cell Permeability and Successful Protein Degradation. Front. Chem. 2021, 9, 672267. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Cui, D.; Chen, X.; Xiong, X.; Zhao, Y. PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. BioEssays 2018, 40, 1700247. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Zhao, E.; Hou, J.; Ke, X.; Abbas, M.N.; Kausar, S.; Zhang, L.; Cui, H. The Roles of Sirtuin Family Proteins in Cancer Progression. Cancers 2019, 11, 1949. [Google Scholar] [CrossRef]
- Sharma, C.; Donu, D.; Curry, A.M.; Barton, E.; Cen, Y. Multifunctional Activity-Based Chemical Probes for Sirtuins. RSC Adv. 2023, 13, 11771–11781. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, F.; Afrin, S.; Forbes-Hernandez, T.Y.; Gasparrini, M.; Cianciosi, D.; Reboredo-Rodriguez, P.; Varela-Lopez, A.; Quiles, J.L.; Battino, M. Autophagy in Human Health and Disease: Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 577–634. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting Autophagy to Overcome Drug Resistance: Further Developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Li, X.; Qiu, K.; Jiao, F.; Liu, Y.; Kong, Q.; Liu, Y.; Wu, Y. Proteasome Inhibition Activates Autophagy-Lysosome Pathway Associated with TFEB Dephosphorylation and Nuclear Translocation. Front. Cell Dev. Biol. 2019, 7, 170. [Google Scholar] [CrossRef]
- Kocak, M.; Ezazi Erdi, S.; Jorba, G.; Maestro, I.; Farrés, J.; Kirkin, V.; Martinez, A.; Pless, O. Targeting Autophagy in Disease: Established and New Strategies. Autophagy 2022, 18, 473–495. [Google Scholar] [CrossRef]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10. [Google Scholar] [CrossRef] [PubMed]
- Noblejas-López, M.d.M.; Nieto-Jimenez, C.; Burgos, M.; Gómez-Juárez, M.; Montero, J.C.; Esparís-Ogando, A.; Pandiella, A.; Galán-Moya, E.M.; Ocaña, A. Activity of BET-Proteolysis Targeting Chimeric (PROTAC) Compounds in Triple Negative Breast Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 383. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Oviedo, L.; Nuncia-Cantarero, M.; Morcillo-Garcia, S.; Nieto-Jimenez, C.; Burgos, M.; Corrales-Sanchez, V.; Perez-Peña, J.; Győrffy, B.; Ocaña, A.; Galán-Moya, E.M. Identification of a Stemness-Related Gene Panel Associated with BET Inhibition in Triple Negative Breast Cancer. Cell. Oncol. 2020, 43, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pan, X.; Wang, A.; Shuai, W.; Bu, F.; Tang, P.; Zhang, S.; Zhang, Y.; Wang, G.; Ouyang, L. Developing Potent LC3-Targeting AUTAC Tools for Protein Degradation with Selective Autophagy. Chem. Commun. 2021, 57, 13194–13197. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef]
- De Vita, E.; Lucy, D.; Tate, E.W. Beyond Targeted Protein Degradation: LD·ATTECs Clear Cellular Lipid Droplets. Cell Res. 2021, 31, 945–946. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, N.; Wang, Z.; Luo, S.; Ding, Y.; Lu, B. Degradation of Lipid Droplets by Chimeric Autophagy-Tethering Compounds. Cell Res. 2021, 31, 965–979. [Google Scholar] [CrossRef]
- Trelford, C.B.; Di Guglielmo, G.M. Molecular Mechanisms of Mammalian Autophagy. Biochem. J. 2021, 478, 3395–3421. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-Targeting Chimaeras for Degradation of Extracellular Proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Ramadas, B.; Kumar Pain, P.; Manna, D. LYTACs: An Emerging Tool for the Degradation of Non-Cytosolic Proteins. ChemMedChem 2021, 16, 2951–2953. [Google Scholar] [CrossRef]
- Zhou, Y.; Teng, P.; Montgomery, N.T.; Li, X.; Tang, W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci. 2021, 7, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs That Engage the Asialoglycoprotein Receptor for Targeted Protein Degradation. Nat. Chem. Biol. 2021, 17, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Guo, Z.-Q.; Wang, B.-F.; Zhao, M. EGFR Alterations in Glioblastoma Play a Role in Antitumor Immunity Regulation. Front. Oncol. 2023, 13, 1236246. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Scaltriti, M.; Baselga, J. The Epidermal Growth Factor Receptor Pathway: A Model for Targeted Therapy. Clin. Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef]
- Sheng, Q.; Liu, J. The Therapeutic Potential of Targeting the EGFR Family in Epithelial Ovarian Cancer. Br. J. Cancer 2011, 104, 1241–1245. [Google Scholar] [CrossRef]
- Liam, C.-K.; Pang, Y.-K.; Poh, M.-E. EGFR Mutations in Asian Patients with Advanced Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, e70–e71. [Google Scholar] [CrossRef]
- Krishnamurti, U.; Silverman, J.F. HER2 in Breast Cancer: A Review and Update. Adv. Anat. Pathol. 2014, 21, 100–107. [Google Scholar] [CrossRef]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 Expression Status in Diverse Cancers: Review of Results from 37,992 Patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.-C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 Maintains the Expression of PD-L1 and Regulates Anti-Tumour Immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Teh, W.P.; Zhu, H.; Marto, J.A.; Buhrlage, S.J. DUB to the Rescue. Mol. Cell 2022, 82, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Chi, F.; Wu, H.; Liu, Y.; Xie, Z.; Huang, W.; Shi, W.; Qian, H. Emerging Targeted Protein Degradation Tools for Innovative Drug Discovery: From Classical PROTACs to the Novel and Beyond. Eur. J. Med. Chem. 2022, 231, 114142. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.; Cheng, M.; Park, K.-S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Sincere, N.I.; Anand, K.; Ashique, S.; Yang, J.; You, C. PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules 2023, 28, 4014. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Samarasinghe, K.T.G.; Crews, C.M. Targeted Protein Degradation: A Promise for Undruggable Proteins. Cell Chem. Biol. 2021, 28, 934–951. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Liu, H.-J.; Wu, Y.; Jin, J.-M.; Zhou, Y.-D.; Zhang, H.; Nagle, D.G.; Chen, H.-Z.; Zhang, W.-D.; Luan, X. Targeted Protein Degradation Technology and Nanomedicine: Powerful Allies against Cancer. Small 2023, 19, 2207778. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Type | Target | Pathway |
|---|---|---|
| AUTAC | Intracellular proteins and damaged organelles | Autophagy-lysosome |
| ATTEC | Intracellular proteins and non-proteic substrates | Autophagy-lysosomes |
| LYTAC | Extracellular and transmembrane proteins | Endosome-lysosome |
| PROTAC | Intracellular proteins | Ubiquitin-proteasome |
| Trivalent PROTAC | Intracellular proteins | Ubiquitin-proteasome |
| PHOTAC | Variety of targets: including BRD2-4 and FKBP12 | Ubiquitin-proteasome |
| CLIPTAC | Intracellular proteins | Ubiquitin-proteasome |
| DUBTAC | Intracellular proteins | Ubiquitin-proteasome |
| Drug Degrader | Compound | Ligase | Target | Reference |
|---|---|---|---|---|
| Heterofunctional PROTAC | KRIBB11 | Pomalidomide | HSF1 | [17] |
| A-1155463 | VHL | Bcl-xL | [18] | |
| AZD1775 | VHL, CRBN | Wee1 | [19] | |
| BI 1701963 (Phase I) | VHL | SOS1 | [20] | |
| ARV-110 (Phase I) | CRBN | AR | [21] | |
| ARV-471 (Phase II) | CRBN | ER | [21] | |
| PHOTAC | Opto-pomalidomide | CRBN | IKZF1/3 | [22] |
| Opto-dBET1 | CRBN | BRD4 | [23] | |
| Opto-dALK | CRBN | EML-ALK | [24] | |
| PHOTAC-II-5 | CRBN | FKBP12 | [25] | |
| PHOTAC-II-6 | CRBN | FKBP12 | [25] | |
| Trivalent PROTAC | VZ185 | CRBN | BRD9 | [26] |
| BRD7 | ||||
| CLIPTAC | Tetrazine-tagged thalidomide | CRBN | BRD4 ERK1/2 | [27] |
| Degrader | Type | Company | Target | E3 Ligase | Phase | Tumor Type | Identifier |
|---|---|---|---|---|---|---|---|
| ARV-110 | Heterobifunctional | Arvinas | AR | CRBN | I | Prostate cancer | NCT03888612 |
| ARV-471 | Heterobifunctional | Arvinas, Pfizer | ER alpha | CRBN | II | Prostate cancer Breast cancer (ER+/HER2−) | NCT04072952 |
| ARV-766 | Heterobifunctional | Arvinas | AR | CRBN | I | Prostate cancer | NTC05067140 |
| AC682 | Heterobifunctional | Accutar Biotech | ER | CRBN | I | Locally Advanced or Metastatic ER+ Breast Cancer | NCT05080842 |
| AR-LDD (CC-94676) | Heterobifunctional | Bristol Myers Squibb | AR | CRBN | I | Metastatic Castration-Resistant Prostate Cancer | NCT04428788 |
| DT2216 | Heterobifunctional | Dialectic | BCL-XL | VHL | I | Solid tumor Hematologic malignancy | NCT04886622 |
| FHD-609 | Heterobifunctional | Foghorn Therapeutics | BRD9 | Undisclosed | I | Advanced Synovial Sarcoma or Advanced SMARCB1-Loss Tumors | NCT04965753 |
| KT-333 | Heterobifunctional | Kymera | STAT3 | Undisclosed | I | Refractory Lymphoma Large Granular Lymphocytic Leukemia Solid Tumors | NCT05225584 |
| KT-413 | Heterobifunctional | Kymera | IRAK4 | Undisclosed | I | Relapsed or refractory diffuse large B-cell lymphoma Marginal zone lymphoma Follicular lymphoma Primary central nervous system lymphoma Waldenstrom macroglobulinemia Nodular lymphocyte-predominant Hodgkin lymphoma. | NA |
| KT-474 | Heterobifunctional | Kymera | IRAK4 | Undisclosed | I | Healthy volunteers Atopic dermatitis Hidradenitis Suppurativa | NCT04772885 |
| NX-2127 | Heterobifunctional | Nurix Therapeutics | BTK | CRBN | I | Chronic lymphocytic leukemia Small lymphocytic lymphoma Mantle cell lymphoma Marginal zone lymphoma Waldenstrom macroglobulinemia Follicular lymphoma Diffuse Large B-cell Lymphoma | NCT04830137 |
| NX-5948 | Molecular glue | Nurix Therapeutics | BTK | CRBN | I | Hematological malignancies | Enter in late 2021 |
| CFT7455 | Molecular glue | C4 Therapeutics | IKZF 1/3 | CRBN | I/II | Multiple myeloma and non-Hodgkin’s lymphomas | NCT04756726 |
| CC92480 | Molecular glue | Bristol-Myers Squibb | IKZF 1/3 | CRBN | II | Multiple myeloma | NCT03989414 |
| CC9982 | Molecular glue | Bristol-Myers Squibb | IKZF 1/3 | CRBN | II | Lymphoma non-Hodgkin’s Lymphoma large B-cell diffuse Lymphoma follicular | NCT03310619 |
| CFT8634 | Heterobifunctional | C4 Therapeutics | BRD9 | CRBN | I/II | Synovial Sarcoma SMARCB1-Null Tumors | NCT05355753 |
| CFT8919 | Heterobifunctional | C4 Therapeutics | EGFR | CRBN | Non-small cell lung cancer | NA | |
| DT2216 | Heterobifunctional | Dialectic therapeutics | Bcl-Xl | VHL | I | Solid tumor Hematologic malignancy | NCT04886622 |
| BGB-16673 | Heterobifunctional | BeiGene | BTK | Undisclosed | I | B-cell malignancy marginal zone lymphoma Follicular lymphoma non-Hodgkin’s Lymphoma Waldenström macroglobulinemia | NCT05006716 |
| FHD-609 | Heterobifunctional | Foghorn Therapeutics Inc | BRD9 | Undisclosed | I | Advanced synovial sarcoma | NCT04965753 |
| CC220 | Molecular glue | Bristol-Myers Squibb | IKZF1/3 | CRBN | II | Multiple Myeloma | NCT02773030 |
| CC90009 | Molecular glue | Bristol-Myers Squibb | GSPT1 | CRBN | II | Acute myeloid leukemia | NCT02848001 NCT04336982 |
| CC99282 | Molecular glue | Bristol-Myers Squibb | IKZF1/3 | CRBN | I | Chronic myeloid leukemia, and non-Hodgkin’s lymphoma | NCT04434196 NCT03930953 |
| CFT7455 | Molecular glue | C4 Therapeutics | IKZF1/3 | CRBN | I | Multiple Myeloma | NCT04756726 |
| DKY709 | Molecular glue | Novartis | Helios | CRBN | I | Solid tumors (Non-small-cell lung carcinoma) | NCT03891953 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noblejas-López, M.d.M.; Tébar-García, D.; López-Rosa, R.; Alcaraz-Sanabria, A.; Cristóbal-Cueto, P.; Pinedo-Serrano, A.; Rivas-García, L.; Galán-Moya, E.M. TACkling Cancer by Targeting Selective Protein Degradation. Pharmaceutics 2023, 15, 2442. https://doi.org/10.3390/pharmaceutics15102442
Noblejas-López MdM, Tébar-García D, López-Rosa R, Alcaraz-Sanabria A, Cristóbal-Cueto P, Pinedo-Serrano A, Rivas-García L, Galán-Moya EM. TACkling Cancer by Targeting Selective Protein Degradation. Pharmaceutics. 2023; 15(10):2442. https://doi.org/10.3390/pharmaceutics15102442
Chicago/Turabian StyleNoblejas-López, María del Mar, David Tébar-García, Raquel López-Rosa, Ana Alcaraz-Sanabria, Pablo Cristóbal-Cueto, Alejandro Pinedo-Serrano, Lorenzo Rivas-García, and Eva M. Galán-Moya. 2023. "TACkling Cancer by Targeting Selective Protein Degradation" Pharmaceutics 15, no. 10: 2442. https://doi.org/10.3390/pharmaceutics15102442
APA StyleNoblejas-López, M. d. M., Tébar-García, D., López-Rosa, R., Alcaraz-Sanabria, A., Cristóbal-Cueto, P., Pinedo-Serrano, A., Rivas-García, L., & Galán-Moya, E. M. (2023). TACkling Cancer by Targeting Selective Protein Degradation. Pharmaceutics, 15(10), 2442. https://doi.org/10.3390/pharmaceutics15102442