

Epidrugs as Promising Tools to Eliminate Plasmodium falciparum Artemisinin-Resistant and Quiescent Parasites
 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Compounds
2.2. Parasite Culture
2.3. Standard Chemosensitivity Assay
2.4. Cytotoxicity Evaluation
2.5. Quiescent-Stage Survival Assay (QSA)
2.6. Determination of Abundance of Methylated and Acetylated Histone
3. Results and discussion
3.1. Antiplasmodial Activity of Some Epidrugs
3.2. Specific Antiplasmodial Activity of Certain Epidrugs
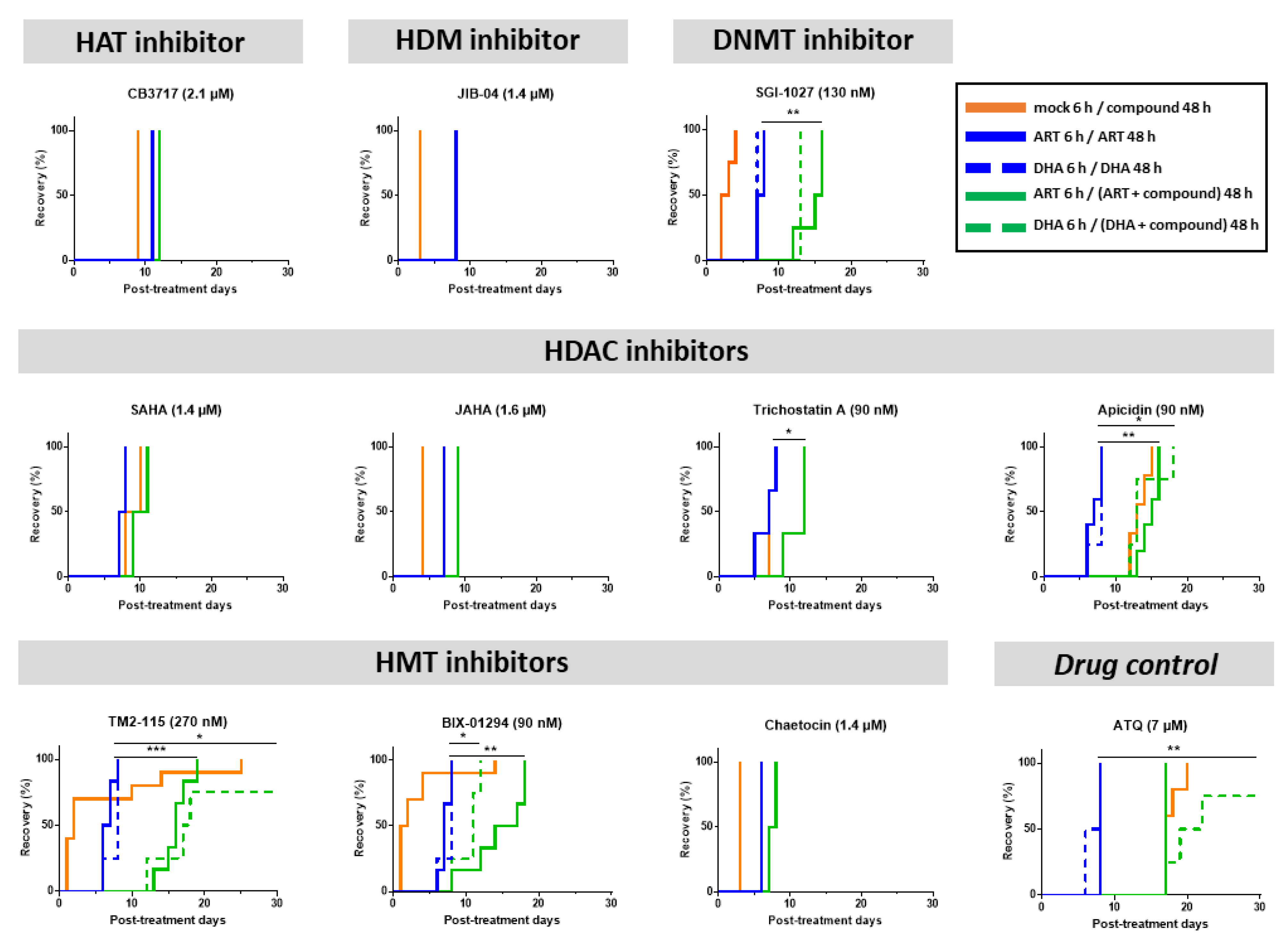
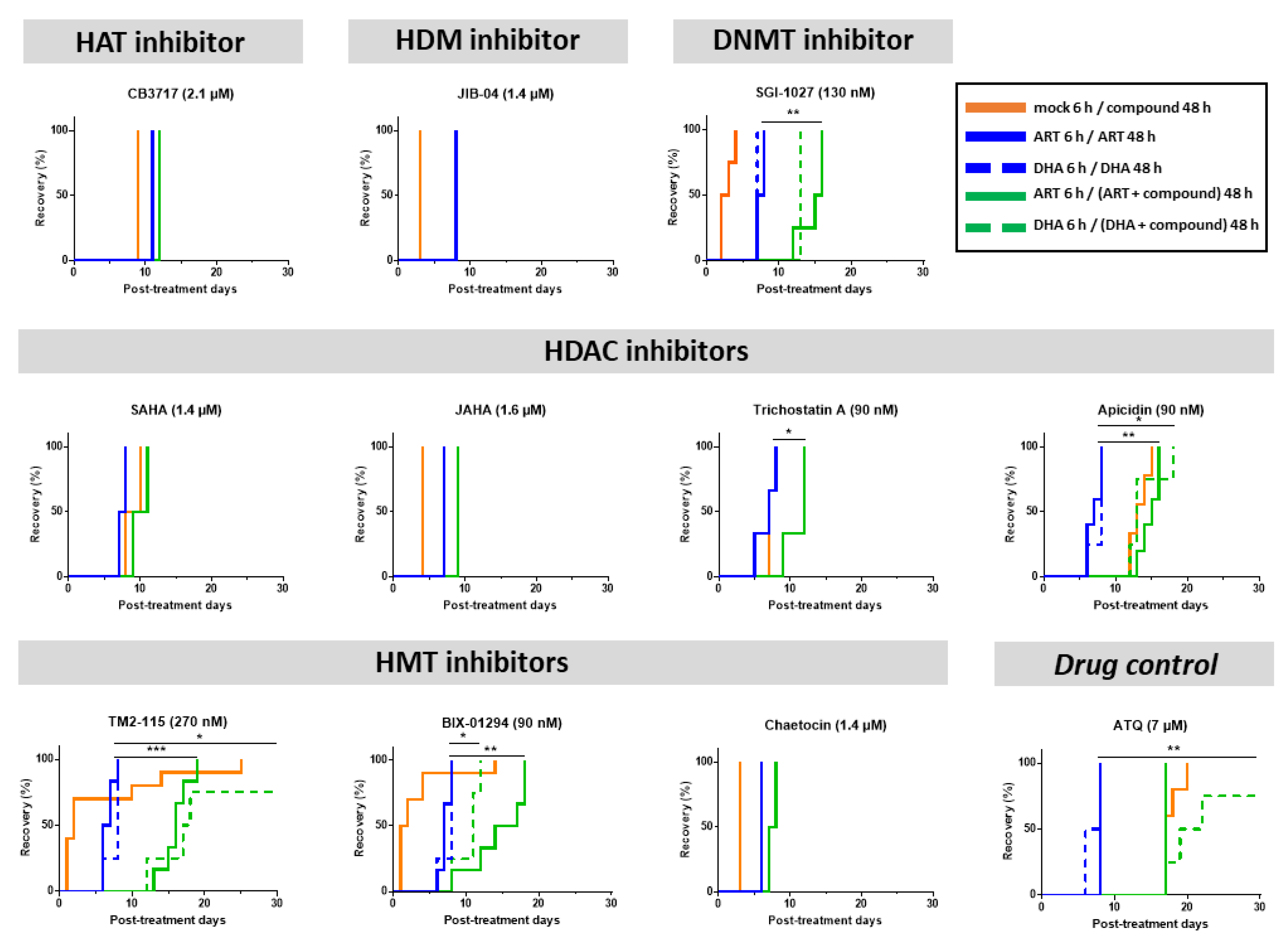
3.3. Some Epidrugs Are Active against ART-Resistant Parasites in a Quiescent State
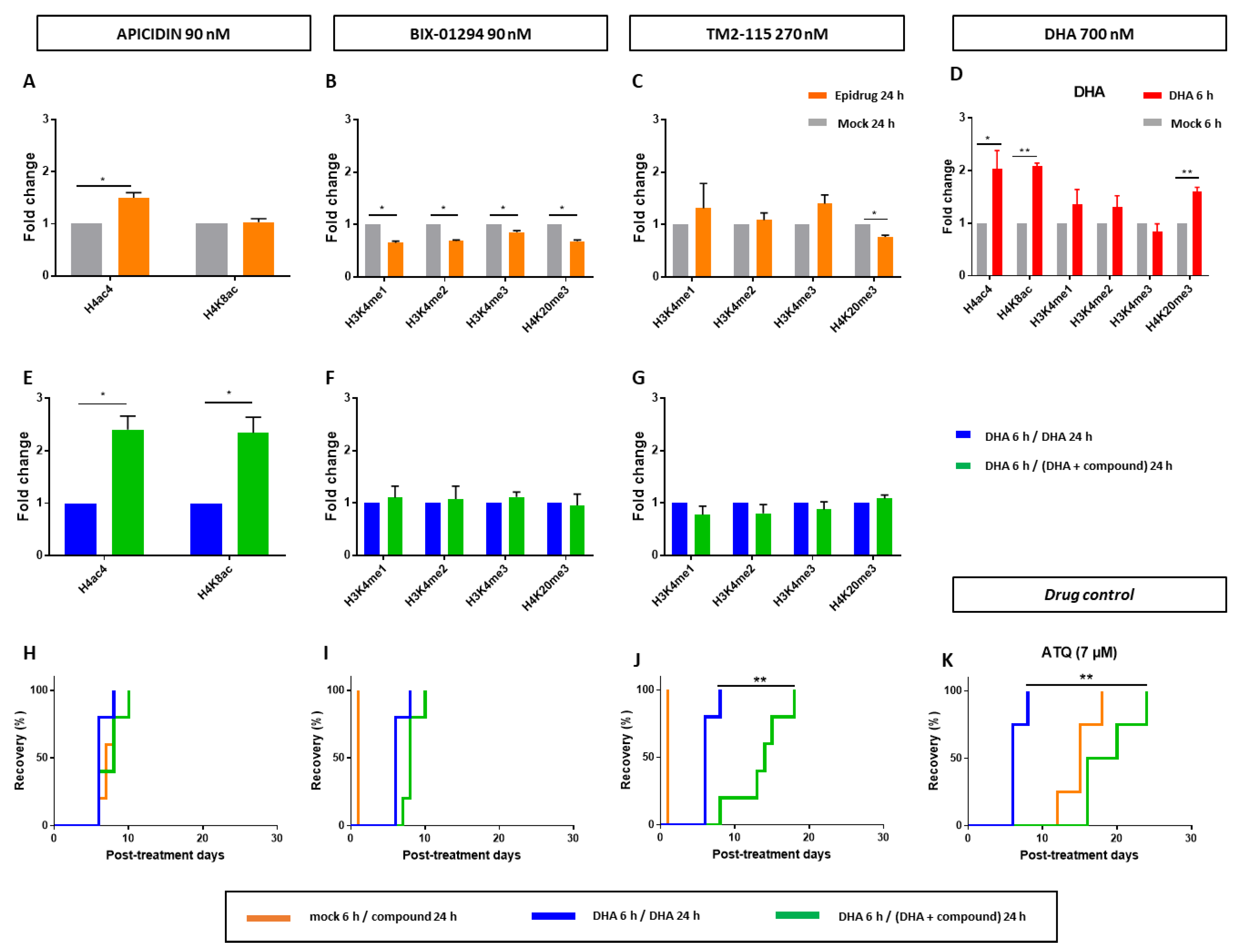
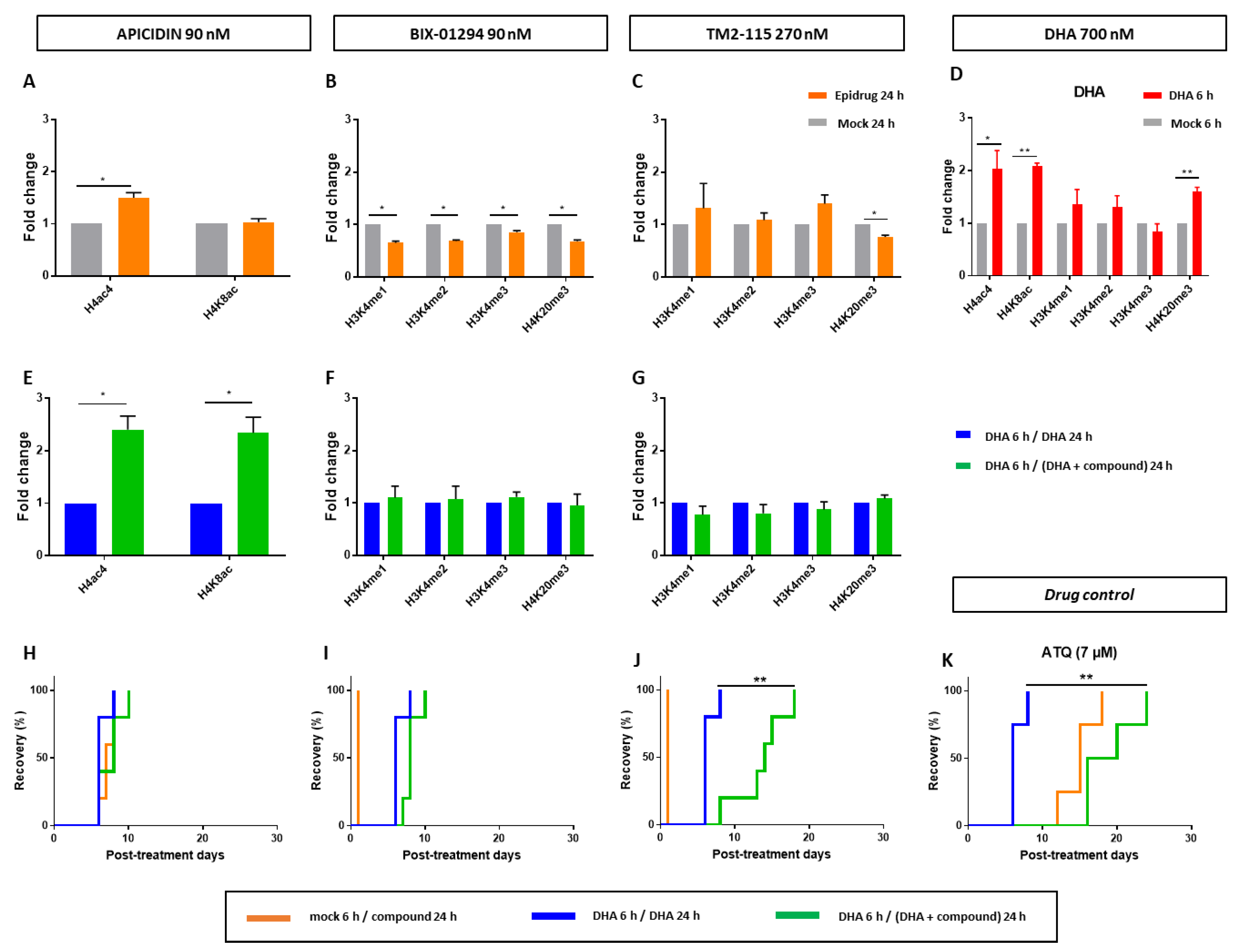
3.4. Effects of Some Epidrugs on Histone Methylation and Acetylation in DHA-Treated Parasites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Malaria Report 2022; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Amato, R.; Pearson, R.D.; Almagro-Garcia, J.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Drury, E.; Stalker, J.; Miotto, O.; et al. Origins of the current outbreak of multidrug-resistant malaria in southeast Asia: A retrospective genetic study. Lancet Infect. Dis. 2018, 18, 337–345. [Google Scholar] [CrossRef]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Haldar, K.; Bhattacharjee, S.; Safeukui, I. Drug resistance in Plasmodium. Nat. Rev. Microbiol. 2018, 16, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Paloque, L.; Ramadani, A.P.; Mercereau-Puijalon, O.; Augereau, J.M.; Benoit-Vical, F. Plasmodium falciparum: Multifaceted resistance to artemisinins. Malar. J. 2016, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.E.; Fidock, D.A.; Bridgford, J.L. Plasmodium falciparum resistance to artemisinin-based combination therapies. Curr. Opin. Microbiol. 2022, 69, 102193. [Google Scholar] [CrossRef]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef]
- Birnbaum, J.; Scharf, S.; Schmidt, S.; Jonscher, E.; Hoeijmakers, W.A.M.; Flemming, S.; Toenhake, C.G.; Schmitt, M.; Sabitzki, R.; Bergmann, B.; et al. A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 2020, 367, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Egwu, C.O.; Perio, P.; Augereau, J.M.; Tsamesidis, I.; Benoit-Vical, F.; Reybier, K. Resistance to artemisinin in falciparum malaria parasites: A redox-mediated phenomenon. Free Radic. Biol. Med. 2022, 179, 317–327. [Google Scholar] [CrossRef]
- Mok, S.; Ashley, E.A.; Ferreira, P.E.; Zhu, L.; Lin, Z.; Yeo, T.; Chotivanich, K.; Imwong, M.; Pukrittayakamee, S.; Dhorda, M.; et al. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 2015, 347, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gallego-Delgado, J.; Fernandez-Arias, C.; Waters, N.C.; Rodriguez, A.; Tsuji, M.; Wek, R.C.; Nussenzweig, V.; Sullivan, W.J., Jr. Inhibiting the Plasmodium eIF2alpha Kinase PK4 Prevents Artemisinin-Induced Latency. Cell Host Microbe 2017, 22, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.; Stokes, B.H.; Gnadig, N.F.; Ross, L.S.; Yeo, T.; Amaratunga, C.; Allman, E.; Solyakov, L.; Bottrill, A.R.; Tripathi, J.; et al. Artemisinin-resistant K13 mutations rewire Plasmodium falciparum’s intra-erythrocytic metabolic program to enhance survival. Nat. Commun. 2021, 12, 530. [Google Scholar] [CrossRef]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.V.; Rizk, S.S.; Njimoh, D.L.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Coppens, I.; Mbengue, A.; Suresh, N.; Ghorbal, M.; Slouka, Z.; Safeukui, I.; Tang, H.Y.; Speicher, D.W.; Stahelin, R.V.; et al. Remodeling of the malaria parasite and host human red cell by vesicle amplification that induces artemisinin resistance. Blood 2018, 131, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.A.; Gresty, K.J.; Chen, N.; Zhang, V.; Gutteridge, C.E.; Peatey, C.L.; Chavchich, M.; Waters, N.C.; Cheng, Q. Correlation between Cyclin Dependent Kinases and Artemisinin-Induced Dormancy in Plasmodium falciparum In Vitro. PLoS ONE 2016, 11, e0157906. [Google Scholar] [CrossRef]
- Tucker, M.S.; Mutka, T.; Sparks, K.; Patel, J.; Kyle, D.E. Phenotypic and Genotypic Analysis of In Vitro-Selected Artemisinin-Resistant Progeny of Plasmodium falciparum. Antimicrob. Agents Chemother. 2012, 56, 302–314. [Google Scholar] [CrossRef]
- Witkowski, B.; Khim, N.; Chim, P.; Kim, S.; Ke, S.; Kloeung, N.; Chy, S.; Duong, S.; Leang, R.; Ringwald, P.; et al. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob. Agents Chemother. 2013, 57, 914–923. [Google Scholar] [CrossRef]
- Witkowski, B.; Amaratunga, C.; Khim, N.; Sreng, S.; Chim, P.; Kim, S.; Lim, P.; Mao, S.; Sopha, C.; Sam, B.; et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: In-vitro and ex-vivo drug-response studies. Lancet Infect. Dis. 2013, 13, 1043–1049. [Google Scholar] [CrossRef]
- Dogovski, C.; Xie, S.C.; Burgio, G.; Bridgford, J.; Mok, S.; McCaw, J.M. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol. 2015, 13, e1002132. [Google Scholar] [CrossRef]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef]
- Peatey, C.; Chen, N.; Gresty, K.; Anderson, K.; Pickering, P.; Watts, R.; Gatton, M.L.; McCarthy, J.; Cheng, Q. Dormant Plasmodium falciparum Parasites in Human Infections Following Artesunate Therapy. J. Infect. Dis. 2021, 223, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Peatey, C.L.; Chavchich, M.; Chen, N.; Gresty, K.J.; Gray, K.A.; Gatton, M.L.; Waters, N.C.; Cheng, Q. Mitochondrial Membrane Potential in a Small Subset of Artemisinin-Induced Dormant Plasmodium falciparum Parasites In Vitro. J. Infect. Dis. 2015, 212, 426–434. [Google Scholar] [CrossRef]
- Teuscher, F.; Gatton, M.L.; Chen, N.; Peters, J.; Kyle, D.E.; Cheng, Q. Artemisinin-induced dormancy in plasmodium falciparum: Duration, recovery rates, and implications in treatment failure. J. Infect. Dis. 2010, 202, 1362–1368. [Google Scholar] [CrossRef]
- Connelly, S.V.; Manzella-Lapeira, J.; Levine, Z.C.; Brzostowski, J.; Krymskaya, L.; Rahman, R.S.; Ellis, A.C.; Amin, S.N.; Sa, J.M.; Wellems, T.E. Restructured Mitochondrial-Nuclear Interaction in Plasmodium falciparum Dormancy and Persister Survival after Artemisinin Exposure. mBio 2021, 12, e0075321. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, B.; Lelievre, J.; Barragan, M.J.; Laurent, V.; Su, X.Z.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef]
- Reyser, T.; Paloque, L.; Ouji, M.; Nguyen, M.; Menard, S.; Witkowski, B.; Augereau, J.M.; Benoit-Vical, F. Identification of compounds active against quiescent artemisinin-resistant Plasmodium falciparum parasites via the quiescent-stage survival assay (QSA). J. Antimicrob. Chemother. 2020, 75, 2826–2834. [Google Scholar] [CrossRef]
- Bozdech, Z.; Llinas, M.; Pulliam, B.L.; Wong, E.D.; Zhu, J.; DeRisi, J.L. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1, E5. [Google Scholar] [CrossRef]
- Nardella, F.; Halby, L.; Hammam, E.; Erdmann, D.; Cadet-Daniel, V.; Peronet, R.; Menard, D.; Witkowski, B.; Mecheri, S.; Scherf, A.; et al. DNA Methylation Bisubstrate Inhibitors Are Fast-Acting Drugs Active against Artemisinin-Resistant Plasmodium falciparum Parasites. ACS Cent. Sci. 2020, 6, 16–21. [Google Scholar] [CrossRef]
- Nardella, F.; Halby, L.; Dobrescu, I.; Viluma, J.; Bon, C.; Claes, A.; Cadet-Daniel, V.; Tafit, A.; Roesch, C.; Hammam, E.; et al. Procainamide-SAHA Fused Inhibitors of hHDAC6 Tackle Multidrug-Resistant Malaria Parasites. J. Med. Chem. 2021, 64, 10403–10417. [Google Scholar] [CrossRef]
- Wang, M.; Tang, T.; Li, R.; Huang, Z.; Ling, D.; Zheng, L.; Ding, Y.; Liu, T.; Xu, W.; Zhu, F.; et al. Drug Repurposing of Quisinostat to Discover Novel Plasmodium falciparum HDAC1 Inhibitors with Enhanced Triple-Stage Antimalarial Activity and Improved Safety. J. Med. Chem. 2022, 65, 4156–4181. [Google Scholar] [CrossRef]
- Bouchut, A.; Rotili, D.; Pierrot, C.; Valente, S.; Lafitte, S.; Schultz, J.; Hoglund, U.; Mazzone, R.; Lucidi, A.; Fabrizi, G.; et al. Identification of novel quinazoline derivatives as potent antiplasmodial agents. Eur. J. Med. Chem. 2019, 161, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, A.K.; Oakes, C.S.; Hassell-Hart, S.; Bruyère, D.; Tizzard, G.J.; Coles, S.J.; Felix, R.; Maple, H.J.; Marsh, G.P.; Spencer, J. Scale-up and optimization of the synthesis of dual CBP/BRD4 inhibitor ISOX-DUAL. Org. Biomol. Chem. 2022, 20, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, A.; Rajavelu, A.; Champion, C.; Rampon, C.; Jurkowska, R.; Jankevicius, G.; Sénamaud-Beaufort, C.; Ponger, L.; Gagey, N.; Dali Ali, H.; et al. C5-DNA Methyltransferase Inhibitors: From Screening to Effects on Zebrafish Embryo Development. Chembiochem 2011, 12, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Dauzonne, D.; Folléas, B.; Martinez, L.; Chabot, G.G. Synthesis and in vitro cytotoxicity of a series of 3-aminoflavones. Eur. J. Med. Chem. 1997, 32, 71–82. [Google Scholar] [CrossRef]
- Pechalrieu, D.; Dauzonne, D.; Arimondo, P.B.; Lopez, M. Synthesis of novel 3-halo-3-nitroflavanones and their activities as DNA methyltransferase inhibitors in cancer cells. Eur. J. Med. Chem. 2020, 186, 111829. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Andrews, K.T.; Tran, T.N.; Lucke, A.J.; Kahnberg, P.; Le, G.T.; Boyle, G.M.; Gardiner, D.L.; Skinner-Adams, T.S.; Fairlie, D.P. Potent Antimalarial Activity of Histone Deacetylase Inhibitor Analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. [Google Scholar] [CrossRef]
- Huber, K.; Doyon, G.; Plaks, J.; Fyne, E.; Mellors, J.W.; Sluis-Cremer, N. Inhibitors of Histone Deacetylases. J. Biol. Chem. 2011, 286, 22211–22218. [Google Scholar] [CrossRef]
- Spencer, J.; Amin, J.; Wang, M.; Packham, G.; Alwi, S.S.; Tizzard, G.J.; Coles, S.J.; Paranal, R.M.; Bradner, J.E.; Heightman, T.D. Synthesis and Biological Evaluation of JAHAs: Ferrocene-Based Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.M.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Leyk, J.; Daly, C.; Janssen-Bienhold, U.; Kennedy, B.N.; Richter-Landsberg, C. HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 2017, 8, e3028. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef]
- Krishna, B.A.; Lau, B.; Jackson, S.E.; Wills, M.R.; Sinclair, J.H.; Poole, E. Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci. Rep. 2016, 6, 24674. [Google Scholar] [CrossRef]
- Malmquist, N.A.; Moss, T.A.; Mecheri, S.; Scherf, A.; Fuchter, M.J. Small-molecule histone methyltransferase inhibitors display rapid antimalarial activity against all blood stage forms in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2012, 109, 16708–16713. [Google Scholar] [CrossRef]
- Lai, Y.S.; Chen, J.Y.; Tsai, H.J.; Chen, T.Y.; Hung, W.C. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. 2015, 5, e313. [Google Scholar] [CrossRef]
- Kumar, A.; Bhowmick, K.; Vikramdeo, K.S.; Mondal, N.; Subbarao, N.; Dhar, S.K. Designing novel inhibitors against histone acetyltransferase (HAT: GCN5) of Plasmodium falciparum. Eur. J. Med. Chem. 2017, 138, 26–37. [Google Scholar] [CrossRef]
- Valerio, D.G.; Xu, H.; Chen, C.W.; Hoshii, T.; Eisold, M.E.; Delaney, C.; Cusan, M.; Deshpande, A.J.; Huang, C.H.; Lujambio, A.; et al. Histone Acetyltransferase Activity of MOF Is Required for MLL-AF9 Leukemogenesis. Cancer Res. 2017, 77, 1753–1762. [Google Scholar] [CrossRef]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Parrish, J.K.; McCann, T.S.; Sechler, M.; Sobral, L.M.; Ren, W.; Jones, K.L.; Tan, A.C.; Jedlicka, P. The Jumonji-domain histone demethylase inhibitor JIB-04 deregulates oncogenic programs and increases DNA damage in Ewing Sarcoma, resulting in impaired cell proliferation and survival, and reduced tumor growth. Oncotarget 2018, 9, 33110–33123. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A New Class of Quinoline-Based DNA Hypomethylating Agents Reactivates Tumor Suppressor Genes by Blocking DNA Methyltransferase 1 Activity and Inducing Its Degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, C. Hydralazine inhibits human cervical cancer cell growth in vitro in association with APC demethylation and re-expression. Cancer Chemother. Pharmacol. 2008, 63, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.E.; Lee, J.W.; Bohmer, M.J.; Welden, J.D.; Arshadi, A.K.; Du, L.; Cichewicz, R.H.; Chakrabarti, D. Cyclic Tetrapeptide HDAC Inhibitors with Improved Plasmodium falciparum Selectivity and Killing Profile. ACS Infect. Dis. 2021, 7, 2889–2903. [Google Scholar] [CrossRef]
- Vanheer, L.N.; Zhang, H.; Lin, G.; Kafsack, B.F. Activity of Epigenetic Inhibitors against Plasmodium falciparum Asexual and Sexual Blood Stages. Antimicrob. Agents Chemother. 2020, 64, e02523-19. [Google Scholar] [CrossRef]
- Espinoza-Chávez, R.M.; Salerno, A.; Liuzzi, A.; Ilari, A.; Milelli, A.; Uliassi, E.; Bolognesi, M.L. Targeted Protein Degradation for Infectious Diseases: From Basic Biology to Drug Discovery. ACS Bio. Med. Chem. Au. 2023, 3, 32–45. [Google Scholar] [CrossRef]
- Chaal, B.K.; Gupta, A.P.; Wastuwidyaningtyas, B.D.; Luah, Y.H.; Bozdech, Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 2010, 6, e1000737. [Google Scholar] [CrossRef]
- Ménard, S.; Ben Haddou, T.; Ramadani, A.P.; Ariey, F.; Iriart, X.; Beghain, J.; Bouchier, C.; Witkowski, B.; Berry, A.; Mercereau-Puijalon, O.; et al. Induction of multidrug tolerance in Plasmodium falciparum by extended Artemisinin pressure. Emerg. Infect. Dis. J. 2015, 21, 1733–1741. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Antiplasmodial Activity: IC50 (nM) | p-Value 1 (F32-ART vs. F32-TEM) | Cytotoxicity: CC50 (µM) (Cell Type) | Selectivity Index 2 CC50/IC50 | ||
|---|---|---|---|---|---|---|
| F32-ART | F32-TEM | |||||
| HDAC inhibitors | Trichostatin A | 29 ± 12 | 30 ± 3 | 0.8 | 0.2 (NFF) [40] 3 | 7 |
| Apicidin | 36 ± 9 | 36 ± 5 | >0.99 | 10 (Jurkat) [41] 3 | 280 | |
| JAHA | 460 ± 30 | 690 ± 80 | 0.1 | 2.4 (MCF7 cancer) [42] 3 | 5 | |
| SAHA | 540 ± 55 | 390 ± 10 | 0.1 | 5.5 (NFF) [43] 3 | 10 | |
| Tubastatin A | 7 × 103 * | 8 × 103 * | >10 (661W) [44] 3 | >1.4 | ||
| PCI 34051 | 8 × 103 * | 9 × 103 * | >20 (MCF7) [45] 3 | >2.5 | ||
| TC-H 106 | >10 × 103 * | >10 × 103 * | 6.3 (GM15850) [46] 3 | <0.6 | ||
| MC 1568 | >10 × 103 * | >10 × 103 * | 10 (monocytes) [47] 3 | <1 | ||
| HMT inhibitors | BI × -01294 | 56 ± 8 | 55 ± 14 | >0.99 | 6.1 (HFF) [48] 3 | 110 |
| TM2-115 | 128 ± 30 | 104 ± 32 | 0.4 | 5.7 (HFF) [48] 3 | 45 | |
| Chaetocin | 640 ± 160 | 730 ± 90 | >0.99 | 0.13 (HL-60) [49] 3 | 0.2 | |
| HAT inhibitors | CB3717 | 1 × 103 ± 0.16 × 103 | 1 × 103 ± 0.11 × 103 | >0.99 | >20 (NIH3T3) [50] 3 | >20 |
| MG149 | >10 × 103 | >10 × 103 | 51 (NOMO1) [51] 3 | <1.7 | ||
| C646 | >10 × 103 | >10 × 103 | Between 10 and 20 (WM983A) [52] 3 | <1 | ||
| HDM inhibitor | JIB-04 | 560 ± 80 | 940 ± 30 | 0.1 | >10 (human mesenchymal stem) [53] 3 | >18 |
| DNMT inhibitors | SGI-1027 | 63 ± 13 | 54 ±22 | 0.7 | >50 (H4IIErat) [54] 3 | >790 |
| MLo1302 | 7 × 103 ± 2 × 103 | 8 × 103 * | 6.5 * (Vero) | 0.9 | ||
| Hydralazine | >10 × 103 | >10 × 103 | 0.4 | 40 (HeLa) [55] 3 | <4 | |
| Flv69 | >10 × 103 * | >10 × 103 * | >50 * (Vero) | |||
| Flv880 | >10 × 103 * | >10 × 103 * | >50 * (Vero) | |||
| MLo1401 | >10 × 103 * | >10 × 103 * | 20 * (Vero) | <2 | ||
| MLo1406 | >10 × 103 * | >10 × 103 * | >50 * (Vero) | |||
| MLo1502 | >10 × 103 | >10 × 103 * | >50 * (Vero) | |||
| MLo1507 | >10 × 103 * | >10 × 103 * | 40 * (Vero) | <4 | ||
| MLo1508 | >10 × 103 * | >10 × 103 * | 36 * (Vero) | <3.6 | ||
| MLo1509 | >10 × 103 * | >10 × 103 * | >50 (Vero) | |||
| NFlav2018 | >10 × 103 * | >10 × 103 * | >50 * (Vero) | |||
| Bromodomain inhibitors | (+)-JD-1 | 6 × 103 * | 4.5 × 103 * | 0.6 * (Vero) | 0.1 | |
| SHH-878-1 | 10 × 103 * | 10 × 103 * | 3 * (Vero) | 0.3 | ||
| (+)-JQ-1 | >10 × 103 * | >10 × 103 * | 0.2 * (Vero) | <0.02 | ||
| AKE-040 | >10 × 103 * | >10 × 103 * | 24 * (Vero) | <2.4 | ||
| PROTAC BRD4 degrader | MZ1 | 1.7 × 103 ± 0.3 × 103 | 1.3 × 103 * | >50 * (Vero) | >30 | |
| Control drug | artemisinin | 14 ± 3 | 13±3 | 0.5 | >100 (Vero) | >7500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyser, T.; Paloque, L.; Nguyen, M.; Augereau, J.-M.; Fuchter, M.J.; Lopez, M.; Arimondo, P.B.; Hassell-Hart, S.; Spencer, J.; Di Stefano, L.; et al. Epidrugs as Promising Tools to Eliminate Plasmodium falciparum Artemisinin-Resistant and Quiescent Parasites. Pharmaceutics 2023, 15, 2440. https://doi.org/10.3390/pharmaceutics15102440
Reyser T, Paloque L, Nguyen M, Augereau J-M, Fuchter MJ, Lopez M, Arimondo PB, Hassell-Hart S, Spencer J, Di Stefano L, et al. Epidrugs as Promising Tools to Eliminate Plasmodium falciparum Artemisinin-Resistant and Quiescent Parasites. Pharmaceutics. 2023; 15(10):2440. https://doi.org/10.3390/pharmaceutics15102440
Chicago/Turabian StyleReyser, Thibaud, Lucie Paloque, Michel Nguyen, Jean-Michel Augereau, Matthew John Fuchter, Marie Lopez, Paola B. Arimondo, Storm Hassell-Hart, John Spencer, Luisa Di Stefano, and et al. 2023. "Epidrugs as Promising Tools to Eliminate Plasmodium falciparum Artemisinin-Resistant and Quiescent Parasites" Pharmaceutics 15, no. 10: 2440. https://doi.org/10.3390/pharmaceutics15102440
APA StyleReyser, T., Paloque, L., Nguyen, M., Augereau, J.-M., Fuchter, M. J., Lopez, M., Arimondo, P. B., Hassell-Hart, S., Spencer, J., Di Stefano, L., & Benoit-Vical, F. (2023). Epidrugs as Promising Tools to Eliminate Plasmodium falciparum Artemisinin-Resistant and Quiescent Parasites. Pharmaceutics, 15(10), 2440. https://doi.org/10.3390/pharmaceutics15102440