

A Micro-Configured Multiparticulate Reconstitutable Suspension Powder of Fixed Dose Rifampicin and Pyrazinamide: Optimal Fabrication and In Vitro Quality Evaluation

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the FDC Reconstitutable Oral Suspension Formulations
2.2.1. Building the Box–Behnken Experimental Design Template
2.2.2. Fabrication of the FDC Reconstitutable Suspension Utilizing Laboratory Methods
2.3. Measurement of Response Parameters
2.3.1. Formulation Yield
2.3.2. Angle of Repose (Flowability)
2.3.3. Measurement of the Degree of Redispersibility
2.3.4. Determination of Sedimentation Volume (Sedimentation Indicator)
2.4. Optimization of the FDC Reconstitutable Dry Suspension Formulation
2.5. In Vitro Evaluation of the Optimized FDC Oral Suspension Formulation
2.5.1. Quantification of Yield, Flowability, Resuspension Rate and Sedimentation Indicator
2.5.2. Drug Loading Efficiency
2.5.3. Dissolution Studies and Drug Release Kinetics
2.6. Optimized Formulation Characterization
2.6.1. Differential Scanning Calorimetry
2.6.2. Thermal Gravimetric Analysis
2.6.3. Structural Elucidation
2.6.4. X-ray Diffractometry
2.6.5. Rheological Behaviour
2.7. Microscopic Analyses
2.7.1. Particle Surface Topography and Shape
2.7.2. Particle Size and Distribution
2.8. Quantification of Surface Area and Porosity Parameters
2.9. Organoleptic Evaluations
2.10. Effect of Changes in Environmental Conditions on the Stability of the Dry and Reconstituted Suspension Powder
2.11. In Vitro Cytobiocompatibility Evaluation of the Optimized Suspension Formulation
3. Results and Discussion
3.1. Design of Experiments
3.2. Process Optimization and Validation
3.3. In Vitro Assessment of the Optimized Suspension Quality and Release Behaviour
3.3.1. Quantification of Physical Parameters
3.3.2. Drug Content Uniformity
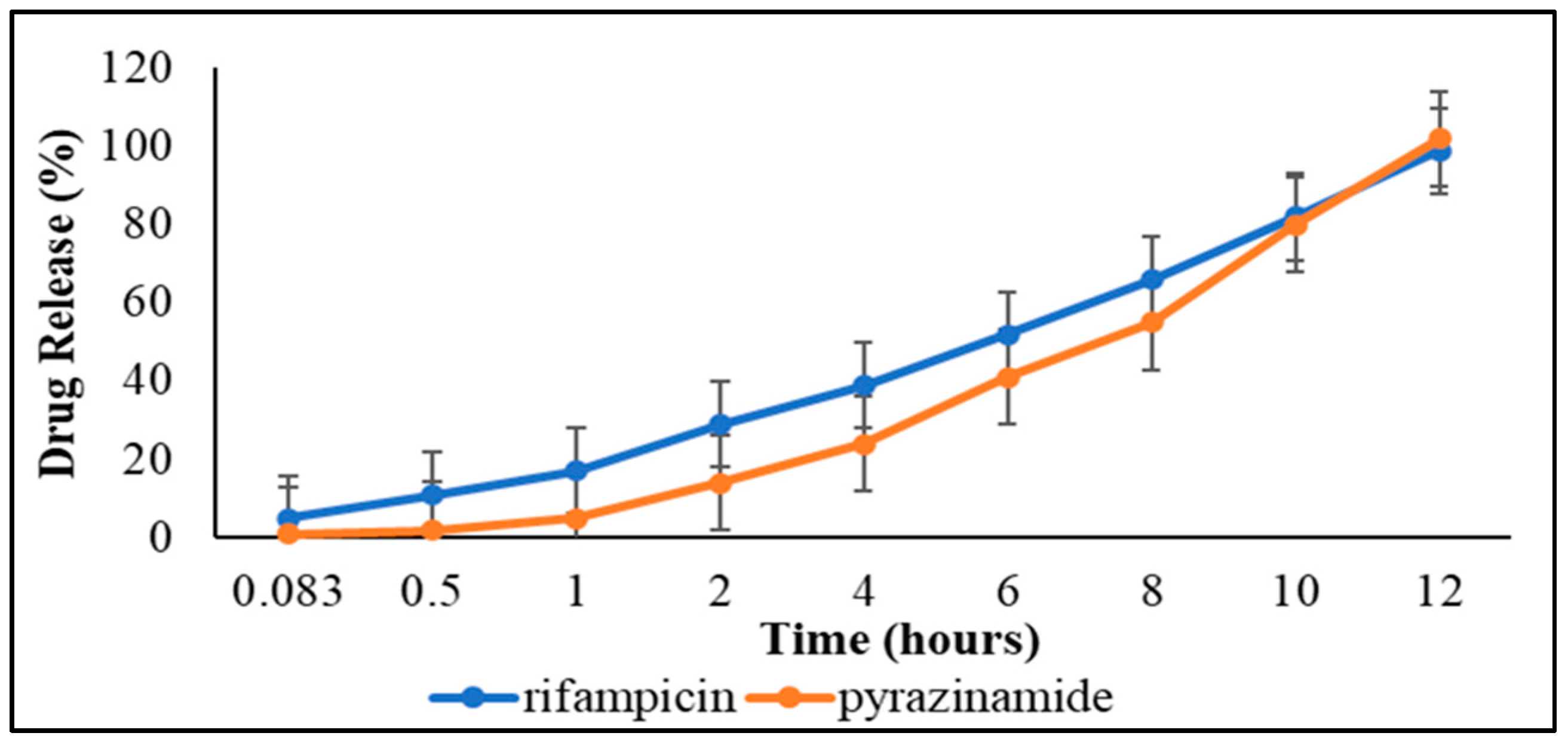
3.3.3. Dissolution Studies and Drug Release Kinetics
3.4. Physicochemical Characterization
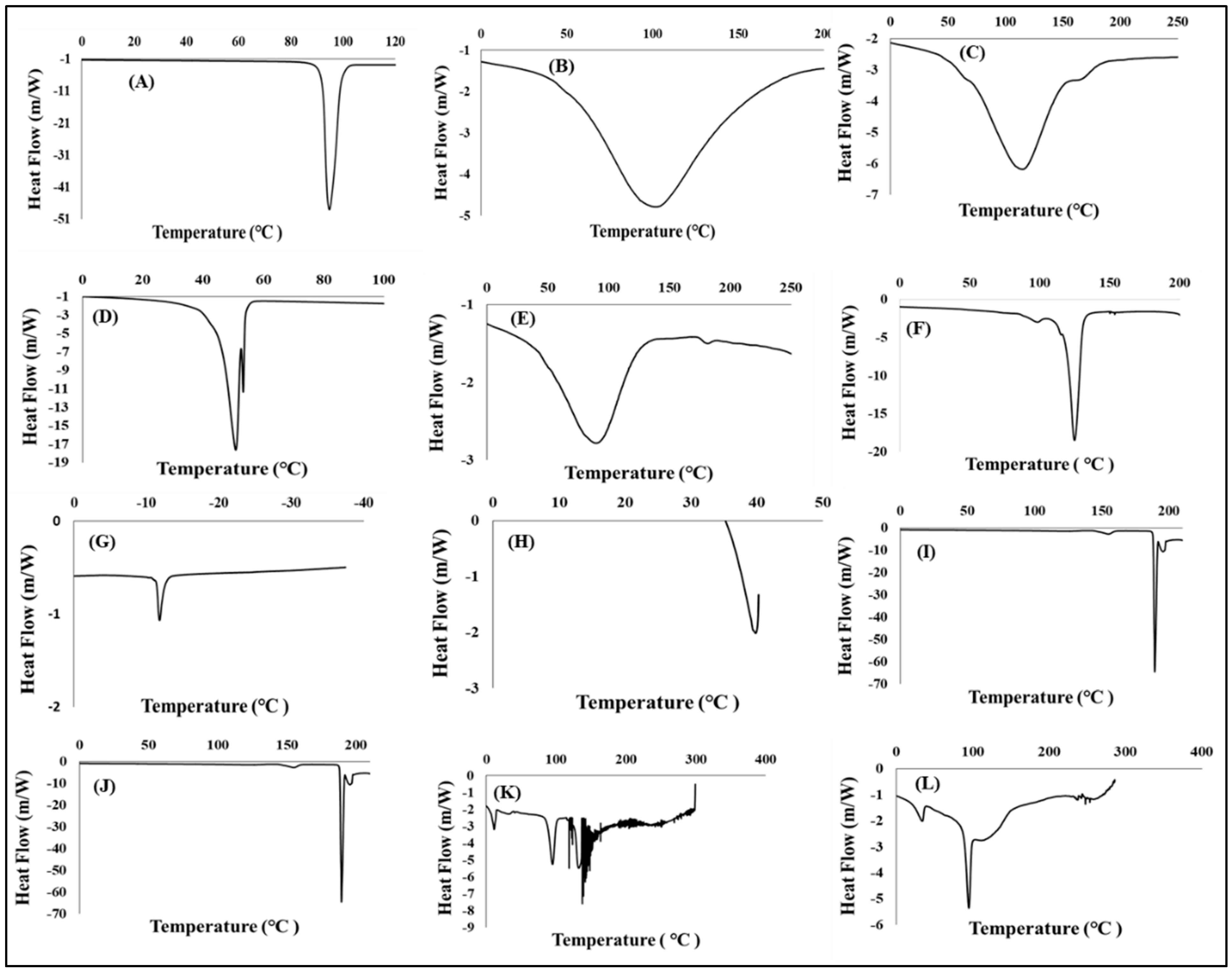
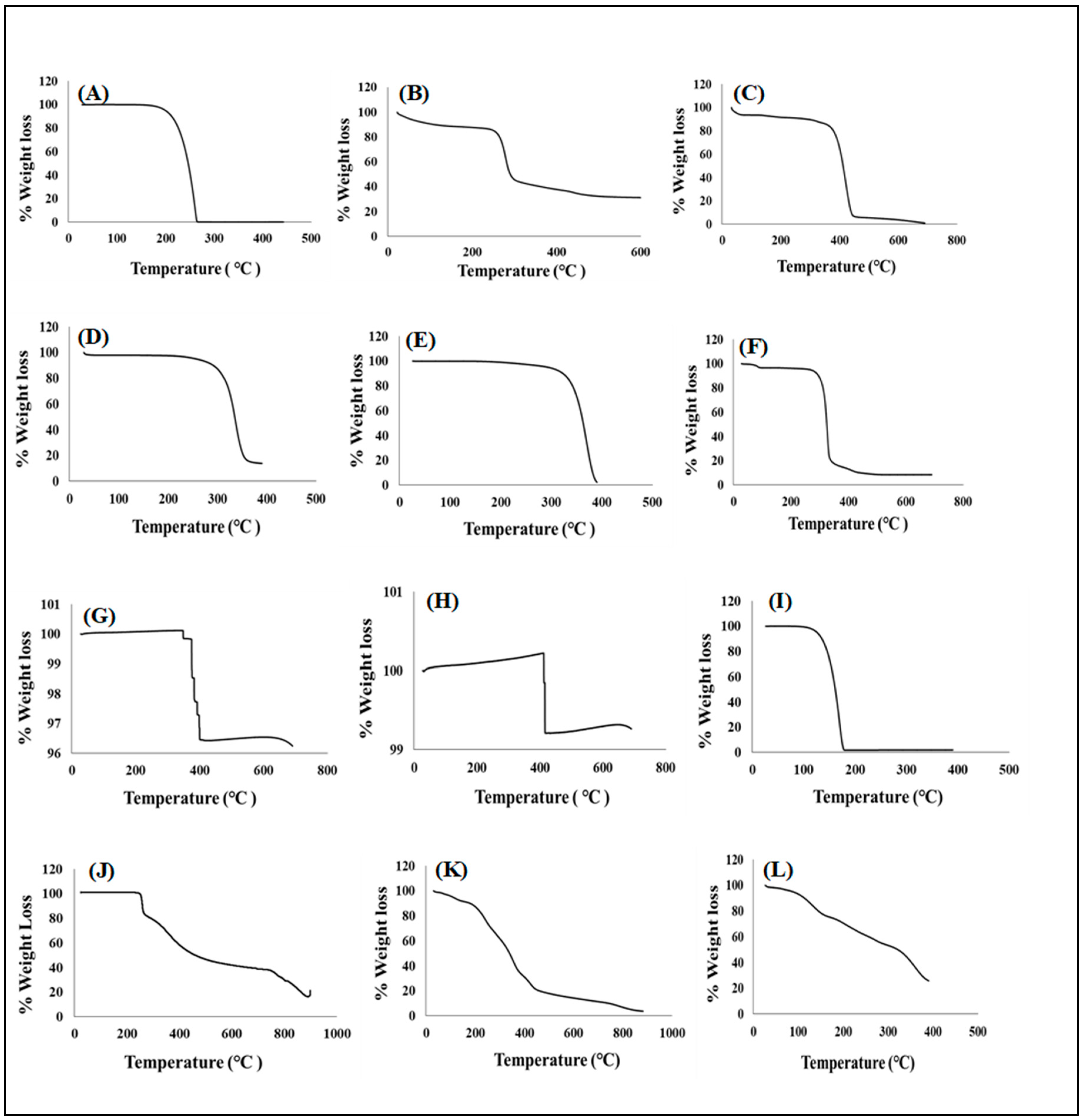
3.4.1. Determination of Thermal Stability
Differential Scanning Calorimetric Analysis
Temperature Dependent Gravimetry
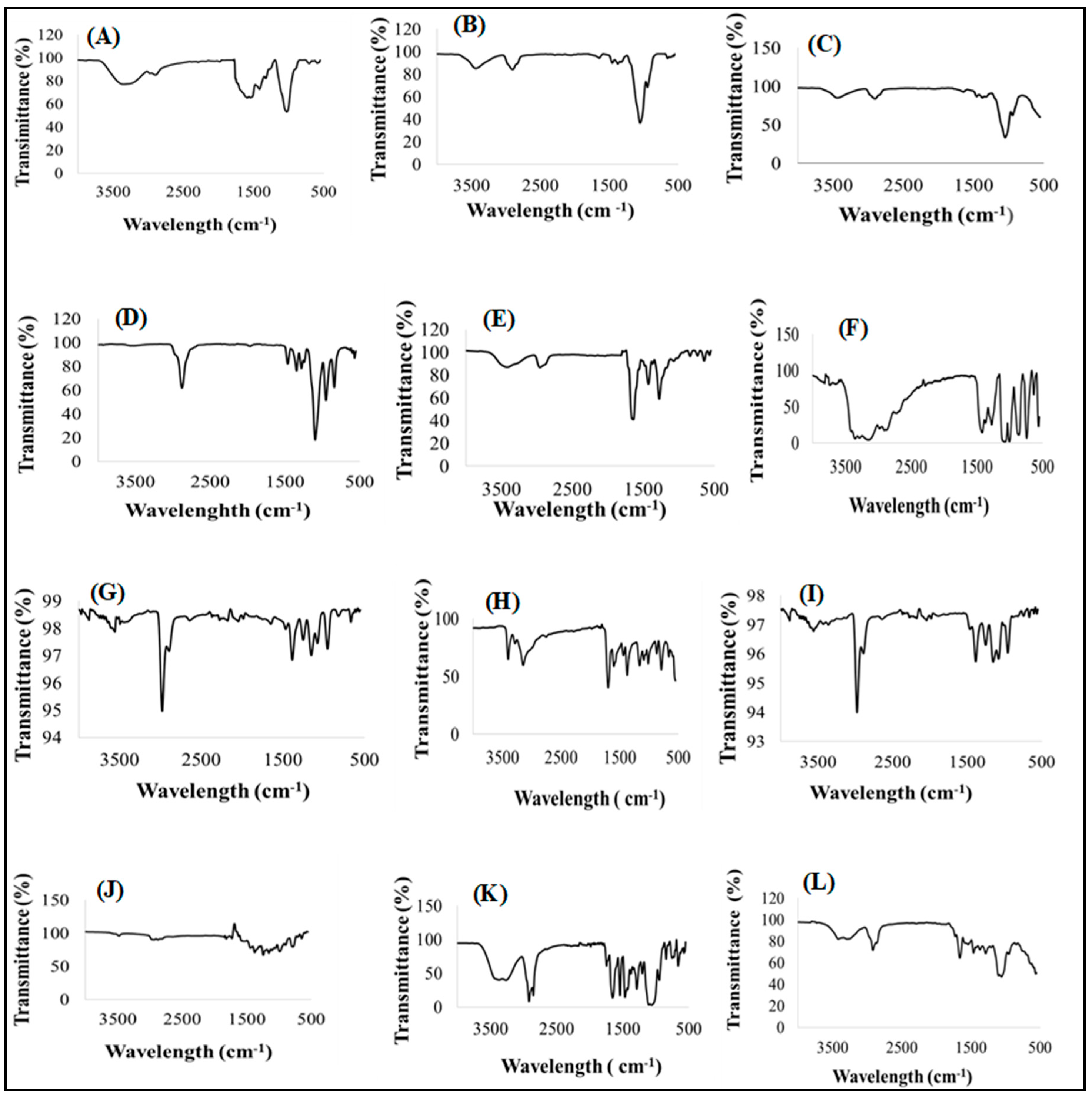
3.4.2. Infrared Absorption Spectroscopy
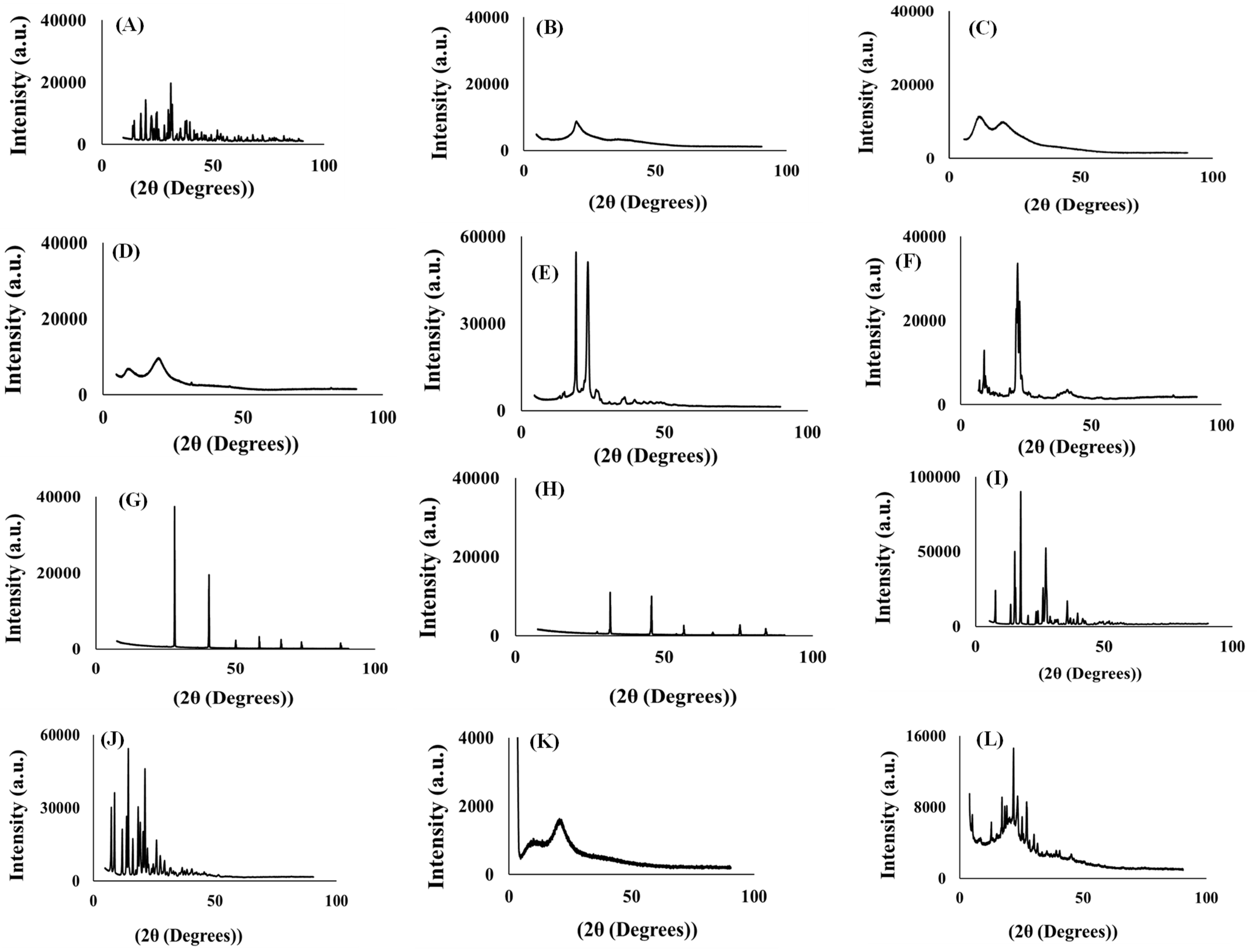
3.4.3. Powder X-ray Crystallography
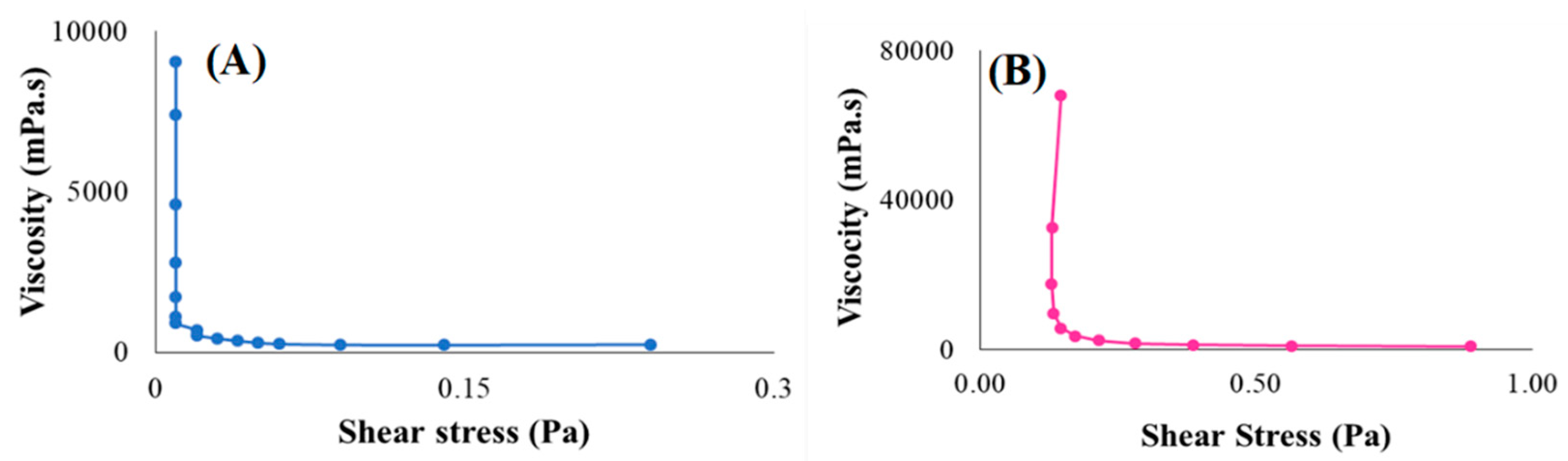
3.4.4. Rheological Behaviour
3.5. Microscopic Analyses
3.5.1. Surface Topography and Shape of Dry Suspension Powder
3.5.2. Microscopy Based Particle Size and Distribution Analysis
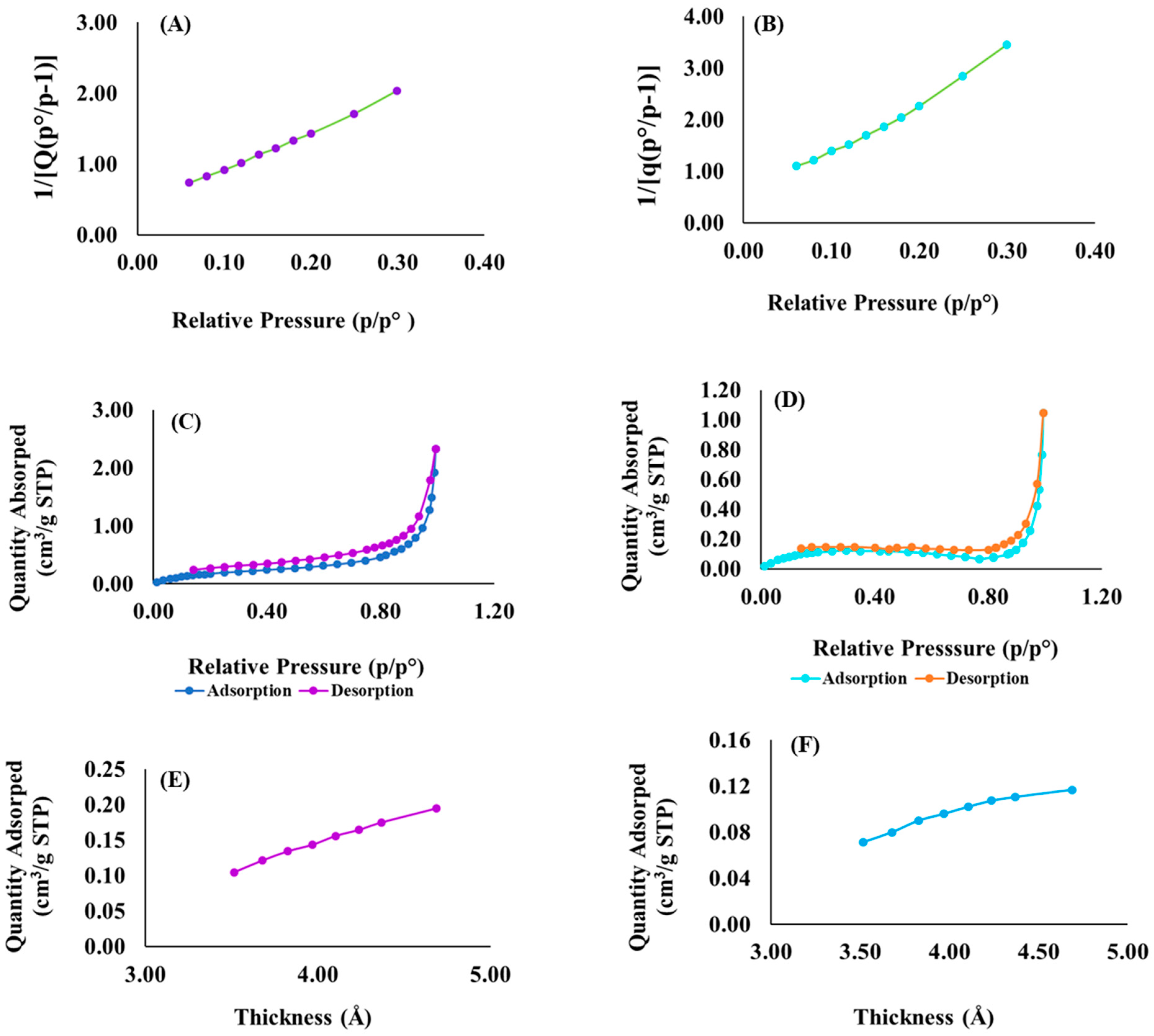
3.6. Surface Area and Porosimetery Analyses
3.7. Organoleptic Evaluations
3.8. Environmental Stability of the Dry Powder and Reconstituted Suspension
3.8.1. Stability Evaluation of Dry Suspension Powder
3.8.2. Stability Testing of Reconstituted Suspension
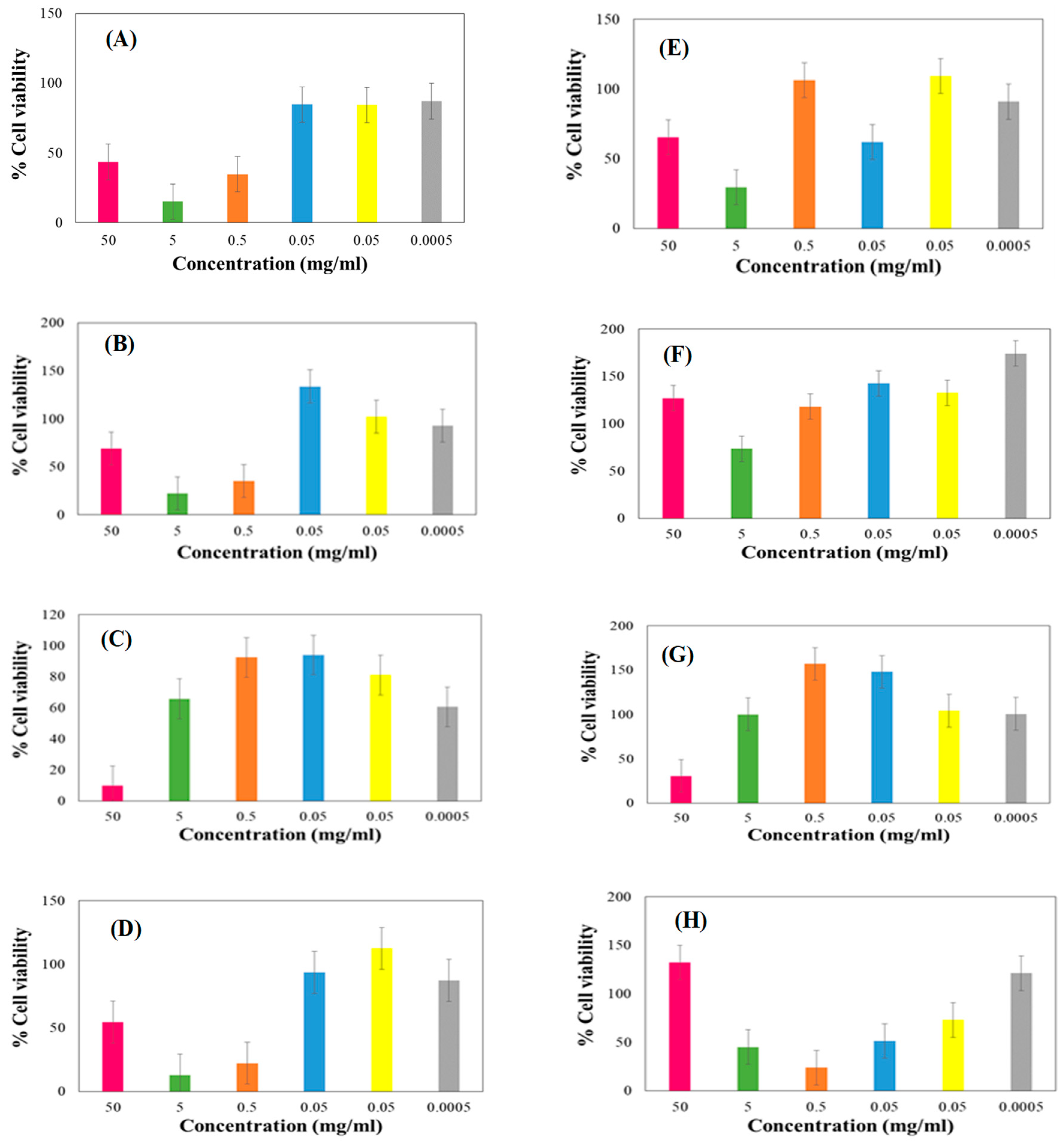
3.9. Biocompatibility Assay on Select Cell Lines
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fukunaga, R.; Glaziou, P.; Harris, J.B.; Date, A.; Floyd, K.; Kasaeva, T. Epidemiology of Tuberculosis and Progress Toward Meeting Global Targets—Worldwide, 2019. Morb. Mortal. Wkly. Rep. 2021, 70, 427. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Global Tuberculosis Report. 2022. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 4 December 2022).
- Hargreaves, J.R.; Boccia, D.; Evans, C.; Adato, M.; Petticrew, M.; Porter, J.D. The social determinants of tuberculosis: From evidence to action. Am. J. Public Health 2011, 101, 654–662. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Tuberculosis Global Report; World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 15 October 2022).
- World Health Organization. Report of the High-Level Consultation on Accelerating the Development of the M72/AS01E Tuberculosis Vaccine Candidate; World Health Organization: Geneva, Switzerland, 2019; Available online: https://www.who.int/tb/areas-of-work/research/meeting_report_m72_vaccine.pdf (accessed on 4 October 2022).
- Cogo, H.; Caseris, M.; Lachaume, N.; Cointe, A.; Faye, A.; Pommelet, V. Tuberculosis in Children Hospitalized in a Low-burden Country: Description and Risk Factors of Severe Disease. Paediatr. Infect. Dis. J. 2021, 40, 199–204. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Tuberculosis Report 2020: Executive Summary. 2020. Available online: https://apps.who.int/iris/handle/10665/337538 (accessed on 29 August 2022).
- Turkova, A.; Wills, G.H.; Wobudeya, E.; Chabala, C.; Palmer, M.; Kinikar, A.; Hissar, S.; Choo, L.; Musoke, P.; Mulenga, V.; et al. Shorter treatment for Nonsevere Tuberculosis in African and Indian children. N. Engl. J. Med. 2022, 386, 911–922. [Google Scholar] [CrossRef]
- Carvalho, A.C.C.; Kritski, A.L. What is the global burden of tuberculosis among children? Lancet Glob. Health 2022, 10, 159–160. [Google Scholar] [CrossRef]
- Anderson, B.J. My child is unique; the pharmacokinetics are universal. Paediatr. Anesth. 2012, 22, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, H.K.; Marriott, J. Paediatric pharmacokinetics: Key considerations. Br. J. Clin. Pharmacol. 2015, 79, 395–404. [Google Scholar] [CrossRef]
- Verkuijl, S.; Sekadde, M.P.; Dodd, P.J.; Arinaitwe, M.; Chiang, S.S.; Brands, A.; Viney, K.; Sismanidis, C.; Jenkins, H.E. Addressing the data gaps on child and adolescent tuberculosis. Pathogens 2022, 11, 352. [Google Scholar] [CrossRef]
- Ernest, T.B.; Elder, D.P.; Martini, L.G.; Roberts, M.; Ford, J.L. Developing paediatric medicines: Identifying the needs and recognizing the challenges. J. Pharmacol. 2007, 59, 1043–1105. [Google Scholar] [CrossRef]
- Cella, M.; Knibbe, C.; Danhof, M.; Della Pasqua, O. What is the right dose for children? J. Clin. Pharmacol. 2010, 70, 597–603. [Google Scholar] [CrossRef]
- Kendra, R.K.; Dooley, K.E.; Dodd, P.J.; Garcia-Prats, A.J.; McKenna, L.; Hesseling, A.C.; Savic, R.M. Alternative dosing guidelines to improve outcomes in childhood tuberculosis: A mathematical modelling study. Lancet Child Adolesc. Health 2019, 3, 636–645. [Google Scholar]
- Bangalore, S.; Kamalakkannan, G.; Parkar, S.; Messerli, F.H. Fixed-dose combinations improve medication compliance: A meta-analysis. Am. J. Med. 2007, 120, 713–719. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2015; Available online: https://www.google.com/search?q=World+Health+Organization.+2015.+Global+Tuberculosis+Report.+Geneva% (accessed on 5 September 2022).
- Svensson, E.M.; Yngman, G.; Denti, P.; McIlleron, H.; Kjellsson, M.C.; Karlsson, M.O. Evidence-based design of fixed-dose combinations: Principles and application to paediatric anti-tuberculosis therapy. Clin. Pharmacokinet. 2018, 57, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Weon, K.Y. Pharmaceutical application and development of fixed-dose combination: Dosage form review. J. Pharm. Investig. 2021, 51, 555–570. [Google Scholar] [CrossRef]
- Blanco, J.L.; Montaner, J.S.G.; Marconi, V.C.; Santoro, M.M.; Campos-Loza, A.E.; Shafer, R.W. Lower prevalence of drug resistance mutations at first-line virological failure to first-line therapy with atripla vs. tenofovir? emtricitabine/lamivudine? efavirenz administered on a multiple tablet therapy. AIDS 2014, 28, 2531–2539. [Google Scholar] [CrossRef]
- Tsiligiannis, A.; Sfouni, M.; Nalda-Molina, R.; Dokoumetzidis, A. Optimization of a paediatric fixed-dose combination mini-tablet and dosing regimen for the first line treatment of tuberculosis. Eur. J. Pharm. Sci. 2019, 138, 105016. [Google Scholar] [CrossRef]
- Chabala, C.; Turkova, A.; Hesseling, A.C.; Zimba, K.M.; van der Zalm, M.; Kapasa, M.; Palmer, M.; Chirehwa, M.; Wiesner, L.; Wobudeya, E.; et al. Pharmacokinetics of First-Line Drugs in Children With Tuberculosis, Using World Health Organization–Recommended Weight Band Doses and Formulations. Clin. Infect. Dis. 2022, 74, 1767–1775. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report; WHO/HTM/TB/2016.13; World Health Organization: Geneva, Switzerland, 2016; Available online: http://www.who.int/tb/publications/2016/global-TB-report/ (accessed on 31 August 2022).
- Suárez-González, J.; Santoveña-Estévez, A.; Soriano, M.; Fariña, J.B. Design and optimization of a child-friendly dispersible tablet containing isoniazid, pyrazinamide, and rifampicin for treating tuberculosis in paediatrics. Drug Dev. Ind. Pharm. 2020, 46, 309–317. [Google Scholar] [CrossRef]
- Lopez, F.L.; Ernest, T.B.; Tuleu, C.; Gul, M.O. Formulation approaches to paediatric oral drug delivery: Benefits and limitations of current platforms. Expert Opin. Drug Deliv. 2015, 12, 1727–1740. [Google Scholar] [CrossRef]
- Adeleke, O.A.; Rose, K.H.; Hajierah, D. Development and Evaluation of a Reconstitutable Dry Suspension Containing Isoniazid for Flexible Paediatric Dosing. J. Pharm. 2020, 12, 286. [Google Scholar]
- Pongsamart, K.; Kleinebudde, P.; Puttipipatkhachorn, S. Preparation of fenofibrate dry emulsion and dry suspension using octenyl succinic anhydride starch as emulsifying agent and solid carrier. Int. J. Pharm. 2016, 498, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.N.; Li, X.H.; Xu, J.H.; Chen, L.Q.; Ahmed, A.M.Q.; Cao, D.; Du, H.H.; Deng, Y.; Cao, Q.R. Optimization and characterization of novel sustained release supermicro-pellet based dry suspensions that load dexibuprofen. J. Drug Deliv. Sci. Technol. 2020, 55, 101420. [Google Scholar] [CrossRef]
- Rautamo, M.; Kvarnström, K.; Sivén, M.; Airaksinen, M.; Lahdenne, P.; Sandler, N. A Focus Group Study about Oral Drug Administration Practices at Hospital Wards—Aspects to Consider in Drug Development of Age-Appropriate Formulations for Children. Pharmaceutics 2020, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Vats, V.; Verma, P.K. A Review on Pharmaceutical Suspension and Its Advancement. Ann. Clin. Med. Case Rep. 2022, 7, 2321. [Google Scholar]
- Araújo, F.; Pedro, J.; Granja, P.L.; Santos, H.A.; Sarmento, B. Functionalized materials for multistage platforms in the oral delivery of biopharmaceuticals. Program. Mater. Sci. 2017, 89, 306–344. [Google Scholar] [CrossRef]
- Hu, Q.; Luo, Y. Recent advances of polysaccharide-based nanoparticles for oral insulin delivery. Int. J. Biol. Macromol. 2018, 120, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Gang, W.; Lichun, Z.; Qikun, J.; Yixin, S.; Dongyang, Z.; Mengchi, S.; Zhonggui, H.; Jin, S.; Yang, W. Intestinal OCTN2- and MCT1-targeted drug delivery to improve oral bioavailability. Asian J. Pharm. Sci. 2020, 15, 158–173. [Google Scholar]
- Matawo, N.; Adeleke, O.A.; Wesley-Smith, J. Optimal Design, Characterization and Preliminary Safety Evaluation of an Edible Orodispersible Formulation for Paediatric Tuberculosis Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 5714. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.M.; Rosa, V.J.S.; Palma, A.J.; Felipe, V.; de Carvalho, V.F.T.M.; Badra, B.M.V.L.; Marlus, C.; Maldonado, M.J.; Rita, T.B.D. Design of experiments (DoE) to develop and to optimize nanoparticles as Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2021, 165, 127–148. [Google Scholar] [CrossRef]
- Patel, G.C.; Prajapati, J.; Morthana, K.M.; Khunt, D.M. Formulation and evalution of oral reconstitutable suspension of cefpodoxime proxetil. J. Pharm. Drug Dev. 2015, 3, 101. [Google Scholar]
- Thakur, B.; Kushawaha, S.K.; Pandit, V.; Ashawat, M.S.; Kumar, P. Formulation and Evaluation of Fixed-dose Combination Tablets of Antifungal Drugs for Candida albicans Resistant to Fluconazole. J. Drug Deliv. Ther. 2020, 10, 26–36. [Google Scholar] [CrossRef]
- Rao, M.R.; Bhingole, R.C. Nanosponge-based paediatric-controlled release dry suspension of Gabapentin for reconstitution. Drug Dev. Ind. Pharm. 2015, 41, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Akre, H.S.; Mundhada, D.R.; Bhaskaran, S.; Asghar, S.; Gandhi, G.S. Dry suspension formulation of taste masked antibiotic drug for paediatric use. J. Appl. Pharm. Sci. 2012, 2, 166–171. [Google Scholar]
- Ebenezer, A.O.; Chioma, C.; Omoegbe, I.M. Instability of cefpodoxime proxetil oral suspension at different temperature storage conditions. J. Pharm. Pharm. 2017, 5, 6. [Google Scholar]
- Patil, J.S.; Devi, V.K.; Devi, K.; Sarasija, S. A novel approach for lung delivery of rifampicin-loaded liposomes in dry powder form for the treatment of tuberculosis. Lung India Off. Organ Indian Chest Soc. 2015, 32, 331. [Google Scholar] [CrossRef] [PubMed]
- Harsha, S. Pharmaceutical suspension containing both immediate/sustained-release amoxicillin-loaded gelatin nanoparticles: Preparation and in vitro characterization. Drug Des. Dev. Ther. 2013, 7, 1027. [Google Scholar] [CrossRef][Green Version]
- Nair, R.S.; Rahman, H.; Kong, M.X.; Tan, X.Y.; Chen, K.Y.; Shanmugham, S. Development and rheological evaluation of DEET (N, N-diethyL-3-methylbenzamide) microparticles loaded hydrogel for topical application. Turk. J. Pharm. Sci. 2021, 18, 352. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.H.; Galaz, A.; Jara, M.O.; Gamboa, A.; Morales, J.O. Drug-loaded lipid-core micelles in mucoadhesive films as a novel dosage form for buccal administration of poorly water-soluble and biological drugs. Pharmaceutics 2020, 12, 1168. [Google Scholar] [CrossRef]
- Xu, W.; Deng, Z.; Xiang, Y.; Zhu, D.; Yi, D.; Mo, Y.; Liu, Y.; Qin, L.; Huang, L.; Wan, B.; et al. Preparation, Characterization and Pharmacokinetics of Tolfenamic Acid-Loaded Solid Lipid Nanoparticles. Pharmaceutics 2022, 14, 1929. [Google Scholar] [CrossRef]
- Zakaria, M.Y.; Georghiou, P.E.; Banoub, J.H.; Beshay, B.Y. Inclusion of a Phytomedicinal Flavonoid in Biocompatible Surface-Modified Chylomicron Mimic Nanovesicles with Improved Oral Bioavailability and Virucidal Activity: Molecular Modeling and Pharmacodynamic Studies. Pharmaceutics 2022, 14, 905. [Google Scholar] [CrossRef]
- Unagolla, J.M.; Jayasuriya, A.C. Drug transport mechanisms and in vitro release kinetics of vancomycin encapsulated chitosan-alginate polyelectrolyte microparticles as a controlled drug delivery system. Eur. J. Pharm. Sci. 2018, 114, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Park, S.Y.; Park, S.J.; Lee, J.; Cho, K.H.; Jee, J.P.; Kim, H.C.; Maeng, H.J.; Jang, D.J. Development and evaluation of a reconstitutable dry suspension to improve the dissolution and oral absorption of poorly water-soluble celecoxib. Pharmaceutics 2018, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, Y.; Wang, X.; Li, M.; Lei, H.; Xu, H. Cellulose nanocrystals prepared from wheat bran: Characterization and cytotoxicity assessment. Int. J. Biol. Macromol. 2019, 140, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Khatib, I.; Khanal, D.; Ruan, J.; Cipolla, D.; Dayton, F.; Blanchard, J.D.; Chan, H.K. Ciprofloxacin nanocrystals liposomal powders for controlled drug release via inhalation. Int. J. Pharm. 2019, 566, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Kassaee, S.N.; Mahboobian, M.M. Besifloxacin-loaded ocular nanoemulsions: Design, formulation, and efficacy evaluation. Drug Deliv. Transl. Res. 2022, 12, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, R.M.; Arantes, J.T.; Santos, S.F. Synthesis, structure, properties and applications of MXenes: Current status and perspectives. Ceram. Int. 2019, 45, 18167–18188. [Google Scholar] [CrossRef]
- Jain, D.K.; Darwhekar, G.N.; Choudhary, N. Formulation and Evaluation of Reconstitutable Oral Suspension of AmbroxolHCl and Azithromycin. Int. J. PharmTech Res. 2011, 3, 741–746. [Google Scholar]
- Doifode, R.A. Design and Development of Oral Reconstituable System of Dry Syrup Containing Ciprofloxacin. Sch. Acad. J. Pharm. 2021, 2, 28–35. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Wang, L.; Sun, Y.; Kuang, C.; Zhang, X. Preparation and evaluation of taste masked oral suspension of arbidol hydrochloride. Asian J. Pharm. Sci. 2015, 10, 73–79. [Google Scholar] [CrossRef]
- Wadud, A.; Ansari, M.I.; Perveen, S.; Ahmad, A.; Akbari, N.; Anwar, D. Quality assessment of market samples of crude drugs with reference to Asrol (Rauvolfia serpentina Benth ex. Kurz). J. Phytopharm. 2020, 9, 197–201. [Google Scholar] [CrossRef]
- Bajaj, S.; Singla, D.; Sakhuja, N. Stability testing of pharmaceutical products. J. Appl. Pharm. Sci. 2012, 2, 129–138. [Google Scholar]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Tostmann, A.; Boeree, M.J.; Peters, W.H.; Roelofs, H.M.; Aarnoutse, R.E.; van der Ven, A.J.; Dekhuijzen, P.R. Isoniazid and its toxic metabolite hydrazine induce in vitro rifampicin toxicity. Int. J. Antimicrob. Agents 2008, 31, 577–580. [Google Scholar] [CrossRef]
- Sen, C.; Patra, C.; Mondol, S.; Datta, A.; Mallick, D.; Mondal, T.K.; Askun, T.; Celikboyun, P.; Cantürk, Z.; Sinha, C. Platinum (II)-azoimidazole drugs against TB and cancer: Structural studies, cytotoxicity and anti-mycobacterial activity. Polyhedron 2018, 152, 1–10. [Google Scholar] [CrossRef]
- Carazo, E.; Sandri, G.; Cerezo, P.; Lanni, C.; Ferrari, F.; Bonferoni, C.; Viseras, C.; Aguzzi, C. Halloysite nanotubes as tools to improve the actual challenge of fixed doses combinations in tuberculosis treatment. J. Biomed. Mater. Res. Part A 2019, 107, 1513–1521. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.Y. Phenomenology of the initial burst release of drugs from PLGA microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Ciro, Y.; Rojas, J.; Alhajj, M.J.; Carabali, G.A.; Salamanca, C.H. Production and Characterization of Chitosan–Polyanion Nanoparticles by Polyelectrolyte Complexation Assisted by High-Intensity Sonication for the Modified Release of Methotrexate. Pharmaceuticals 2020, 13, 11. [Google Scholar] [CrossRef]
- Medeiros, A.; Correia, L.; Simões, M.; Macedo, R. Technological quality determination of pharmaceutical disintegrant by DSC cooling and DSC photo visual. J. Therm. Anal. Calorim. 2007, 88, 311–315. [Google Scholar] [CrossRef]
- Tan, Z.; Shi, Q.; Liu, X.u. Thermodynamic properties of the polyols as phase change materials for thermal energy storage. In Phase Change Materials and Their Applications; IntechOpen: London, UK, 2018; p. 71894. [Google Scholar]
- Shavi, G.V.; Kumar, A.R.; Usha, Y.N.; Armugam, K.; Ranjan, O.P.; Ginjupalli, K.; Pandey, S.; Udupa, N. Enhanced dissolution and bioavailability of gliclazide using solid dispersion techniques. Int. J. Drug Deliv. 2010, 2, 49–57. [Google Scholar] [CrossRef]
- Fahad, M.; Khan, M.A.; Gilbert, M. Investigation of Thermal Gel Formation of Methylcellulose in Glycols Using DSC and XRD. Gels 2021, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.B.; Liang, Y.Z.; Chen, Y.L.; Xie, J.H.; Liu, W.H.; Chen, J.N.; Lai, X.P.; Su, Z.R. Novel patchouli alcohol ternary solid dispersion pellets prepared by poloxamers. Iran. J. Pharm. Res. IJPR 2015, 14, 15. [Google Scholar] [PubMed]
- Nokhodchi, A.; Okwudarue, O.N.; Valizadeh, H.; Momin, M.N. Cogrinding as a tool to produce sustained release behaviour for theophylline particles containing magnesium stearate. AAPS PharmSciTech 2009, 10, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, D.; Kobertz, D.; Müller, M. Thermodynamics of the NaCl–KCl system. Thermochim. Acta 2015, 606, 25–33. [Google Scholar] [CrossRef]
- Ortin, J. Thermodynamics and Kinetics of Phase Transitions. In Mechanics of Solids with Phase Changes; Springer: Vienna, Austria, 1997; pp. 1–52. [Google Scholar]
- Castro, R.A.E.; Maria TM, R.; Évora AO, L.; Feiteira, J.C.; Silva, M.; Ramos, B.; Matos, A.; Canotilho, J.; Eusébio, M.; Ermelinda, S. A New Insight into pyrazinamide polymorphic forms and their thermodynamic relationships. Cryst. Growth Des. 2010, 10, 274–282. [Google Scholar] [CrossRef]
- Alves, R.; Reis, T.V.D.S.; Silva, L.C.C.D.; Storpírtis, S.; Mercuri, L.P.; Matos, J.D.R. Thermal behaviour and decomposition kinetics of RIF polymorphs under isothermal and non-isothermal conditions. Braz. J. Pharm. Sci. 2010, 46, 343–351. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.; Jiang, L.; Kang, Y.; Cheng, C.; Guo, Z.; Zhang, C. Development of carbon adsorbents with high surface acidic and basic group contents from phosphoric acid activation of xylitol. RSC Adv. 2015, 5, 81220–81228. [Google Scholar] [CrossRef]
- Nadour, M.; Boukraa, F.; Ouradi, A.; Benaboura, A. Effects of methylcellulose on the properties and morphology of polysulfone membranes prepared by phase inversion. Mater. Res. 2017, 20, 339–348. [Google Scholar] [CrossRef]
- Nanaki, S.; Eleftheriou, R.M.; Barmpalexis, P.; Kostoglou, M.; Karavas, E.; Bikiaris, D. Evaluation of dissolution enhancement of aprepitant drug in ternary pharmaceutical solid dispersions with Soluplus® and Poloxamer 188 prepared by melt mixing. Sciences 2019, 1, 48. [Google Scholar]
- Pereira, M.A.V.; Fonseca, G.D.; Silva-Júnior, A.A.; Fernandes-Pedrosa, M.F.; de FV de Moura, M.; Barbosa, E.G.; Gomes, A.P.B.; Dos Santos, K.S.C.R. Compatibility study between chitosan and pharmaceutical excipients used in solid dosage forms. J. Therm. Anal. Calorim. 2014, 116, 1091–1100. [Google Scholar] [CrossRef]
- Hotaby, W.; Sherif, H.H.A.; Hemdan, B.A.; Khalil, W.A.; Khalil, S.K.H. Assessment of in situ-Prepared Polyvinylpyrrolidone-Silver Nanocomposite for Antimicrobial Applications. Acta Phys. Pol. 2017, 131, 1554–1560. [Google Scholar] [CrossRef]
- Ngilirabanga, J.B.; Aucamp, M.; Pires Rosa, P.; Samsodien, H. Mechanochemical synthesis and physicochemical characterization of isoniazid and pyrazinamide co-crystals with glutaric acid. Front. Chem. 2020, 1051, 595908. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Cooney, R.P. Thermal degradation of polymer and polymer composites. In Handbook of Environmental Degradation of Materials; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 185–206. [Google Scholar]
- Król-Morkisz, K.; Pielichowska, K. Thermal decomposition of polymer nanocomposites with functionalized nanoparticles. In Polymer Composites with Functionalized Nanoparticles; Elsevier: Amsterdam, The Netherlands, 2019; pp. 405–435. [Google Scholar]
- Remya, K.S.; Beena, P.; Bijesh, P.V.; Sheeba, A. Formulation development, evaluation and comparative study of effects of super disintegrants in cefixime oral disintegrating tablets. J. Young Pharm. 2010, 2, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Bhavani, B.; Chadalawada, P.K.; Gupta, T.R. Formulation and evaluation of extended release mucoadhesive microspheres of atorvastatin. Indo Am. J. Pharm. Sci. 2015, 2, 1423–1436. [Google Scholar]
- Nep, E.I.; Conway, B.R. Preformulation studies on grewia gum as a formulation excipient. J. Therm. Anal. Calorim. 2012, 108, 197–205. [Google Scholar] [CrossRef]
- Garala, K.; Joshi, P.; Shah, M.; Ramkishan, A.; Patel, J. Formulation and evaluation of periodontal in situ gel. Int. J. Pharm. Investig. 2013, 3, 29. [Google Scholar] [CrossRef]
- Bryaskova, R.; Pencheva, D.; Nikolov, S.; Kantardjiev, T. Synthesis and comparative study on the antimicrobial activity of hybrid materials based on silver nanoparticles (AgNps) stabilized by polyvinylpyrrolidone (PVP). J. Chem. Biol. 2011, 4, 185–191. [Google Scholar] [CrossRef]
- Mosquera, M.; Orozco, F.; Benítez, R.; Martin, J.; Rojas, G. Controlled Branching by Step-Growth Polymerization of Xylitol and Succinic Acid via Microwave Irradiation. ACS Omega 2021, 6, 13987–13994. [Google Scholar] [CrossRef]
- Mamba, B.; Nyembe, D.; Mulaba-Bafubiandi, A. The effect of conditioning with NaCl, KCl and HCl on the performance of natural clinoptilolite’s removal efficiency of Cu2+ and Co2+ from Co/Cu synthetic solutions. Water Sanit. 2010, 36, 4. [Google Scholar]
- Anggarini, U.; Sukmana, N.C.; Prasetya, F.A. Synthesis and Characterization of Fly Ash Geopolymer for Water Absorbent Material. MATEC Web Conf. 2017, 97, 01024. [Google Scholar] [CrossRef]
- Al-Samarrai, M.N.; Hamzah, R.; Sam, S.T.; Noriman, N.Z.; Dahham, O.S.; Al-rashdi, A.A.; Alakrach, A.M. A slow-release fertilizer from urea and rice straw coated by ENR-50 for agricultural application. AIP Conf. Proc. 2018, 2030, 020026. [Google Scholar]
- Quaresma, S.; Alves, P.C.; Rijo, P.; Duarte, M.T.; André, V. Antimicrobial Activity of Pyrazinamide Coordination Frameworks Synthesized by Mechanochemistry. Molecules 2021, 26, 1904. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Tomila, T.; Ulyanchich, N.; Yarmola, T.; Uvarova, I. Fourier-Transform Infrared Spectroscopy of antibiotic loaded Ag-free and Ag-doped hydroxyapatites. Adv. Sci. Eng. Med. 2014, 6, 193–202. [Google Scholar] [CrossRef]
- Farooq, U.; Ahmad, T.; Khan, A.; Sarwar, R.; Shafiq, J.; Raza, Y.; Ahmed, A.; Ullah, S.; Rehman, N.U.; Al-Harrasi, A. Rifampicin conjugated silver nanoparticles: A new arena for development of antibiofilm potential against methicillin resistant Staphylococcus aureus and Klebsiella pneumoniae. Int. J. Nanomed. 2019, 14, 3983. [Google Scholar] [CrossRef]
- Mehanna, M.M.; Mohyeldin, S.M.; Elgindy, N.A. Rifampicin-carbohydrate spray-dried nanocomposite: A futuristic multiparticulate platform for pulmonary delivery. Int. J. Nanomed. 2019, 14, 9089. [Google Scholar] [CrossRef] [PubMed]
- Firoozi, N.; Kang, Y. A Highly Elastic and Autofluorescent Poly (xylitol-dodecanedioic Acid) for Tissue Engineering. ACS Biomater. Sci. Eng. 2019, 5, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Windriyati, Y.N.; Sumirtapura, Y.C.; Pamudji, J.S. Dissolution enhancement and physicochemical characterization of fenofibric acid in surface solid dispersion with croscarmellose sodium. J. Respir. Pharm. 2019, 23, 315–325. [Google Scholar] [CrossRef]
- Nik Aziz, N.A.; Idris, N.K.; Isa, M.I.N. Solid polymer electrolytes based on methylcellulose: FT-IR and ionic conductivity studies. Int. J. Polym. Charact. 2010, 15, 319–327. [Google Scholar] [CrossRef]
- Ramadhani, N.; Shabir, M.; McConville, C. Preparation and characterisation of Kolliphor® P 188 and P 237 solid dispersion oral tablets containing the poorly water-soluble drug disulfiram. Int. J. Pharm. 2014, 475, 514–522. [Google Scholar] [CrossRef]
- Delaney, S.P.; Nethercott, M.J.; Mays, C.J.; Winquist, N.T.; Arthur, D.; Calahan, J.L.; Sethi, M.; Pardue, D.S.; Kim, J.; Amidon, G.; et al. Characterization of synthesized and commercial forms of magnesium stearate using differential scanning calorimetry, thermogravimetric analysis, powder X-ray diffraction, and solid-state NMR spectroscopy. J. Pharm. Sci. 2017, 106, 338–347. [Google Scholar] [CrossRef]
- Ahda, S.; Misfadhila, S.; Parikin, P.; Putra, T.Y.S.P. Molten salt synthesis and structural characterization of BaTiO3 nanocrystal ceramics. Mater. Sci. Eng. 2017, 176, 12048. [Google Scholar] [CrossRef]
- Bao, N.; Miao, X.; Hu, X.; Zhang, Q.; Jie, X.; Zheng, X. Novel synthesis of plasmonic Ag/AgCl@ TiO2 continues fibers with enhanced broadband photocatalytic performance. Catalysts 2017, 7, 117. [Google Scholar] [CrossRef]
- Varma, J.R.; Kumar, T.S.; Prasanthi, B.; Ratna, J.V. Formulation and characterization of pyrazinamide polymeric nanoparticles for pulmonary tuberculosis: Efficiency for alveolar macrophage targeting. Indian J. Pharm. Sci. 2015, 77, 258. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Latha, M.; Thomas, K.K. Alginate/Chitosan nanoparticles for improved oral delivery of rifampicin: Optimization, characterization, and in vitro evaluation. Asian J. Chem. 2018, 30, 736–740. [Google Scholar] [CrossRef]
- Teixeira, G.L.; Züge, L.C.B.; Silveira, J.L.M.; Scheer, A.D.P.; Ribani, R.H. The impact of polyoxyethylene sorbitan surfactants in the microstructure and rheological behaviour of emulsions made with melted fat from Cupuassu (Theobroma grandiflorum). J. Surfactants Deterg. 2016, 19, 725–738. [Google Scholar] [CrossRef]
- Auch, C.; Harms, M.; Golitsyn, Y.; Reichert, D.; Mäder, K. Miniaturized measurement of drug–polymer interactions via viscosity increase for polymer selection in amorphous solid dispersions. Mol. Pharm. 2019, 16, 2214–2225. [Google Scholar] [CrossRef]
- Aremu, O.I.; Oduyela, O.O. Evaluation of metronidazole suspensions. Afr. J. Pharm. Pharmacol. 2015, 9, 439–450. [Google Scholar] [CrossRef]
- Kumar, R.S.; Yagnesh, T.N.S. Pharmaceutical suspensions: Patient compliance oral dosage forms. World J. Pharm. Pharm. Sci. 2016, 7, 1471–1537. [Google Scholar]
- Kumar, V.; Sharma, N.; Maitra, S.S. In vitro and in vivo toxicity assessment of nanoparticles. Int. Nano Lett. 2017, 7, 243–256. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P. How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech. Dis. 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Calabrese, E.J. Hormesis: Path and progression to significance. Int. J. Mol. Sci. 2018, 19, 2871. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Levels/Limits | ||
|---|---|---|---|
| Lower (−1) | Middle (0) | Upper (+1) | |
| Factor A * | 0 | 1 | 2 |
| Factor B ** | 3 | 4 | 5 |
| Factor C *** | 6 | 7 | 8 |
| Reconstitutable Suspension | Factor A (X1) | Factor B (X2) | Factor C (X3) |
|---|---|---|---|
| RS 1 * | 1 | 4 | 7 |
| RS 2 | 2 | 4 | 6 |
| RS 3 | 1 | 5 | 6 |
| RS 4 | 0 | 3 | 7 |
| RS 5 * | 1 | 4 | 7 |
| RS 6 | 1 | 3 | 6 |
| RS 7 | 2 | 5 | 7 |
| RS 8 | 2 | 4 | 8 |
| RS 9 | 1 | 5 | 8 |
| RS 10 | 0 | 5 | 7 |
| RS 11 * | 1 | 4 | 7 |
| RS 12 | 0 | 4 | 8 |
| RS 13 | 2 | 3 | 7 |
| RS 14 | 1 | 3 | 8 |
| RS 15 | 0 | 4 | 6 |
| Response Parameters | Optimization Goal | Lower | Target | Upper | R2 (%) | p-Value |
|---|---|---|---|---|---|---|
| Y1 * (%) | Target | 68.700 | 80.000 | 94.300 | 84.800 | 0.000 |
| Y2 * (degrees) | Minimum | 11.300 | 15.700 | 24.300 | 89.460 | 0.019 |
| Y3 * (strokes/second) | Target | 1.675 | 1.800 | 1.990 | 78.790 | 0.001 |
| Y4 * | Target | 0.400 | 1.000 | 1.060 | 93.400 | 0.007 |
| Formulation | Y1 (%) | Y2 (°) | Y3 (Strokes/Seconds) | Y4 |
|---|---|---|---|---|
| RS 1 * | 93.30 ± 6.51 | 22.30 ± 0.58 | 1.69 ± 0.01 | 1.06 ± 0.05 |
| RS 2 | 79.30 ± 7.51 | 21.00 ± 1.73 | 1.88 ± 0.19 | 0.78 ± 0.08 |
| RS 3 | 83.30 ± 7.77 | 15.30 ± 3.21 | 1.73 ± 0.03 | 0.87 ± 0.13 |
| RS 4 | 83.00 ± 11.27 | 12.70 ± 2.52 | 1.87 ± 0.02 | 0.76 ± 0.27 |
| RS 5* | 92.30 ± 3.51 | 24.00 ± 0.00 | 1.88 ± 0.01 | 1.06 ± 0.00 |
| RS 6 | 75.30 ± 10.69 | 24.30 ± 1.53 | 1.80 ± 0.01 | 0.40 ± 0.05 |
| RS 7 | 68.70 ± 3.51 | 15.70 ± 2.52 | 1.94 ± 0.08 | 0.92 ± 0.14 |
| RS 8 | 76.30 ± 1.15 | 21.70 ± 1.15 | 1.96 ± 0.00 | 1.01 ± 0.02 |
| RS 9 | 81.30 ± 4.93 | 11.30 ± 1.53 | 1.85 ± 0.05 | 0.99 ± 0.02 |
| RS 10 | 88.00 ± 6.24 | 12.00 ± 1.00 | 1.71 ± 0.02 | 0.97 ± 0.00 |
| RS 11 * | 94.30 ± 2.52 | 23.00 ± 1.00 | 1.71 ± 0.01 | 1.05 ± 0.02 |
| RS 12 | 79.30 ± 3.06 | 13.00 ± 0.00 | 1.84 ± 0.13 | 0.99 ± 0.02 |
| RS 13 | 90.70 ± 4.16 | 21.00 ± 1.00 | 1.79 ± 0.06 | 0.48 ± 0.07 |
| RS 14 | 76.70 ± 4.04 | 21.70 ± 2.52 | 1.99 ± 0.12 | 0.44 ± 0.04 |
| RS 15 | 82.70 ± 8.14 | 12.00 ± 1.73 | 1.68 ± 0.03 | 0.74 ± 0.11 |
| Optimized Formular (RSopt) | Indicators of Design Template Validity | |||||
|---|---|---|---|---|---|---|
| Responses | Predicted | Experimental | R2 (%) | p-Value | Desirability Level | |
| Factor A = 0 | Y1 (%) | 91.562 | 96.000 ± 3.271 | 84.800 | 0.042 | 0.891 |
| Factor B = 5 | Y2 (°) | 8.945 | 9.670 ± 1.150 | 89.461 | 0.037 | 1.000 |
| Factor C = 8 | Y3 (strokes/ seconds) | 1.800 | 1.720 ± 0.011 | 93.400 | 0.016 | 1.000 |
| Y4 | 0.999 | 1.000 ± 0.010 | 78.792 | 0050 | 0.982 | |
| Composite desirability level | 0.967 | |||||
| Mathematical Model | Pyrazinamide | Rifampicin | ||
|---|---|---|---|---|
| R2 Value | AIC | R2 Value | AIC | |
| Zero-order | 0.99 | 48.77 | 0.99 | 40.75 |
| First-order | 0.84 | 85.05 | −1.70 | 72.74 |
| Second-order | 0.43 | 94.92 | −2.06 | 112.98 |
| Higuchi | 0.30 | 84.08 | 0.87 | 67.32 |
| Korsmeyer-Peppas | 0.96 | 61.52 | −3.16 | 55.23 |
| Michelis-Menten | 0.90 | 90.01 | −9.05 | 83.18 |
| 95% Confidence Interval for Mean | ||||||
|---|---|---|---|---|---|---|
| Samples | N | Mean (µm) | Standard Deviation | Standard Error | Upper Bound | Lower Bound |
| Placebo | 1685.00 | 307.86 | 231.20 | 5.63 | 296.81 | 318.91 |
| Drug-loaded | 643.00 | 392.79 | 433.20 | 17.08 | 359.24 | 462.34 |
| Pyrazinamide | 339.00 | 205.59 | 108.60 | 5.90 | 193.98 | 217.20 |
| Rifampicin | 322.00 | 144.85 | 60.21 | 3.35 | 138.25 | 151.46 |
| Stability Indicators | Varying Storage Conditions over 8 Weeks | |||
| Week 0 | Week 4 | |||
| I * | II * | III * | ||
| Yield (%) | 93.33 ± 1.67 | 91.63 ± 3.13 | 90.06 ± 0.97 | 90.20 ± 4.45 |
| Angle of repose (°) | 12.37 ± 1.37 | 11.86 ± 0.80 | 12.69 ± 0.81 | 12.44 ± 0.63 |
| Drug content (%) | Rifampicin | |||
| 97.61 ± 0.02 | 97.01 ± 0.01 | 99.05 ± 0.02 | 97.21 ± 0.01 | |
| Pyrazinamide | ||||
| 97.23 ± 2.57 | 98.12 ± 0.03 | 96.23 ± 0.01 | 98.03 ± 0.02 | |
| Colour change | 4.00 ± 0.00 | 3.80 ± 0.45 | 2.40 ± 0.55 | 4.00 ± 0.00 |
| Odour | 5.00 ± 0.00 | 4.60 ± 0.55 | 2.80 ± 0.45 | 4.80 ± 0.45 |
| Stability indicators | Varying storage conditions over 8 weeks | |||
| Week 0 | Week 8 | |||
| I * | II * | III * | ||
| Yield (%) | 93.33 ± 1.67 | 90.13 ± 2.73 | 90.13 ± 2.73 | 88.27 ± 2.28 |
| Angle of repose (°) | 12.37 ± 1.37 | 12.26 ± 1.27 | 12.27 ± 1.07 | 12.27 ± 1.07 |
| Drug content (%) | Rifampicin | |||
| 97.61 ± 0.02 | 96.28 ± 0.01 | 97.11 ± 0.02 | 96.24 ± 0.00 | |
| Pyrazinamide | ||||
| 97.23 ± 2.57 | 93.00 ± 0.01 | 93.02 ± 0.01 | 94.32 ± 0.02 | |
| Colour change | 4.00 ± 0.00 | 3.60 ± 0.55 | 2.40 ± 0.55 | 3.60 ± 0.55 |
| Odour | 5.00 ± 0.00 | 4.40 ± 0.55 | 2.80 ± 0.45 | 4.60 ± 0.55 |
| Stability Indicators | Varying Storage Conditions over 14 Days | |||
| Day 0 | Day 7 | |||
| I * | II * | III * | ||
| Rate of redispersibility (stroke/seconds) | 1.40 ± 0.02 | 1.80 ± 0.00 | 1.57 ± 0.00 | 1.76 ± 0.00 |
| Sedimentation | 2.30 ± 0.06 | 0.81 ± 0.02 | 0.91 ± 0.08 | 0.97 ± 0.02 |
| Drug content (%) | Rifampicin | |||
| 97.61 ± 0.02 | 97.00 ± 0.01 | 99.00 ± 0.02 | 97.22 ± 0.01 | |
| Pyrazinamide | ||||
| 97.23 ± 2.57 | 98.00 ± 0.03 | 96.04 ± 0.01 | 98.34 ± 0.02 | |
| pH | 6.95 ± 0.13 | 7.05 ± 0.01 | 7.79 ± 0.02 | 7.65 ± 0.04 |
| Taste | 5.00 ± 0.00 | 4.40 ± 0.55 | 4.40 ± 0.55 | 4.80 ± 0.45 |
| Smell | 5.00 ± 0.00 | 5.00 ± 0.00 | 4.40 ± 0.55 | 5.00 ± 0.00 |
| Appearance | 4.00 ± 0.00 | 3.80 ± 0.45 | 3.60 ± 0.55 | 3.80 ± 0.45 |
| Colour | 4.00 ± 0.00 | 3.40 ± 0.55 | 3.20 ± 0.45 | 3.60 ± 0.55 |
| Stability indicators | Varying storage conditions over 14 days | |||
| Day 0 | Day 14 | |||
| I * | II * | III * | ||
| Rate of redispersibility (stroke/seconds) | 1.40 ± 0.02 | 2.00 ± 0.00 | 2.00 ± 0.00 | 2.11 ± 0.19 |
| Sedimentation | 2.30 ± 0.06 | 0.7 ± 0.08 | 0.91 ± 0.08 | 0.86 ± 0.02 |
| Drug content (%) | Rifampicin | |||
| 97.61 ± 0.02 | 96.34 ± 0.01 | 97.00 ± 0.02 | 96.00 ± 0.00 | |
| Pyrazinamide | ||||
| 97.23 ± 2.57 | 93.19 ± 0.01 | 93.00 ± 0.01 | 94.00 ± 0.02 | |
| pH | 6.95 ± 0.13 | 8.29 ± 0.05 | 7.59 ± 0.05 | 8.16 ± 0.04 |
| Taste | 5.00 ± 0.00 | 4.40 ± 0.55 | 4.00 ± 0.00 | 4.60 ± 0.55 |
| Smell | 5.00 ± 0.00 | 4.00 ± 0.00 | 4.20 ± 0.45 | 4.60 ± 0.55 |
| Appearance | 4.00 ± 0.00 | 3.60 ± 0.55 | 3.00 ± 0.00 | 3.40 ± 0.55 |
| Colour | 4.00 ± 0.00 | 3.60 ± 0.50 | 3.00 ± 0.00 | 3.60 ± 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampedi, P.N.; Ogunrombi, M.O.; Wesley-Smith, J.; Adeleke, O.A. A Micro-Configured Multiparticulate Reconstitutable Suspension Powder of Fixed Dose Rifampicin and Pyrazinamide: Optimal Fabrication and In Vitro Quality Evaluation. Pharmaceutics 2023, 15, 64. https://doi.org/10.3390/pharmaceutics15010064
Rampedi PN, Ogunrombi MO, Wesley-Smith J, Adeleke OA. A Micro-Configured Multiparticulate Reconstitutable Suspension Powder of Fixed Dose Rifampicin and Pyrazinamide: Optimal Fabrication and In Vitro Quality Evaluation. Pharmaceutics. 2023; 15(1):64. https://doi.org/10.3390/pharmaceutics15010064
Chicago/Turabian StyleRampedi, Penelope N., Modupe O. Ogunrombi, James Wesley-Smith, and Oluwatoyin A. Adeleke. 2023. "A Micro-Configured Multiparticulate Reconstitutable Suspension Powder of Fixed Dose Rifampicin and Pyrazinamide: Optimal Fabrication and In Vitro Quality Evaluation" Pharmaceutics 15, no. 1: 64. https://doi.org/10.3390/pharmaceutics15010064
APA StyleRampedi, P. N., Ogunrombi, M. O., Wesley-Smith, J., & Adeleke, O. A. (2023). A Micro-Configured Multiparticulate Reconstitutable Suspension Powder of Fixed Dose Rifampicin and Pyrazinamide: Optimal Fabrication and In Vitro Quality Evaluation. Pharmaceutics, 15(1), 64. https://doi.org/10.3390/pharmaceutics15010064