

Oligonucleotide Therapeutics for Age-Related Musculoskeletal Disorders: Successes and Challenges
 , , and
, , and
Abstract
1. Introduction
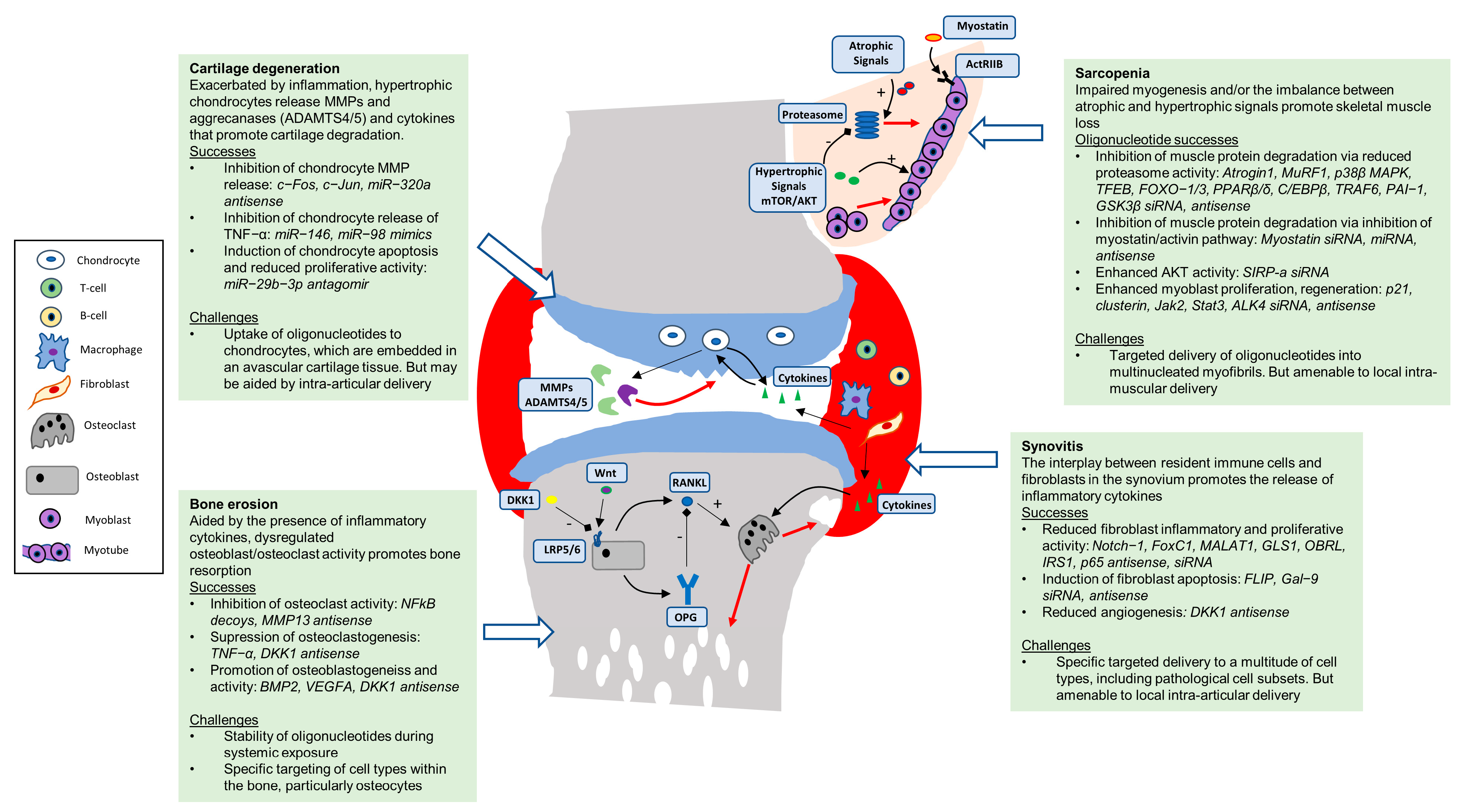
1.1. Sarcopenia
1.2. Osteoporosis
1.3. Arthritis
2. Oligonucleotides to Target the Cells and Tissues of the Musculoskeletal System
2.1. Skeletal Muscle
2.2. Bone
2.3. Articular Cartilage
2.4. Synovium

{kind=link}
| Tissue | Target | Oligonucleotide | Study Model | Administration | Function | Reference |
|---|---|---|---|---|---|---|
| Muscle | atrogin-1 and MuRF1 | siRNA | In vitro, L6 myotubes | - | Combined atrogin-1 and MuRF1 knock-down prevents dexamethasone-induced myotube atrophy | [79] |
| PPARβ/δ | siRNA | In vitro, L6 myotubes | - | PPARβ/δ knock-down reduces FOXO1 and MuRF1 expression and protein degradation in dexamethasone-treated myotubes | [78] | |
| p38β MAPK | siRNA | In vitro, C2C12 | - | MAPK knock-down prevents activin A induced catabolic activity | [70] | |
| TFEB | siRNA | In vitro, C2C12 | - | TFEB knock-down prevented angiotensin II-induced MuRF1 expression and atrophy | [80] | |
| C/EBPβ | siRNA | In vitro, L6 myoblasts and myotubes | - | C/EBPβ knock-down inhibited dexamethasone-induced increase in protein degradation, atrogin-1 expression and muscle cell atrophy | [81] | |
| p21 | siRNA | In vitro, primary mouse satellite cells | - | p21 knock-down restored proliferative capacity of uraemic serum-treated muscle progenitor cells | [75] | |
| FOXO3 | siRNA | In vitro, C2C12 | - | FOXO3 knock-down prevented iron-induced upregulation of atrogin-1 and MuRF1 and reduction in myotube diameter | [82] | |
| myostatin | antisense oligonucleotide | In vivo, MF1 or C57Bl10 mice | IM (ineffective); IV, weekly for 5 weeks (effective) | repeated intravenous administration of myostatin targeting vivo-PMO induced soleus muscle hypertrophy | [63] | |
| myostatin | siRNA | In vivo, caveolin-3 transgenic mouse dystrophy model | IM, single injection | nanoparticle complex with myostatin-siRNA increased muscle size and fibre size | [67] | |
| myostatin | cholesterol-conjugated siRNA | In vivo, CD-1 mouse | IV, single injection | myostatin knock-down increased skeletal muscle mass and strength | [66] | |
| Myostatin, miR-1, miR-206 | siRNA and/or miRNA | In vivo, Lewis rat, barium chloride-induced muscle injury | IM, single injection | combined administration of myostatin-siRNA and microRNAs improves in situ skeletal muscle regeneration | [68] | |
| myostatin | 2′-O-methyl antisense RNA | In vivo, BALB/c mouse, cancer cachexia model | oral or IV, twice weekly for 4 weeks | myostatin knock-down induced skeletal muscle hypertrophy | [62] | |
| Foxo-1 | 2′-O-methyl antisense RNA | In vivo, BALB/c mouse, cancer cachexia model | IV, twice weekly for 4 weeks | Foxo1 knock-down suppressed myostatin and induced MyoD and skeletal muscle hypertrophy | [69] | |
| myostatin | antisense chimeras | In vitro, human primary myoblasts | - | antisense oligonucleotide-mediated myostatin knock-down enhanced myoblast proliferation | [64] | |
| FOXO4 | siRNA | In vitro, C2C12 | - | FOXO4 knock-down reversed myotube atrophy from siRNA-mediated WNK1 silencing | [83] | |
| ALK4 | antisense oligonucleotide | In vivo, C57BL/6Jico and mdx dystrophy model mouse | IM, daily injections for 2 days | Alk4 knock-down stimulated muscle regeneration, but decreased muscle mass | [71] | |
| Jak2, Stat3 | siRNA | In vivo, Pax7-ZsGreen mice, mdx mice, and SV129 mice | IM, single injection of siRNA-transfected satellite cells | Knock-down of Jak2 and/or stat3 enhances regenerative capacity of muscle pre-cursor cells | [73] | |
| In vitro, aged mouse satellite cells | ||||||
| Clusterin | siRNA | In vitro, human primary myoblasts | - | Clusterin knock-down enhanced proliferation in myoblasts from older patients with osteoporosis, but in myoblasts from younger patients with osteoarthritis | [74] | |
| myostatin | siRNA | In vitro, differentiated mouse primary muscle cells, C2C12 and H2K mdx tsA58 cells | - | Description of epigenetic myostatin silencing, no functional readouts | [65] | |
| TRAF6 | siRNA | In vivo, ICR mice dexamethasone-induced muscle atrophy | IM, every 3 days for 2 weeks | TRAF6 knock-down attenuated dexamethasone-induced muscle atrophy and reduced expression of atrogin-1 and MuRF1 | [77] | |
| PAI-1 | siRNA | In vitro, C2C12 | - | PAI-1 knock-down prevents dexamethasone-induced atrogin 1 and MuRF1 upregulation | [84] | |
| SIRP-a | siRNA | In vitro, C2C12 | - | SIRPα knock-down promoted insulin signal transduction, pAkt and suppressed protein degradation | [85] | |
| GSK3β | siRNA | In vitro, C2C12 | - | GSK3β knock-down prevented atrogin-1/MuRF1 upregulation and myosin loss with IGF-1 blockade or dexamethasone | [86] | |
| Bone | DKK1 | Phosphorothioate antisense oligonucleotide | In vivo, ovariectomized rats. In vitro, primary rat osteoclasts | IP, for 4 weeks (5 consecutive days per week) | Alleviated loss of bone mass and biomechanical property, reduced osteoclast formation | [102] |
| NFKB | Decoy oligodeoxynucleotide | In vivo, ovariectomized rat and rats with osteogenic disorder Shionogi. In vitro, rabbit/rat osteoclasts | oral and implantation | Inhibited the formation and activity of osteoclasts, Increased bone length and bone mineral density | [88] | |
| NFKB | Decoy oligodeoxynucleotide | In vivo, C57BL/J6 mice osteolysis model | SQ, every other day for 14 days | Mitigated the expression of TNF-α, RANKL, and induced the expression of Il-1 receptor antagonist and OPG | [89] | |
| TNF-alpha | Antisense oligonucleotide | In vivo, C57BL/J6 mice osteolysis model | SQ, single injection | 90% of metal particulates induced osteoclastogenesis was suppressed by oligonucleotide delivery | [90] | |
| TNF-alpha | Phosphorothioate antisense oligonucleotides | In vivo, mouse model of edotoxin-induced bone resorption. In vitro, raw 264.7 cells | implantation | Oligonucleotide delivery via a bio-degradable cationic hydrogel suppressed the expression of TNF-α and osteoclastogenesis | [91] | |
| MMP13 | Antisense oligonucleotide | In vivo, mouse model of tumour induced osteolysis | IP, daily for 5 days, 2 days off, then daily for an additional 4 days | Significantly reduced bone destruction and the number of activated osteoclasts | [94] | |
| Cannabinoid receptor 1 | Phosphorothioate antisense oligonucleotide | In vivo, rats with glucocorticoid induced osteoporosis. In vitro, primary rat osteoblasts | IP, for 5 weeks | Attenuated the deleterious effects of glucocorticoid treatment on bone mineral density, trabecular microarchitecture, and mechanical properties | [100] | |
| BMP-2 and VEGF-A | Antisense oligonucleotides | In vivo ovariectomized rats | implantation | Significantly improved implant integration with local bone | [103] | |
| Synovium | DKK1 | Phosphorothioate antisense oligonucleotide | In vivo, SD rats ACLT OA model. In vitro, primary human SF | IP, weekly for 2, 6 or 12 weeks | Reduced proteinases and angiogenic factors, vessel distribution and formation and cartilage injury | [124] |
| FoxC1 | siRNA | In vivo, DMM mouse. In vitro, primary human SF | IA, twice weekly for 8 weeks | Inhibited IL-6, IL-8, TNF, ADAMTS-5, fibronectin, MMP3 and MMP13 and proliferation of OA SF and prevented OA development in vivo | [125] | |
| Notch-1 | Antisense oligonucleotide | In vitro, primary human synoviocytes | - | Partially blocked proliferation of RA synoviocytes and inhibited TNFα-induced proliferation in both OA and RA synoviocytes | [123] | |
| MALAT1 | Locked nucleic acid | In vitro, primary human SF | - | Inhibited proliferative and inflammatory phenotype of obese OA SF, reduced CXCL8 expression and secretion and increased expression of TRIM6, IL7R, HIST1H1C and MAML3 | [50] | |
| GLS1 | siRNA | In vitro, primary human SF | - | Reduced IL6 expression in OA SF | [117] | |
| Galectin-9 | siRNA | In vitro, primary human SF | - | Increased apoptosis of human RA SF | [122] | |
| OBRI | Phosphorothioate antisense oligonucleotide | In vitro, primary human SF | - | Reduced leptin-mediated IL-8 secretion via JAK2/STAT3 pathway, whilst activating the IRS1/PI3K/Akt/NF-kappaB-dependent pathway and recruitment of p300 | [118] | |
| IRS1 | Phosphorothioate antisense oligonucleotide | In vitro, primary human SF | - | Leptin mediated by inflammatory OA fibroblast phenotype was inhibited resulting in reduced IL-6 via IRS-1/PI3K/Akt/ AP-1 pathway | [119] | |
| FLIP | Phosphorothioate antisense oligonucleotide | In vitro, primary human synoviocytes | - | Sensitized RA FLS to increased fas-mediated apoptosis by 3-fold | [121] | |
| p65 | Phosphorothioate antisense oligonucleotide | In vitro, primary human synoviocytes | - | Decreased binding of NF-KB to COX-2 promoter and COX-2 protein expression | [120] | |
| Cartilage | c-Fos | Phosphorothioate antisense oligonucleotide | In vitro, primary human chondrocytes | - | Silenced the potentiating action of SDF-1α on MMP-13 promoter activity | [105] |
| c-Jun | Phosphorothioate antisense oligonucleotide | In vitro, primary human chondrocytes | - | Silenced the potentiating action of SDF-1α on MMP-13 promoter activity | [105] | |
| mir-146 | pre-miRNA mimics | In vitro, primary human chondrocytes | - | Overexpression significantly attenuated IL-1β induced reduced TNFα production. | [52] | |
| miR-320a | antisense oligonucleotides | In vitro, primary human chondrocytes | - | Reduced IL-1β mediated release of MMP13 and sGAG whilst enhancing expression of COL2A1 and ACAN | [106] | |
| miR-98 | pre-miRNA mimics | In vitro, primary human chondrocytes | - | Overexpression significantly attenuated IL-1β induced reduced TNFα production | [52] | |
| miR-29b-3p | antagomir | In vivo, OA rat model, SD rats ACLT/PCLT. In vitro, primary SD rat chondrocytes and SW1353 cells | - | Blocked PGRN, induced apoptosis, inhibited proliferation, and scratch wound closure of chondrocytes. In vivo, ameliorated articular chondrocytes apoptosis and cartilage loss | [107] | |
| miR-3680–3p | antagomir | In vivo, DMM rat OA model. In vitro, primary human chondrocytes | - | Reversed IL-1β induced chondrocyte injury and delayed OA progression via targeting OGG1. In vivo, attenuated cartilage destruction and loss of cartilage | [108] |
3. Challenges for Clinical Development
3.1. Stability
3.2. Tissue Penetration and Targeted Cellular Uptake
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylavsky, F.A.; Visser, M.; Newman, A.B.; et al. The Loss of Skeletal Muscle Strength, Mass, and Quality in Older Adults: The Health, Aging and Body Composition Study. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Hughes, V.A.; Fielding, R.A.; Fiatarone, M.A.; Evans, W.J.; Roubenoff, R. Aging of skeletal muscle: A 12-yr longitudinal study. J. Appl. Physiol. 2000, 88, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Rantanen, T.; Masaki, K.; Foley, D.; Izmirlian, G.; White, L.; Guralnik, J.M. Grip strength changes over 27 yr in Japanese-American men. J. Appl. Physiol. 1998, 85, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.; Church, C.; Baker, D.J.; Jones, S.W. The role of adipokines in skeletal muscle inflammation and insulin sensitivity. J. Inflamm. 2018, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Utriainen, T.; Takala, T.; Luotolahti, M.; Rönnemaa, T.; Laine, H.; Ruotsalainen, U.; Haaparanta, M.; Nuutila, P.; Yki-Järvinen, H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998, 41, 555–559. [Google Scholar] [CrossRef]
- Pendergrass, M.; Bertoldo, A.; Bonadonna, R.; Nucci, G.; Mandarino, L.; Cobelli, C.; DeFronzo, R.A. Muscle glucose transport and phosphorylation in type 2 diabetic, obese nondiabetic, and genetically predisposed individuals. Am. J. Physiol. Metab. 2007, 292, E92–E100. [Google Scholar] [CrossRef]
- Thiebaud, D.; Jacot, E.; Defronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.-P. The Effect of Graded Doses of Insulin on Total Glucose Uptake, Glucose Oxidation, and Glucose Storage in Man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef]
- Pinedo-Villanueva, R.; Westbury, L.D.; Syddall, H.E.; Sanchez-Santos, M.T.; Dennison, E.M.; Robinson, S.M.; Cooper, C. Health Care Costs Associated with Muscle Weakness: A UK Population-Based Estimate. Calcif. Tissue Int. 2019, 104, 137–144. [Google Scholar] [CrossRef]
- Wilhelmsen, A.; Tsintzas, K.; Jones, S.W. Recent advances and future avenues in understanding the role of adipose tissue cross talk in mediating skeletal muscle mass and function with ageing. Geroscience 2021, 43, 85–110. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Wallace, G.R.; Davis, E.T.; Murphy, D.P.; Nicholson, T.; Bennett, A.J.; Tsintzas, K.; Jones, S.W. Obese subcutaneous adipose tissue impairs human myogenesis, particularly in old skeletal muscle, via resistin-mediated activation of NFkappaB. Sci. Rep. 2018, 8, 15360. [Google Scholar] [CrossRef]
- Jones, S.W.; Hill, R.J.; Krasney, P.A.; O’Conner, B.; Peirce, N.; Greenhaff, P. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. 2004, 18, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Fenton, C.G.; Langen, R.; Hardy, R.S. Exploring the Interface between Inflammatory and Therapeutic Glucocorticoid Induced Bone and Muscle Loss. Int. J. Mol. Sci. 2019, 20, 5768. [Google Scholar] [CrossRef] [PubMed]
- Benz, E.; Trajanoska, K.; LaHousse, L.; Schoufour, J.D.; Terzikhan, N.; De Roos, E.; De Jonge, G.B.; Williams, R.; Franco, O.H.; Brusselle, G.; et al. Sarcopenia in COPD: A systematic review and meta-analysis. Eur. Respir. Rev. 2019, 28, 190049. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, A.; Cuppari, L.; Stenvinkel, P.; Lindholm, B.; Avesani, C.M. Sarcopenia in chronic kidney disease: What have we learned so far? J. Nephrol. 2021, 34, 1347–1372. [Google Scholar] [CrossRef]
- Allen, S.L.; Quinlan, J.I.; Dhaliwal, A.; Armstrong, M.J.; Elsharkawy, A.M.; Greig, C.A.; Lord, J.M.; Lavery, G.G.; Breen, L. Sarcopenia in chronic liver disease: Mechanisms and countermeasures. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G241–G257. [Google Scholar] [CrossRef]
- Le Grand, F.; Rudnicki, M.A. Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 2007, 19, 628–633. [Google Scholar] [CrossRef]
- Yamakawa, H.; Kusumoto, D.; Hashimoto, H.; Yuasa, S. Stem Cell Aging in Skeletal Muscle Regeneration and Disease. Int. J. Mol. Sci. 2020, 21, 1830. [Google Scholar] [CrossRef]
- Wilkinson, D.; Piasecki, M.; Atherton, P. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res. Rev. 2018, 47, 123–132. [Google Scholar] [CrossRef]
- Breen, L.; Phillips, S.M. Skeletal muscle protein metabolism in the elderly: Interventions to counteract the ‘anabolic resistance’ of ageing. Nutr. Metab. 2011, 8, 68. [Google Scholar] [CrossRef]
- Wall, B.T.; Gorissen, S.; Pennings, B.; Koopman, R.; Groen, B.B.L.; Verdijk, L.; van Loon, L.J. Aging Is Accompanied by a Blunted Muscle Protein Synthetic Response to Protein Ingestion. PLoS ONE 2015, 10, e0140903. [Google Scholar] [CrossRef] [PubMed]
- Beaudart, C.; McCloskey, E.; Bruyère, O.; Cesari, M.; Rolland, Y.; Rizzoli, R.; Araujo De Carvalho, I.; Amuthavalli Thiyagarajan, J.; Bautmans, I.; Bertière, M.-C.; et al. Sarcopenia in daily practice: Assessment and management. BMC Geriatr. 2016, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Langsetmo, L.; Joseph, L.; Hanley, D.A.; Davison, K.S.; Josse, R.; Kreiger, N.; Tenenhouse, A.; Goltzman, D. The Canadian Multicentre Osteoporosis Study Research Group Change in bone mineral density as a function of age in women and men and association with the use of antiresorptive agents. Can. Med. Assoc. J. 2008, 178, 1660–1668. [Google Scholar] [CrossRef]
- Pouresmaeili, F.; Dehghan, B.K.; Kamarehei, M.; Meng, G.Y. A comprehensive overview on osteoporosis and its risk factors. Ther. Clin. Risk Manag. 2018, 14, 2029–2049. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-J.; Liu, Y.-J.; Liu, P.-Y.; Hamilton, J.; Recker, R.R.; Deng, H.-W. Relationship of Obesity with Osteoporosis. J. Clin. Endocrinol. Metab. 2007, 92, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.; Scott, A.; Ede, M.N.; Jones, S.W. Do E-cigarettes and vaping have a lower risk of osteoporosis, nonunion, and infection than tobacco smoking? Bone Jt. Res. 2021, 10, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Yoon, V.; Maalouf, N.M.; Sakhaee, K. The effects of smoking on bone metabolism. Osteoporos. Int. 2012, 23, 2081–2092. [Google Scholar] [CrossRef]
- Hardy, R.S.; Raza, K.; Cooper, M.S. Therapeutic glucocorticoids: Mechanisms of actions in rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 133–144. [Google Scholar] [CrossRef]
- Leboime, A.; Confavreux, C.B.; Mehsen, N.; Paccou, J.; David, C.; Roux, C. Osteoporosis and mortality. Joint. Bone Spine 2010, 77 (Suppl. S2), S107–S112. [Google Scholar] [CrossRef]
- Bliuc, D.; Nguyen, N.D.; Milch, V.E.; Nguyen, T.V.; Eisman, J.A.; Center, J.R. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA 2009, 301, 513–521. [Google Scholar] [CrossRef]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of Action and Role in Clinical Practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Kennel, K.A.; Drake, M.T. Adverse effects of bisphosphonates: Implications for osteoporosis management. Mayo Clin. Proc. 2009, 84, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Diab, D.L.; Watts, N.B. Bisphosphonate drug holiday: Who, when and how long. Ther. Adv. Musculoskelet. Dis. 2013, 5, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.Y.; Mason, A.; Cooper, C.; Dennison, E. Novel advances in the treatment of osteoporosis. Br. Med. Bull. 2016, 119, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Tai, N.; Inoue, D. Anti-Dickkopf1 (Dkk1) antibody as a bone anabolic agent for the treatment of osteoporosis. Clin. Calcium 2014, 24, 75–83. [Google Scholar]
- Clynes, M.A.; Harvey, N.C.; Curtis, E.M.; Fuggle, N.R.; Dennison, E.M.; Cooper, C. The epidemiology of osteoporosis. Br. Med. Bull. 2020, 133, 105–117. [Google Scholar] [CrossRef]
- Gullberg, B.; Johnell, O.; Kanis, J.A. World-wide Projections for Hip Fracture. Osteoporos. Int. 1997, 7, 407–413. [Google Scholar] [CrossRef]
- Poudel, P.; Goyal, A.; Lappin, S.L. Inflammatory Arthritis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef]
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; Lin, J.; Guo, A. Prevalence Trends of Site-Specific Osteoarthritis from 1990 to 2019: Findings from the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Sato, K.; Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 2006, 18, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.; Zhou, H.; Seibel, M.J.; Cooper, M.S. Glucocorticoids and Bone: Consequences of Endogenous and Exogenous Excess and Replacement Therapy. Endocr. Rev. 2018, 39, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Oehler, S.; Neureiter, D.; Meyer-Scholten, C.; Aigner, T. Subtyping of osteoarthritic synoviopathy. Clin. Exp. Rheumatol. 2002, 20, 633–640. [Google Scholar]
- Fernandez-Madrid, F.; Karvonen, R.L.; Teitge, R.A.; Miller, P.R.; An, T.; Negendank, W.G. Synovial thickening detected by MR imaging in osteoarthritis of the knee confirmed by biopsy as synovitis. Magn. Reson. Imaging 1995, 13, 177–183. [Google Scholar] [CrossRef]
- Rhodes, L.A.; Conaghan, P.G.; Radjenovic, A.; Grainger, A.J.; Emery, P.; McGonagle, D. Further evidence that a cartilage-pannus junction synovitis predilection is not a specific feature of rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 1347–1349. [Google Scholar] [CrossRef]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef]
- Nanus, D.E.; Wijesinghe, S.N.; Pearson, M.; Hadjicharalambous, M.R.; Rosser, A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Regulation of the Inflammatory Synovial Fibroblast Phenotype by Metastasis-Associated Lung Adenocarcinoma Transcript 1 Long Noncoding RNA in Obese Patients with Osteoarthritis. Arthritis Rheumatol. 2020, 72, 609–619. [Google Scholar] [CrossRef]
- Jones, S.W.; Brockbank, S.M.; Clements, K.M.; Le Good, N.; Campbell, D.; Read, S.J.; Needham, M.R.; Newham, P. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthr. Cartil. 2009, 17, 124–131. [Google Scholar] [CrossRef]
- Jones, S.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.; Brockbank, S.; Needham, M.; Read, S.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-α and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Gerstenfeld, L.; Bishop, G.; Davis, A.D.; Mason, Z.D.; Einhorn, T.A.; Maciewicz, R.A.; Newham, P.; Foster, M.; Jackson, S.; et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res. Ther. 2009, 11, R11. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Jackson, S.G.; Wardale, J.; Jones, S.W. Hypoxia modulates the phenotype of osteoblasts isolated from knee osteoarthritis patients, leading to undermineralized bone nodule formation. Arthritis Rheumatol. 2014, 66, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.A.; McNair, P.J.; Lewis, G.N. Mechanisms of quadriceps muscle weakness in knee joint osteoarthritis: The effects of prolonged vibration on torque and muscle activation in osteoarthritic and healthy control subjects. Arthritis Res. Ther. 2011, 13, R151. [Google Scholar] [CrossRef]
- Shorter, E.; Sannicandro, A.J.; Poulet, B.; Goljanek-Whysall, K. Skeletal Muscle Wasting and Its Relationship with Osteoarthritis: A Mini-Review of Mechanisms and Current Interventions. Curr. Rheumatol. Rep. 2019, 21, 40. [Google Scholar] [CrossRef]
- van Vollenhoven, R. Treat-to-target in rheumatoid arthritis-are we there yet? Nat. Rev. Rheumatol. 2019, 15, 180–186. [Google Scholar] [CrossRef]
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2017, 56, 869–881. [Google Scholar] [CrossRef]
- Maricar, N.; Parkes, M.J.; Callaghan, M.J.; Hutchinson, C.E.; Gait, A.D.; Hodgson, R.; Felson, D.T.; O’Neill, T.W. Structural predictors of response to intra-articular steroid injection in symptomatic knee osteoarthritis. Arthritis Res. Ther. 2017, 19, 88. [Google Scholar] [CrossRef]
- Bourne, R.B.; Chesworth, B.M.; Davis, A.M.; Mahomed, N.N.; Charron, K.D. Patient satisfaction after total knee arthroplasty: Who is satisfied and who is not? Clin. Orthop. Relat. Res. 2010, 468, 57–63. [Google Scholar] [CrossRef]
- Lee, S.-J. Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J. Clin. Invest. 2021, 131, e148372. [Google Scholar] [CrossRef]
- Liu, C.-M.; Yang, Z.; Liu, C.-W.; Wang, R.; Tien, P.; Dale, R.; Sun, L.-Q. Myostatin antisense RNA-mediated muscle growth in normal and cancer cachexia mice. Gene Ther. 2008, 15, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.K.; Malerba, A.; Popplewell, L.; Foster, K.; Dickson, G. Antisense-induced Myostatin Exon Skipping Leads to Muscle Hypertrophy in Mice Following Octa guanidine Morpholino Oligomer Treatment. Mol. Ther. 2011, 19, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Maeta, K.; Farea, M.; Nishio, H.; Matsuo, M. An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor. Int. J. Mol. Sci. 2022, 23, 5016. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Andaloussi, S.E.; Morris, K.V.; McClorey, G.; Wood, M.J. Small RNA-Mediated Epigenetic Myostatin Silencing. Mol. Ther. Nucleic Acids 2012, 1, e23. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Weber, H.; DiMuzio, J.; Matter, A.; Dogdas, B.; Shah, T.; Thankappan, A.; Disa, J.; Jadhav, V.; Lubbers, L.; et al. Silencing Myostatin Using Cholesterol-conjugated siRNAs Induces Muscle Growth. Mol. Ther. Nucleic Acids 2016, 5, e342. [Google Scholar] [CrossRef]
- Kawakami, E.; Kawai, N.; Kinouchi, N.; Mori, H.; Ohsawa, Y.; Ishimaru, N.; Sunada, Y.; Noji, S.; Tanaka, E. Local applications of myostatin-siRNA with atelocollagen increase skeletal muscle mass and recovery of muscle function. PLoS ONE 2013, 8, e64719. [Google Scholar] [CrossRef]
- Kim, N.; Yoo, J.J.; Atala, A.; Lee, S.J. Combination of small RNAs for skeletal muscle regeneration. FASEB J. 2016, 30, 1198–1206. [Google Scholar] [CrossRef]
- Liu, C.-M.; Yang, Z.; Liu, C.W.; Wang, R.; Tien, P.; Dale, R.; Sun, L.-Q. Effect of RNA oligonucleotide targeting Foxo-1 on muscle growth in normal and cancer cachexia mice. Cancer Gene Ther. 2007, 14, 945–952. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.; Liu, Z.; Lin, R.K.; Li, M.; Li, Y.P. Activin A induces skeletal muscle catabolism via p38β mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Pasteuning-Vuhman, S.; Boertje-van der Meulen, J.W.; Van Putten, M.; Overzier, M.; Ten Dijke, P.; Kielbasa, S.M.; Arindrarto, W.; Wolterbeek, R.; Lezhnina, K.V.; Ozerov, I.V.; et al. New function of the myostatin/activin type I receptor (ALK4) as a mediator of muscle atrophy and muscle regeneration. FASEB J. 2017, 31, 238–255. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; García-Prat, L.; Muñoz-Cánoves, P. Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 2022, 23, 204–226. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Pucci, S.; Greggi, C.; Polidoro, C.; Piro, M.C.; Celi, M.; Feola, M.; Gasbarra, E.; Iundusi, R.; Mastrangeli, F.; Novelli, G.; et al. Clusterin silencing restores myoblasts viability and down modulates the inflammatory process in osteoporotic disease. J. Transl. Med. 2019, 17, 118. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, B.; Hassounah, F.; Price, S.R.; Klein, J.; Mohamed, T.M.; Wang, Y.; Park, J.; Cai, H.; Zhang, X.; et al. The impact of senescence on muscle wasting in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2022. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 Inhibition Rescues Dexamethasone-Induced Muscle Atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141. [Google Scholar] [CrossRef]
- Castillero, E.; Alamdari, N.; Aversa, Z.; Gurav, A.; Hasselgren, P.-O. PPARβ/δ Regulates Glucocorticoid- and Sepsis-Induced FOXO1 Activation and Muscle Wasting. PLoS ONE 2013, 8, e59726. [Google Scholar] [CrossRef]
- Castillero, E.; Alamdari, N.; Lecker, S.H.; Hasselgren, P.O. Suppression of atrogin-1 and MuRF1 prevents dexamethasone-induced atrophy of cultured myotubes. Metabolism 2013, 62, 1495–1502. [Google Scholar] [CrossRef]
- Du Bois, P.; Tortola, C.P.; Lodka, D.; Kny, M.; Schmidt, F.; Song, K.; Schmidt, S.; Bassel-Duby, R.; Olson, E.N.; Fielitz, J. Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ. Res. 2015, 117, 424–436. [Google Scholar] [CrossRef]
- Gonnella, P.; Alamdari, N.; Tizio, S.; Aversa, Z.; Petkova, V.; Hasselgren, P.-O. C/EBPβ regulates dexamethasone-induced muscle cell atrophy and expression of atrogin-1 and MuRF1. J. Cell. Biochem. 2011, 112, 1737–1748. [Google Scholar] [CrossRef]
- Ikeda, Y.; Imao, M.; Satoh, A.; Watanabe, H.; Hamano, H.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Miyamoto, L.; Ishizawa, K.; et al. Iron-induced skeletal muscle atrophy involves an Akt-forkhead box O3–E3 ubiquitin ligase-dependent pathway. J. Trace Elem. Med. Biol. 2016, 35, 66–76. [Google Scholar] [CrossRef]
- Mandai, S.; Mori, T.; Nomura, N.; Furusho, T.; Arai, Y.; Kikuchi, H.; Sasaki, E.; Sohara, E.; Rai, T.; Uchida, S. WNK1 regulates skeletal muscle cell hypertrophy by modulating the nuclear localization and transcriptional activity of FOXO4. Sci. Rep. 2018, 8, 9101. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Kawao, N.; Shimoide, T.; Okada, K.; Matsuo, O.; Kaji, H. Role of plasminogen activator inhibitor-1 in glucocorticoid-induced muscle change in mice. J. Bone Miner. Metab. 2018, 36, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Dong, Y.; Zhang, L.; Mitch, W.E. Signal regulatory protein-α interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney Int. 2013, 84, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Verhees, K.J.; Schols, A.M.; Kelders, M.C.; Op den Kamp, C.M.; van der Velden, J.L.; Langen, R.C. Glycogen synthase kinase-3β is required for the induction of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2011, 301, C995–C1007. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y. NF-κB signaling and bone resorption. Osteoporos. Int. 2013, 24, 2377–2386. [Google Scholar] [CrossRef]
- Shimizu, H.; Nakagami, H.; Tsukamoto, I.; Morita, S.; Kunugiza, Y.; Tomita, T.; Yoshikawa, H.; Kaneda, Y.; Ogihara, T.; Morishita, R. NFκB decoy oligodeoxynucleotides ameliorates osteoporosis through inhibition of activation and differentiation of osteoclasts. Gene Ther. 2006, 13, 933–941. [Google Scholar] [CrossRef]
- Sato, T.; Pajarinen, J.; Lin, T.H.; Tamaki, Y.; Loi, F.; Egashira, K.; Yao, Z.; Goodman, S.B. NF-κB decoy oligodeoxynucleotide inhibits wear particle-induced inflammation in a murine calvarial model. J. Biomed. Mater. Res. Part A 2015, 103, 3872–3878. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, R.; Zhu, Y.A.; Wang, C.; Diao, H.; Zhang, C.; Zhao, J.; Zhang, J. Antisense oligonucleotide targeting TNF-α can suppress Co-Cr-Mo particle-induced osteolysis. J. Orthop. Res. 2008, 26, 1114–1120. [Google Scholar] [CrossRef]
- Dong, L.; Huang, Z.; Cai, X.; Xiang, J.; Zhu, Y.-A.; Wang, R.; Chen, J.; Zhang, J. Localized Delivery of Antisense Oligonucleotides by Cationic Hydrogel Suppresses TNF-α Expression and Endotoxin-Induced Osteolysis. Pharm. Res. 2011, 28, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T. Osteoporosis and cancer. Curr. Osteoporos. Rep. 2013, 11, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Nannuru, K.C.; Futakuchi, M.; Varney, M.L.; Vincent, T.M.; Marcusson, E.G.; Singh, R.K. Matrix metalloproteinase (MMP)-13 regulates mammary tumor-induced osteolysis by activating MMP9 and transforming growth factor-β signaling at the tumor-bone interface. Cancer Res. 2010, 70, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.E.; Stone, M.D.; Edwards, P.H.; Duckers, J.M.; Evans, W.D.; Shale, D.J. Circulating matrix metalloproteinase-9 and osteoporosis in patients with chronic obstructive pulmonary disease. Chron. Respir. Dis. 2009, 6, 81–87. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, Y.; Guo, S.; Zhang, W.; Wang, J.; Lin, Y. Dynamic expression of matrix metalloproteinases 2, 9 and 13 in ovariectomy-induced osteoporosis rats. Exp. Ther. Med. 2018, 16, 1807–1813. [Google Scholar] [CrossRef]
- Chotiyarnwong, P.; McCloskey, E.V. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat. Rev. Endocrinol. 2020, 16, 437–447. [Google Scholar] [CrossRef]
- Fenton, C.G.; Doig, C.L.; Fareed, S.; Naylor, A.; Morrell, A.P.; Addison, O.; Wehmeyer, C.; Buckley, C.D.; Cooper, M.S.; Lavery, G.G.; et al. 11β-HSD1 plays a critical role in trabecular bone loss associated with systemic glucocorticoid therapy. Arthritis Res. Ther. 2019, 21, 188. [Google Scholar] [CrossRef]
- Fenton, C.G.; Webster, J.M.; Martin, C.; Fareed, S.; Wehmeyer, C.; Mackie, H.; Jones, R.; Seabright, A.P.; Lewis, J.W.; Lai, Y.-C.; et al. Therapeutic glucocorticoids prevent bone loss but drive muscle wasting when administered in chronic polyarthritis. Arthritis Res. Ther. 2019, 21, 182. [Google Scholar] [CrossRef]
- Ko, J.Y.; Wu, R.W.; Kuo, S.J.; Chen, M.W.; Yeh, D.W.; Ke, H.C.; Wu, S.L.; Wang, F.S. Cannabinoid receptor 1 mediates glucocorticoid-induced bone loss in rats by perturbing bone mineral acquisition and marrow adipogenesis. Arthritis Rheum. 2012, 64, 1204–1214. [Google Scholar] [CrossRef]
- Wang, J.; Shou, J.; Chen, X. Dickkopf-1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene 2000, 19, 1843–1848. [Google Scholar] [CrossRef]
- Wang, F.-S.; Ko, J.-Y.; Lin, C.-L.; Wu, H.-L.; Ke, H.-J.; Tai, P.-J. Knocking down dickkopf-1 alleviates estrogen deficiency induction of bone loss. A histomorphological study in ovariectomized rats. Bone 2007, 40, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Wölfle, J.V.; Fiedler, J.; Dürselen, L.; Reichert, J.; Scharnweber, D.; Förster, A.; Schwenzer, B.; Reichel, H.; Ignatius, A.; Brenner, R.E. Improved Anchorage of Ti6Al4V Orthopaedic Bone Implants through Oligonucleotide Mediated Immobilization of BMP-2 in Osteoporotic Rats. PLoS ONE 2014, 9, e86151. [Google Scholar] [CrossRef] [PubMed]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-C.; Yang, R.-S.; Hsieh, K.-H.; Fong, Y.-C.; Way, T.-D.; Lee, T.-S.; Wu, H.-C.; Fu, W.-M.; Tang, C.-H. Stromal Cell-Derived Factor-1 Induces Matrix Metalloprotease-13 Expression in Human Chondrocytes. Mol. Pharmacol. 2007, 72, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, X.; Gao, Z.Y.; Liu, K.; Hou, Y.; Zheng, J. The role of miR-320a and IL-1β in human chondrocyte degradation. Bone Joint Res. 2017, 6, 196–203. [Google Scholar] [CrossRef]
- Chen, L.; Li, Q.; Wang, J.; Jin, S.; Zheng, H.; Lin, J.; He, F.; Zhang, H.; Ma, S.; Mei, J.; et al. MiR-29b-3p promotes chondrocyte apoptosis and facilitates the occurrence and development of osteoarthritis by targeting PGRN. J. Cell. Mol. Med. 2017, 21, 3347–3359. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Suo, Y.; Li, Y.; Li, Q.; Chen, X. Downregulation of miR-3680–3p inhibits the progression of osteoarthritis via targeting OGG1. Arch. Gerontol. Geriatr. 2022, 100, 104626. [Google Scholar] [CrossRef]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Baker, K.; Grainger, A.; Niu, J.; Clancy, M.; Guermazi, A.; Crema, M.D.; Hughes, L.; Buckwalter, J.; Wooley, A.; Nevitt, M.; et al. Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann. Rheum. Dis. 2010, 69, 1779–1783. [Google Scholar] [CrossRef]
- Nanus, D.E.; Badoume, A.; Wijesinghe, S.N.; Halsey, A.M.; Hurley, P.; Ahmed, Z.; Botchu, R.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Synovial tissue from sites of joint pain in knee osteoarthritis patients exhibits a differential phenotype with distinct fibroblast subsets. EBioMedicine 2021, 72, 103618. [Google Scholar] [CrossRef] [PubMed]
- Dawes, J.M.; Kiesewetter, H.; Perkins, J.R.; Bennett, D.L.; McMahon, S.B. Chemokine expression in peripheral tissues from the monosodium iodoacetate model of chronic joint pain. Mol. Pain 2013, 9, 1744–8069. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Miller, R.J.; Malfait, A.M. Osteoarthritis joint pain: The cytokine connection. Cytokine 2014, 70, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Farah, H.; Young, S.P.; Mauro, C.; Jones, S.W. Metabolic dysfunction and inflammatory disease: The role of stromal fibroblasts. FEBS J. 2020, 288, 5555–5568. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Farah, H.; Wijesinghe, S.N.; Nicholson, T.; Alnajjar, F.; Certo, M.; Alghamdi, A.; Davis, E.T.; Young, S.P.; Mauro, C.; Jones, S.W. Differential Metabotypes in Synovial Fibroblasts and Synovial Fluid in Hip Osteoarthritis Patients Support Inflammatory Responses. Int. J. Mol. Sci. 2022, 23, 3266. [Google Scholar] [CrossRef]
- Tong, K.M.; Shieh, D.C.; Chen, C.P.; Tzeng, C.Y.; Wang, S.P.; Huang, K.C.; Chiu, Y.C.; Fong, Y.C.; Tang, C.H. Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3K, Akt cascade and promotion of NF-κB/p300 binding in human synovial fibroblasts. Cell Signal. 2008, 20, 1478–1488. [Google Scholar] [CrossRef]
- Yang, W.-H.; Liu, S.-C.; Tsai, C.-H.; Fong, Y.-C.; Wang, S.-J.; Chang, Y.-S.; Tang, C.-H. Leptin Induces IL-6 Expression through OBRl Receptor Signaling Pathway in Human Synovial Fibroblasts. PLoS ONE 2013, 8, e75551. [Google Scholar] [CrossRef]
- Crofford, L.J.; Tan, B.; McCarthy, C.J.; Hla, T. Involvement of nuclear factor kB in the regulation of cyclooxygenase-2 expression by interleukin-1 in rheumatoid synoviocytes. Arthritis Rheum. 1997, 40, 226–236. [Google Scholar] [CrossRef]
- Palao, G.; Santiago, B.; Galindo, M.; Payá, M.; Ramirez, J.C.; Pablos, J.L. Down-regulation of FLIP sensitizes rheumatoid synovial fibroblasts to Fas-mediated apoptosis. Arthritis Rheum. 2004, 50, 2803–2810. [Google Scholar] [CrossRef]
- Pearson, M.J.; Bik, M.A.; Ospelt, C.; Naylor, A.J.; Wehmeyer, C.; Jones, S.W.; Buckley, C.D.; Gay, S.; Filer, A.; Lord, J.M. Endogenous Galectin-9 Suppresses Apoptosis in Human Rheumatoid Arthritis Synovial Fibroblasts. Sci. Rep. 2018, 8, 12887. [Google Scholar] [CrossRef]
- Nakazawa, M.; Ishii, H.; Aono, H.; Takai, M.; Honda, T.; Aratani, S.; Fukamizu, A.; Nakamura, H.; Yoshino, S.-I.; Kobata, T.; et al. Role of notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 2001, 44, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.-H.; Ko, J.-Y.; Wang, C.-J.; Sun, Y.-C.; Wang, F.-S. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012, 64, 3267–3277. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zhang, H.; Gao, W.; Lu, M.; Liu, W.; Li, Y.; Yin, Z. Forkhead box C1 promotes the pathology of osteoarthritis by upregulating β-catenin in synovial fibroblasts. FEBS J. 2020, 287, 3065–3087. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Gooding, M.; Malhotra, M.; Evans, J.C.; Darcy, R.; O’Driscoll, C.M. Oligonucleotide conjugates–Candidates for gene silencing therapeutics. Eur. J. Pharm. Biopharm. 2016, 107, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Zhang, Y.-J. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front. Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. Author Correction: The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 841. [Google Scholar] [CrossRef] [PubMed]
- Gökirmak, T.; Nikan, M.; Wiechmann, S.; Prakash, T.P.; Tanowitz, M.; Seth, P.P. Overcoming the challenges of tissue delivery for oligonucleotide therapeutics. Trends Pharmacol. Sci. 2021, 42, 588–604. [Google Scholar] [CrossRef]
- Anderson, B.A.; Freestone, G.C.; Low, A.; De-Hoyos, C.L.; Iii, W.J.D.; Ostergaard, M.E.; Migawa, M.T.; Fazio, M.; Wan, W.B.; Berdeja, A.; et al. Towards next generation antisense oligonucleotides: Mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021, 49, 9026–9041. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.; Williams, A.; Lindsay, M. Cell-penetrating-peptide-mediated siRNA lung delivery. Biochem. Soc. Trans. 2007, 35, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.V.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Onuma, H.; Sato, R.; Sato, Y.; Hashiba, A.; Maeki, M.; Tokeshi, M.; Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K.; et al. Lipid nanoparticles loaded with ribonucleoprotein–oligonucleotide complexes synthesized using a microfluidic device exhibit robust genome editing and hepatitis B virus inhibition. J. Control. Release 2021, 330, 61–71. [Google Scholar] [CrossRef]
- Żak, M.M.; Zangi, L. Lipid Nanoparticles for Organ-Specific mRNA Therapeutic Delivery. Pharmaceutics 2021, 13, 1675. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Patel, A.K.; Rhym, L.H.; Palmiero, U.C.; Bhat, B.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Systemic delivery of mRNA and DNA to the lung using polymer-lipid nanoparticles. Biomaterials 2021, 275, 120966. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Dugal-Tessier, J.; Thirumalairajan, S.; Jain, N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J. Clin. Med. 2021, 10, 838. [Google Scholar] [CrossRef]
- Sacchetti, C.; Liu-Bryan, R.; Magrini, A.; Rosato, N.; Bottini, N.; Bottini, M. Polyethylene-Glycol-Modified Single-Walled Carbon Nanotubes for Intra-Articular Delivery to Chondrocytes. ACS Nano 2014, 8, 12280–12291. [Google Scholar] [CrossRef]
- Ji, M.L.; Jiang, H.; Wu, F.; Geng, R.; Ya, L.K.; Lin, Y.C.; Xu, J.H.; Wu, X.T.; Lu, J. Precise targeting of miR-141/200c cluster in chondrocytes attenuates osteoarthritis development. Ann. Rheum. Dis. 2021, 80, 356–366. [Google Scholar] [CrossRef]
- Xia, C.F.; Boado, R.J.; Pardridge, W.M. Antibody-mediated targeting of siRNA via the human insulin receptor using avidin-biotin technology. Mol. Pharm. 2009, 6, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.-F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB–siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicholson, T.A.; Sagmeister, M.; Wijesinghe, S.N.; Farah, H.; Hardy, R.S.; Jones, S.W. Oligonucleotide Therapeutics for Age-Related Musculoskeletal Disorders: Successes and Challenges. Pharmaceutics 2023, 15, 237. https://doi.org/10.3390/pharmaceutics15010237
Nicholson TA, Sagmeister M, Wijesinghe SN, Farah H, Hardy RS, Jones SW. Oligonucleotide Therapeutics for Age-Related Musculoskeletal Disorders: Successes and Challenges. Pharmaceutics. 2023; 15(1):237. https://doi.org/10.3390/pharmaceutics15010237
Chicago/Turabian StyleNicholson, Thomas A., Michael Sagmeister, Susanne N. Wijesinghe, Hussein Farah, Rowan S. Hardy, and Simon W. Jones. 2023. "Oligonucleotide Therapeutics for Age-Related Musculoskeletal Disorders: Successes and Challenges" Pharmaceutics 15, no. 1: 237. https://doi.org/10.3390/pharmaceutics15010237
APA StyleNicholson, T. A., Sagmeister, M., Wijesinghe, S. N., Farah, H., Hardy, R. S., & Jones, S. W. (2023). Oligonucleotide Therapeutics for Age-Related Musculoskeletal Disorders: Successes and Challenges. Pharmaceutics, 15(1), 237. https://doi.org/10.3390/pharmaceutics15010237