1. Introduction
Antibody–drug conjugates (ADCs) combine a highly potent cytotoxic agent (drug or payload) conjugated through a suitably constructed linker onto a monoclonal antibody (mAb) directed to a tumor-selective antigen [
1,
2,
3,
4]. The aims of this approach are to reduce systemic toxicity while enhancing antitumor efficacy. There are currently 14 ADCs approved worldwide that are successfully implemented in clinical strategies [
1], while more than 200 clinical trials involving ADCs are actually either recruiting or active [
5].
Many of these ADCs are generated using a stochastic bioconjugation process. Site-specific conjugation has been described to improve ADC therapeutic index in comparison to classical random methods [
6]. This attractive strategy led to the recent approvals by the Food and Drug Administration of Enhertu
® and Trodelvy
® (in December 2019 and April 2020, respectively). Despite their recent keen interest, one of the major drawbacks associated with ADCs targeting solid tumors is their insufficient activity at the maximum tolerated dose (MTD) upon repeated doses, leading to many of them being discontinued after failing to progress beyond Phase II. This suggests there are still unmet parameters needing to be optimized in order to reach further translational successes [
5,
7]. To explain this finding, the efficacy of ADCs based on a full immunoglobulin-G (IgG) format is limited by their size (150 kDa), associated with suboptimal tumor penetration and uptake [
8,
9]. Furthermore, their Fc portion is considered to mediate off-target toxicity [
10,
11,
12]. Indeed, the long half-life of ADCs [
13] due to the neonatal Fc receptor (FcRn) is increasing normal tissue exposure, while Fc-gamma receptors (FcγR) cross-react with endothelial cells and the immune system.
To overcome these drawbacks, several smaller formats of drug conjugates [
10,
14] have been explored, including peptides [
15], single-domain antibody fragments (sdAb or VHH) [
16], single-chain fragment variables (scFvs) [
17,
18], antigen-binding fragment (Fabs) [
19] or minibodies (small immunoprotein as scFvs dimerized using a CHε4 domain) [
20,
21]. Surprisingly, among them, only a few examples of efficiently vectorized drugs with smaller antibody formats have been reported, exhibiting subnanomolar activity with a drug-to-antibody ratio (DAR) between 1 and 2 [
19,
22,
23]. As part of this strategy, we recently described the site-specific conjugation of one monomethyl auristatin F (MMAF) at the C-terminal position onto an engineered anti-HER2 scFv of the trastuzumab antibody (4D5.2), generating an antibody fragment–drug conjugate (FDC) with a DAR of 1 [
18,
19]. 4D5.2 was produced in the bacteria
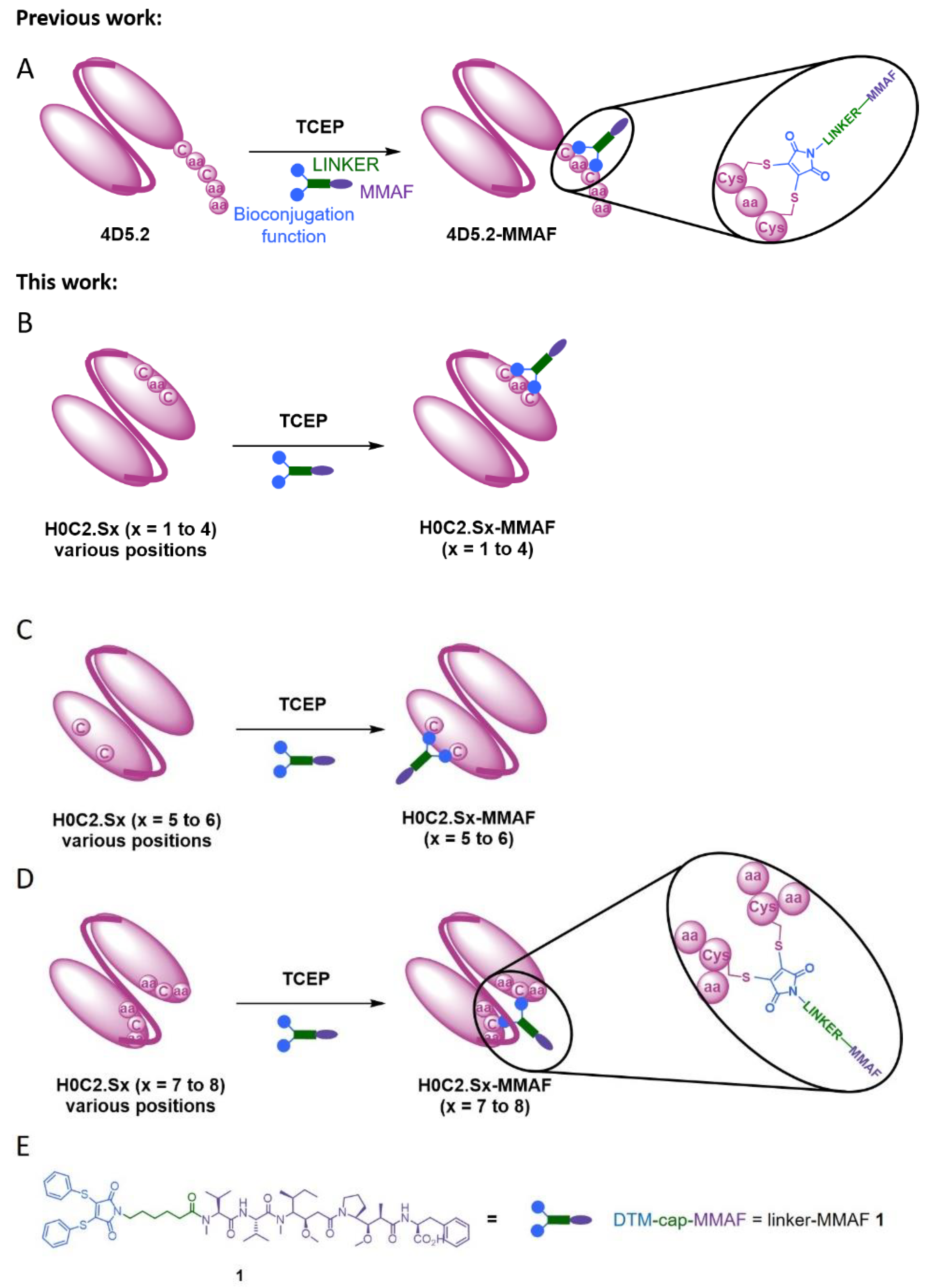
Escherichia coli. A bioconjugation motif including two cysteines (Cys-Gly-Cys) was incorporated at the beginning of the scFv hexahistidine tag, in order to allow controlled bioconjugation of our non-cleavable heterobifunctional linker-MMAF
1 including a diphenylthiomaleimide (DTM) and MMAF [
18,
24,
25,
26]. Satisfyingly, our FDC conserved its affinity to HER2 and was able to kill in vitro HER2-positive SK-BR-3 cells at subnanomolar concentrations (EC
50 of 0.32 nM), while no effect was observed on HER2-negative MCF-7 cells.
However, 4D5.2 was obtained with a relatively low yield classically associated with production in bacteria. Moreover, the intra-tag cysteine (ITC) implantation strategy has shown an important limitation because the tag can be proteolyzed with the loss of the bioconjugation motif. Indeed, when produced in the eukaryotic system (CHO), the scFv 4D5.2 showed a loss of the tag for at least 6% of the fragments produced, characterized by instability over time when stored at 4 °C in PBS. The presence of proteolysis can be a real brake on the future development of biopharmaceuticals. We hypothesize that this phenomenon of proteolysis is due to the length and nature (including cysteines) of the tag, but also to the nature of the amino acid residues present on the surface of the variable domains. Interestingly, the scFv fragment derived from trastuzumab is naturally recognized by protein L; therefore, there is no need to use a tag to purify or detect it [
27]. In addition, the bioconjugation site has been described to have the utmost importance to optimize safety, stability, pharmacokinetics, and the therapeutic index of bioconjugates [
28,
29,
30].
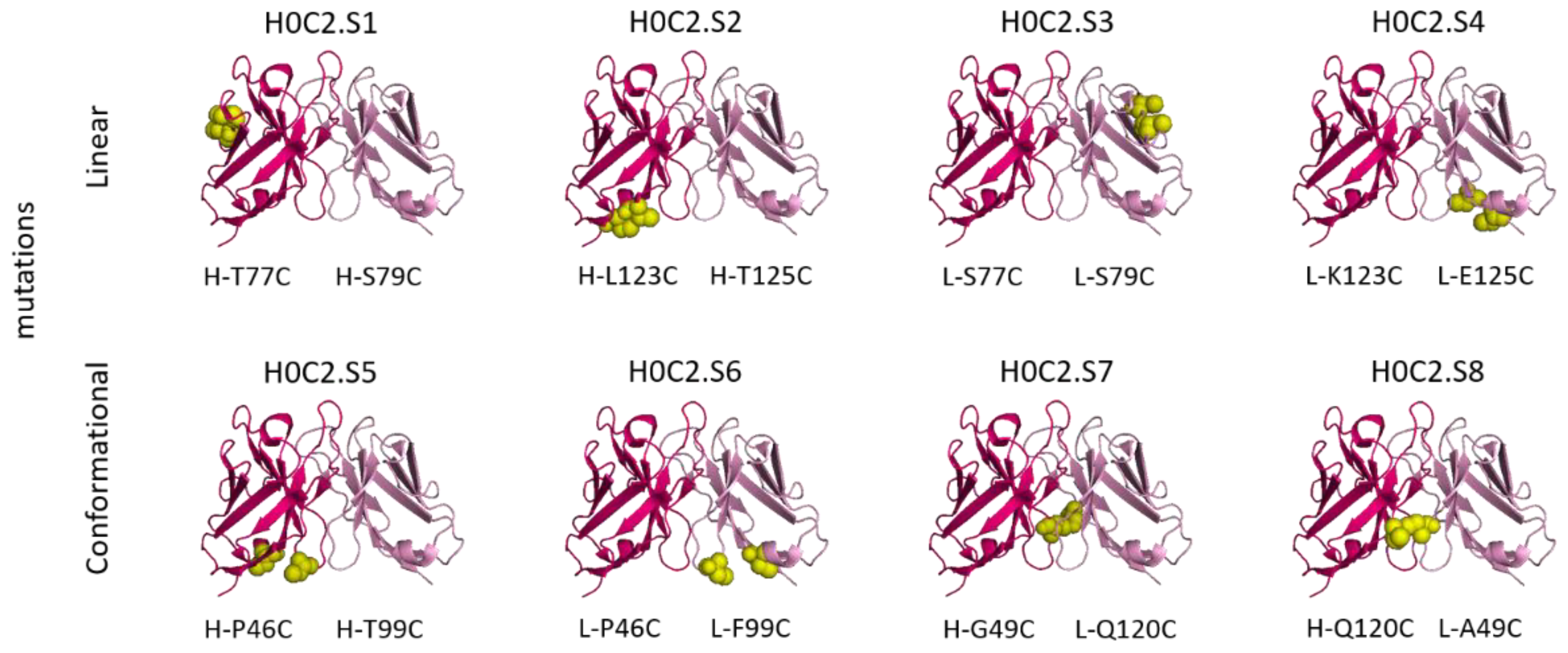
In this context, herein, we compared three intra-domain cysteine (IDC) strategies for the incorporation of a cysteine pair directly in the VH and VL domains of the scFv (
Figure 1). For this purpose, eight original engineered anti-HER2 scFvs were produced in CHO cells (H0C2.Sx, x = 1 to 8). We assessed the impact of the location of mutation sites on the stability and production yield of each clone and the capacity of each position to allow the site-specific conjugation of one MMAF through our non-cleavable heterobifunctional linker-MMAF
1 (
Figure 1). Four clones (H0C2.Sx, x = 3 to 6) were able to afford their respective conjugates H0C2.Sx-MMAF (x = 3 to 6) with an average DAR as close as possible to 1.0 (at least >0.7). We measured the affinity of these four clones to HER2 and their stability, in both their native and conjugated forms. After internalization in HER2-positive cells, the four FDCs H0C2.Sx-MMAF (x = 3 to 6) with a non-cleavable linker are likely to be degraded by lysosomal proteases to release an MMAF metabolite able to kill cancer cells [
1,
2]. Therefore, we evaluated in vitro their cytotoxicity (vs. 4D5.2-MMAF) on two human breast cancer cell lines: SK-BR-3 (HER2 high expression) and MDA-MB-231 (HER2 low expression).
2. Materials and Methods
2.1. Protein Expression and Purification
The H0C2 scFv fragment resulted from the association of the heavy and light variable domains of an antibody via the (Gly4Ser)3 peptide link (without peptide flag in the C-terminal portion). The pcDNA3.4 plasmid was used in the expression of all scFv constructs.
The nucleotide sequences of scFv H0C2 were designed with an optimized codon from Cricetulus griseus. The gene was then synthesized and cloned into the pcDNA3.4 plasmid by GeneArt (Thermo Fisher Scientific, Waltham, MA, USA). For the generation of plasmid pcDNA3.4-H0C2.Sx, mutations were introduced by the golden gate technique. Thus, the gene and the plasmid were amplified by PCR using several primers, which allowed the introduction of mutations, as well as the recognition sequence of the Type IIS restriction enzyme. For digestion and ligation, BsaI-HF®v2 was used (New England Biolabs). Subsequently, TG1 chemically competent bacteria were transformed with the neo-formed plasmids. All constructs were sequenced and thus confirmed.
Thus, scFv H0C2.Sx (x = 1 to 8) were produced using the ExpiCHO-S cell line (ThermoFisher). Briefly, on the day prior to transfection (Day-1), ExpiCHO-S cells were split to a final density of 4 × 10
6 viable cells/mL and incubated overnight at 37 °C and 8% CO
2 under shaking. On the next day (Day 0), cell culture was diluted to 6 × 10
6 cells/mL and transfected by 0.8 μg/mL of plasmid encoding H0C2 (.Sx), previously mixed to ExpiFectamine CHO reagent. On the day after transfection, the max titer protocol was performed. ExpiFectamine CHO Enhancer and ExpiCHO Feed were added, and cells were incubated at 32 °C with 5% CO
2 under shaking. On Day 5 post-transfection, a second volume of ExpiCHO Feed was added to the flask. After ten days post-transfection, the supernatant was harvested and purified with an Akta purifier using a HiScreen™ Capto™ L column (Cytiva Europe GmbH, Velizy-Villacoublay, France, 17-5478-14). scFv was eluted by a linear pH gradient in 0.1 M glycine buffer running from pH 6 to pH 2, and the buffer was removed by a desalting column. Antibody concentration was determined with a UV detector at 280 nm. ScFvs molecular mass and molar extinction coefficient data were all generated by the Protparam tool from
http://web.expasy.org/protparam/ accessed on 24 May 2019.
2.2. Biochemical Characterization and scFv Integrity Analysis
The size and integrity of all purified scFvs were assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on homogeneous 12% polyacrylamide gel, under denaturation and reducing or non-reducing conditions. Purified scFv samples were all loaded at 1 µg for Coomassie Blue staining (0.1% Coomassie Brilliant Blue R-250, 30% methanol, and 10% glacial acetic acid).
The purified scFv preparations were resolved by size-exclusion chromatography (SEC) on a Superdex 75 10/300 GL column (molecular mass range 3000–70,000) (GE Healthcare Life Sciences, 17-5174-01) with an Äkta purifier. The column was loaded with 20 µg of each scFv construct. Proteins were eluted with PBS at a rate of 0.5 mL/min and detected with a UV detector at 280 nm.
2.3. Determination of Thermal Unfolding
Differential scanning fluorimetry experiments were performed on a nanoDSF device (Prometheus NT.48, NanoTemper, München, Germany). All samples were used to a final concentration of 10 µM and loaded into high-sensitivity capillaries. The protein unfolding process was subjected to a thermal ramp (20–95 °C, 1 °C/min). Data analysis was performed using the Prometheus PR ThermControl software. The Tm value was determined by fitting the tryptophan 350/330 nm fluorescence emission ratio using a polynomial function in which the maximum slope is indicated by the peak of its first derivative.
2.4. Affinity Analysis by Microscale Thermophoresis
The antigen labeling was carried out according to the instructions in the His-Tag labeling kit Red-tris-NTA (Nanotemper, München, Germany).
Her2-His (Her2/ERBB2 Protein, Human, Recombinant (ECD, His Tag), sinoBiological) was diluted to 200 nM in PBST buffer (137 mM NaCl, 2.5 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4, 0.05% Tween-20). Tris-NTA dyes were diluted in PBST buffer to a final concentration of 100 nM. A 100 µL volume of protein was then mixed with 100 µL of dye, and the reaction mixtures were incubated for 30 min at room temperature in the dark and then centrifuged for 10 min at 10,000× g. The labeling was verified by following the instructions of the pretest of the MO.Control software (Nanotemper, München, Germany).
For the MST binding experiment, the concentration of fragments was diluted to 2 µM in PBS. This solution was used for a 1:1 serial dilution using 16 dilution steps, with a final volume of 6 µL for each point of the dilution series. Afterwards, 6 µL of HER2-Dye was added to all steps of the dilution series, giving a final ligand concentration of 5 nM. The reaction was incubated for 30 min at room temperature and loaded into Monolith NT.115 MST Premium Capillaries. The MST experiment was carried out using 100% LED power and medium MST power for the NT.115 RED instrument (Nanotemper, München, Germany).
2.5. Synthesis and Mass Spectrometry on Linker-MMAF 1 and Its Precursors
Synthesis of linker-MMAF
1 was performed according to our previously reported procedure [
18]. High-resolution accurate mass measurements (HRAMs) were performed in positive mode with an electrospray ionization (ESI) source on a UHR Q-TOF mass spectrometer (Bruker, Bremen, Germany) with an accuracy tolerance of 2 ppm by the “Fédération de Recherche” ICOA/CBM (FR2708) platform.
2.6. Bioconjugation
To scFv H0C2.Sx (x = 0 to 8) (500 μL, 0.12 mg/mL) in BBS (pH 8.0, 1 mM EDTA, 25 mM NaCl) was added TCEP (1 mM, 12 eq.), and the reaction was incubated at 37 °C for 75 min. Linker-MMAF 1 was added as a solution in DMSO (1 mM, 16 eq.), and the reaction was incubated at 4 °C for 16 h under stirring (600 rpm). Crude mixtures were then purified by the Vivaspin centrifugal concentrator (10 kDa MWCO, Sartorius, GE Healthcare, Tremblay-en-France, France) by three cycles (10,000 rpm, 3 min, 4 °C, each) against phosphate-buffered saline (PBS) pH 7.4 and filtered on 0.22 µm membranes (Millex, Sigma Aldrich; Saint-Quentin-Fallavier, France), to afford the desired conjugates H0C2.Sx-MMAF (x = 0 to 8). The protein concentration of purified FDCs was assessed by UV absorption at 280 nm (Nanodrop, Fisher Scientific SAS, Illkirch, France).
2.7. Mass Spectrometry on Conjugates
Mass spectrometric analyses of AFCs were performed on a Bruker maXis mass spectrometer coupled to a Dionex Ultimate 3000 RSLC system (Dionex, Germering, Germany) by the “Fédération de Recherche” ICOA/CBM (FR2708) platform. Prior to mass spectrometry (MS) analysis, samples (ca. 1 µg) were desalted on a MassPREP desalting cartridge (2.1 × 10 mm) (Waters, Saint-Quentin-en-Yvelines, France) heated at 80 °C using 0.1% formic acid as solvent A and 0.1% formic acid in acetonitrile as solvent B at 500 µL/min. After 1 min, a linear gradient from 5 to 90% B in 1.5 min was applied; the first 1.5 min were diverted to waste. MS data were acquired in positive mode with an ESI source over the
m/
z range from 900 up to 5000 at 1 Hz and processed using DataAnalysis 4.4 SR1 software from Bruker (Bremen, Germany) and the MaxEnt algorithm for spectral deconvolution. Deconvolution was carried out in the range 20–60 kDa. The average drug-to-antibody ratio (
DARaverage) was calculated according to a previously reported method. Briefly, the percentage abundance of each
DARi species represents the relative distribution of each particular drug-loaded FDC species, as monomer and dimer. The
DARaverage was then calculated using the percentage peak areas combined with their respective drug load numbers, according to the corresponding formula:
2.8. Cell Viability
Cell culture. Human breast carcinoma cells MDA-MB-231 were obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK). The cells were grown at 37 °C/5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) with glucose and l-glutamine containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin–streptomycin solution (10,000 U/mL, Gibco®) and 1% of nonessential amino acid 1X (HyClone Laboratories, Logan, UT, USA). SK-BR-3 cells were obtained from Cell Lines Service (CLS Eppelheim, Eppelheim, Germany). SK-BR-3 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin solution, in a humidified atmosphere with 5% CO2 at 37 °C.
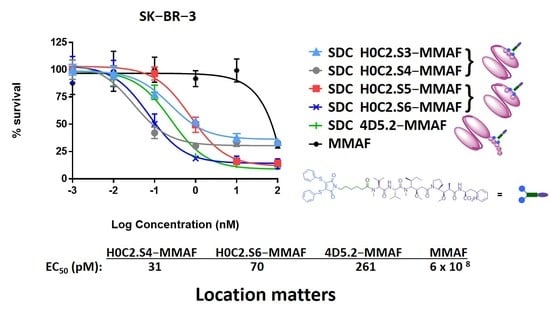
Cytotoxicity assay. SK-BR-3 and MDA-MB-231 cells were first plated at a density of 6 × 103 and 3 × 103 cells/mL in 96-well plates for 24 h and then treated with increasing concentrations of various preparations: H0C2.S3-MMAF, H0C2.S4-MMAF, H0C2.S5-MMAF, H0C2.S6-MMAF, 4D5.2-MMAF, MMAF.
The samples were diluted in complete culture medium to obtain concentration from 100 to 0.001 nM. H2O2 solution at 20 mM and culture medium alone were tested as positive and negative controls. The cells were incubated with 100 µL of each solution at 37 °C/5% CO2 for 5 days. Cell viability was then determined using the MTT reagent (Sigma Aldrich; Saint-Quentin-Fallavier, France). Briefly, 10 µL of MTT solution at 5 mg/mL was added to each well, and the plates were incubated at 37 °C for 4 h. The medium was removed, and 200 µL of dimethyl sulfoxide (DMSO) was added to each well and mixed thoroughly to completely dissolve the dark blue crystals. The optical density values were measured at 550 nm using an absorbance microplate reader (Bio-Tek® instruments, Inc., Winooski, VT, USA). The 50% inhibitory concentration (IC50) was determined as the MMAF (or equivalent) concentration to induce a 50% reduction in cell viability. Three independent repetition experiments were conducted, each with at least 4 repeated samples.
4. Discussion
Armed antibodies currently represent sophisticated targeted therapies. Although the ADC concept is quite simple, its implementation is very challenging, and limitations still exist. Improving ADC can still be achieved by several strategies (e.g., linker, payload, release mechanisms), including the optimization of the antibody format used to vectorize the cytotoxic conjugated compound, to reach better tumor penetration and biodistribution profile. As part of this international effort, we recently described a first strategy, the intra-tag cysteine (ITC) strategy, with the site-specific conjugation of one MMAF through our linker-MMAF
1, onto a bioconjugation motif including two cysteines at the C-terminal position of an anti-HER2 scFv of the humanized antibody trastuzumab (4D5.2), to generate an FDC with a DAR of 1 [
18]. The obtained FDC, 4D5.2-MMAF, conserved its affinity to HER2 and was able to selectively kill in vitro HER2-positive SK-BR-3 cells with an EC
50 of 0.32 nM. However, the ITC implantation strategy was limited by the tag proteolysis associated with the loss of the bioconjugation motif, which constitutes a real brake on the future development of more complex biopharmaceuticals, using the scFv as a building block. Linker-MMAF
1 was first designed to rebridge IgG disulfide bridges, after mild reduction, to lead to an ADC with a DAR 4 [
26]. We also demonstrated that linker-MMAF
1 was efficient for the ITC strategy [
18]. However, in these previous studies, we did not evaluate the minimum or maximum distance required between the two cysteine side chains to afford a bioconjugation when using linker-MMAF
1.
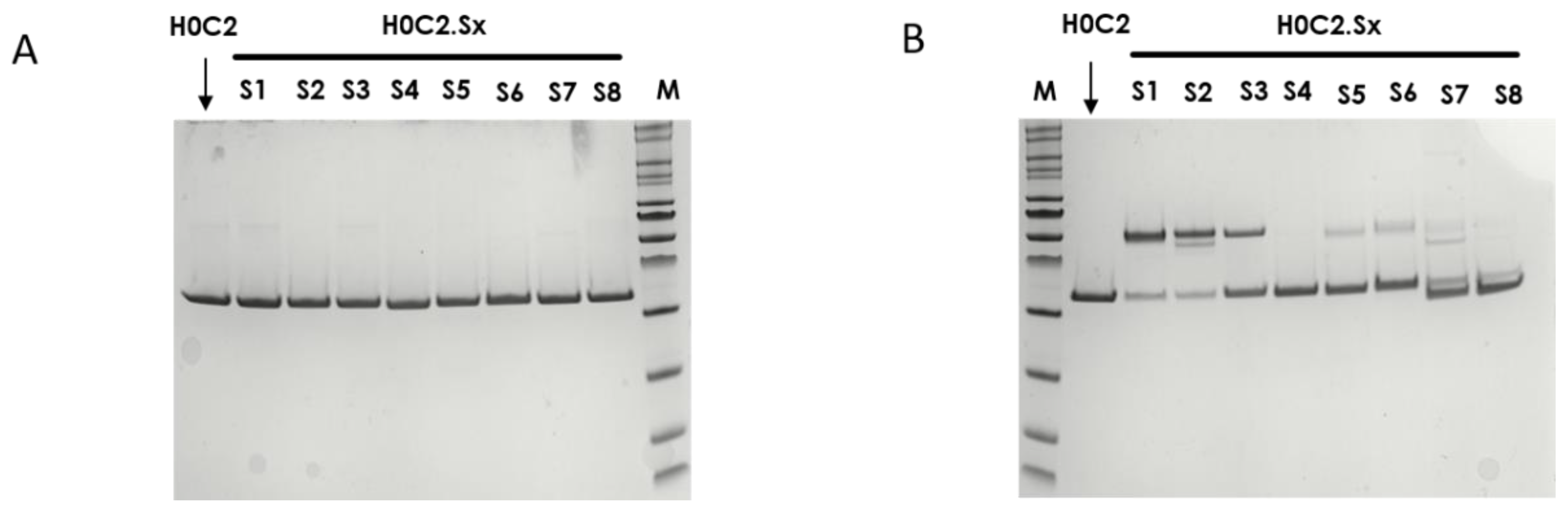
Thus, we developed here a second strategy, the intra-domain cysteine (IDC) strategy, where four solutions have been identified to reproduce this “CxC” linear motif (H0C2.S1 to S4). In parallel, the cysteines could also be a “C.C” conformational motif (H0C2.S5 to S8), where the cysteine residues are nearby in space, while distant in the amino acid sequence, according to the three-dimensional structure of the scFv conformation. The different cysteine positions clearly influenced the covalent oligomerization of the scFv fragments. The “linear” mutations were made in a comparable way on the VH and VL. However, the S1 or S2 positions on the VH, respectively, equivalent to S3 or S4 on the VL, led to a different result, in particular with a large majority of dimer for S1 and S2. For S7 and S8, the mutations were made symmetrically on the VH and VL resulting in the formation of an inter-domain disulfide bridge. This was checked on SDS-PAGE by the greater migration of S7 and S8 compared to the other fragments. When the SDS-PAGE analysis was performed under reductive conditions (
Figure 3A), all fragments appeared completely reduced, suggesting that the formation of disulfide bridges, intra- or inter-domain, should not be a restriction for bioconjugation. Thus, the eight solutions proposed for the implantation of two cysteines directly in the variable domains were validated by obtaining all the purified fragments. For H0C2.S5, a reduced Tm (by 9.3 °C) was intriguing, especially since H0C2.S6 (similar mutations but on the VL) share the same Tm with reference H0C2. The mutated proline in the PG motif, responsible for the formation of a C-C′ loop between CDR1 and CDR2, certainly has a more crucial role in the VH domain than VL.
The bioconjugation results made it possible to classify the fragments into three categories. In the first category, H0C2.S1 and H0C2.S2 were associated with an absence of conjugation with a high proportion of native dimer. This phenomenon has not been observed with H0C2.S3 and H0C2.S4 (on identical positions), which could be surprising because H0C2.S3 is the equivalent of H0C2.S1 and H0C2.S4 is the equivalent of H0C2.S2. Thus, the phenomenon seems to be linked more to the nature of the domain than to the location of the cysteines (S3 similar to S1 and S4 similar to S2). Probably, the environment of the cysteines implanted in the VL domains would not favor a strong dimerization between two VL domains. This phenomenon is probably related to the nature of H0C2 and could be different with another antibody. It is possible to reduce S1 and S2 but with an important number of equivalents of TCEP (more than 200 eq., data not shown) not compatible for further development in a bioconjugation process.
In the second category, H0C2.S7 and H0C2.S8 were associated with a weak to non-existent bioconjugation. The reason is different from the first category, because there are no dimeric forms. Structurally, the implanted cysteines are less exposed on the surface of the scFv, making them good candidates for an intra-domain disulfide bridge (dsFv) but not for a bioconjugation site. It is also very likely that the cysteine environment is crucial, since H0C2.S7 was weakly bioconjugated with an average DAR of 0.5, unlike H0C2.S8.
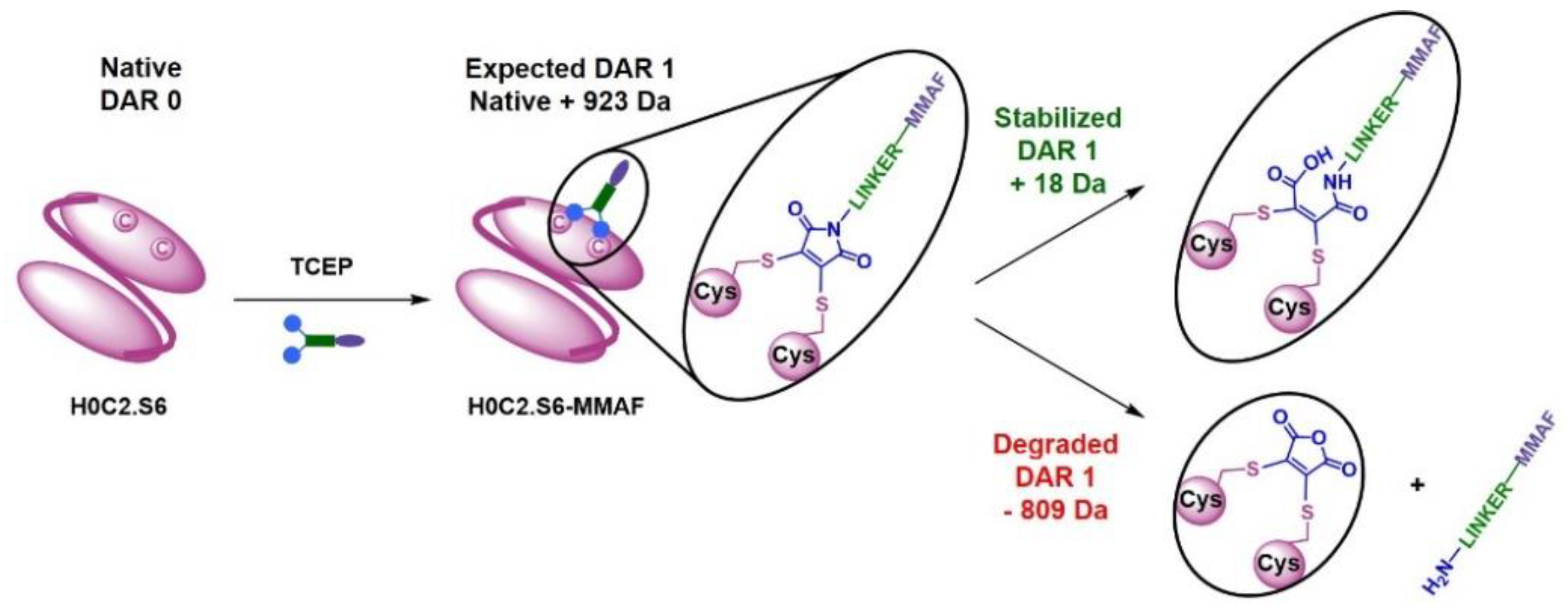
In the third category, the best bioconjugations were observed with H0C2.S3 to H0C2.S6, and following our results, it is possible to classify the conjugation sites from least to most favorable: S6 < S3 < S4 < S5. Although it was not the goal of this study, it is noteworthy to add that the average DAR was probably improvable by (i) a slightly better reduction for S4 and S3, to avoid the remnant native dimer, and (ii) by a number of linker equivalent being optimized to avoid a non-conjugating part of the native monomer. DAR 2 fragments were observed on all the conjugable fragments (H0C2.S3 to H0C2.S6). Concerning the stabilization of the conjugated linker by the close protein environment, on the one hand, we noted that only for H0C2.S6-MMAF of DAR 1, an increment of +18 Da was observed on the DAR 1. On the other hand, for the DAR 2, all the linkers were stabilized, and this was visible by an increment of 2 × 18 = 36 Da. Observing DAR 2 was not necessarily an issue, as it was always in minor proportion, and always observed with this bioconjugation technology in previous publications [
18,
19,
26]. Therefore, we decided that only scFv H0C2.Sx (x = 3 to 6) were suitable to continue the rest of the study, where the two implanted cysteines were correctly incorporated to obtain a satisfactory average DAR (>0.7) for the conjugates.
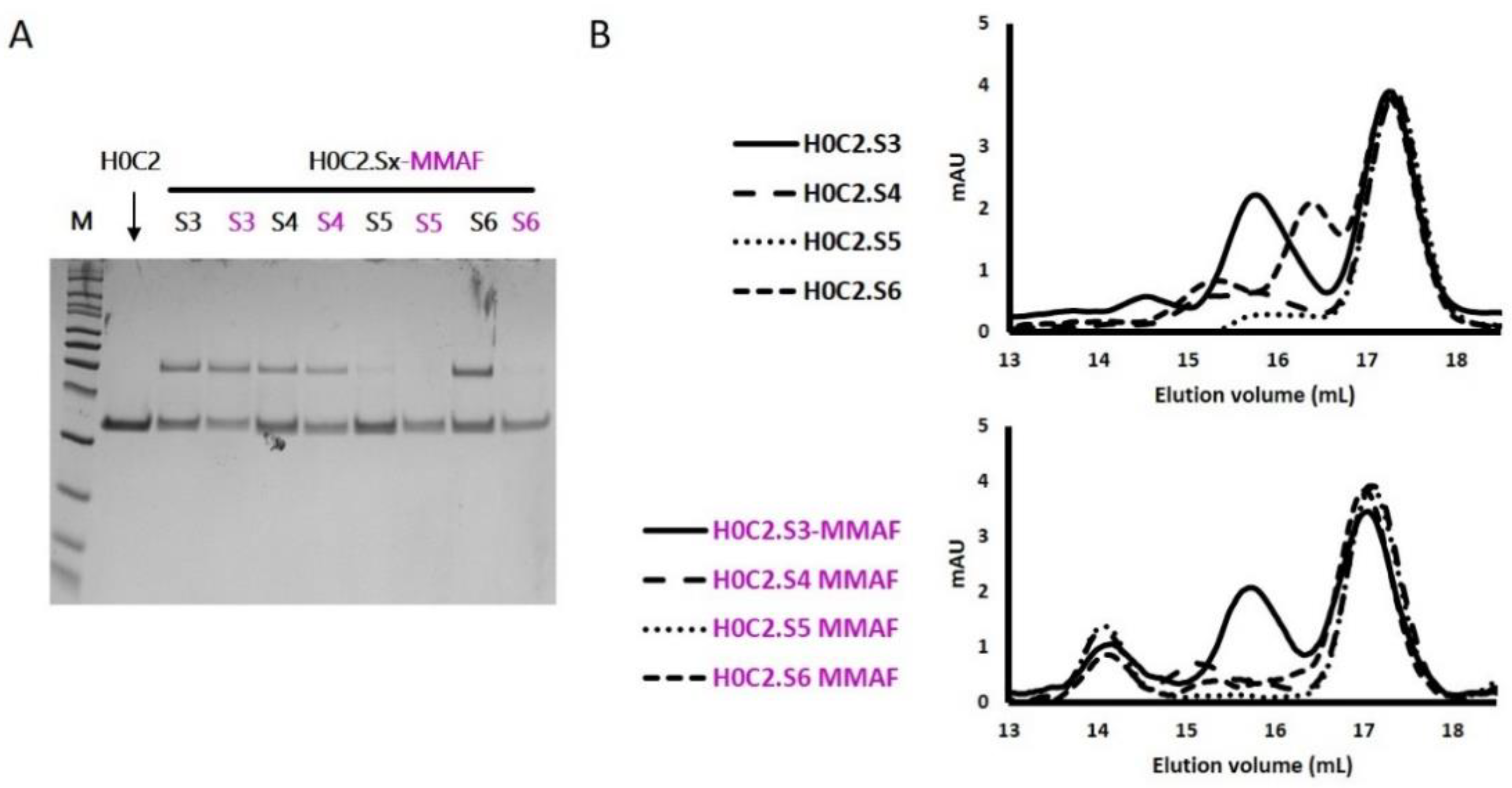
For the four selected conjugates H0C2.Sx-MMAF (x = S3 to S6), SEC analysis showed the presence of a new oligomer (about 15%) at an elution volume of 14 mL, most likely due to aggregation associated with the hydrophobic character of linker-MMAF 1. Concerning the affinity to HER2, for FDCs 4D5.2-MMAF and the H0C2.Sx-MMAF (x = 3 to 6), the presence of the linker slightly decreased the affinity when compared to their respective native scFv (KD of one log lower), except for H0C2.S3-MMAF when compared to H0C2.S3 (KD of two logs lower). The position of the linker on the side of the fragment, therefore closer to the recognition site of HER2, could be responsible for this greater loss. The positioning of the linker, located opposite the paratope, would seem more favorable, even if the loss of one log, associated with all conjugated fragments, was difficult to explain. Probably, the hydrophobic charge of linker-MMAF 1 changed the environment of the fragment and did not facilitate interaction with a large antigen such as HER2. According to the affinity ranking, the least to the most favorable location of MMAF would be: the flank of VH domain (S3) < the bottom of VH domain (S5) < the bottom of VL domain (S4, S6, as well as 4D5.2).
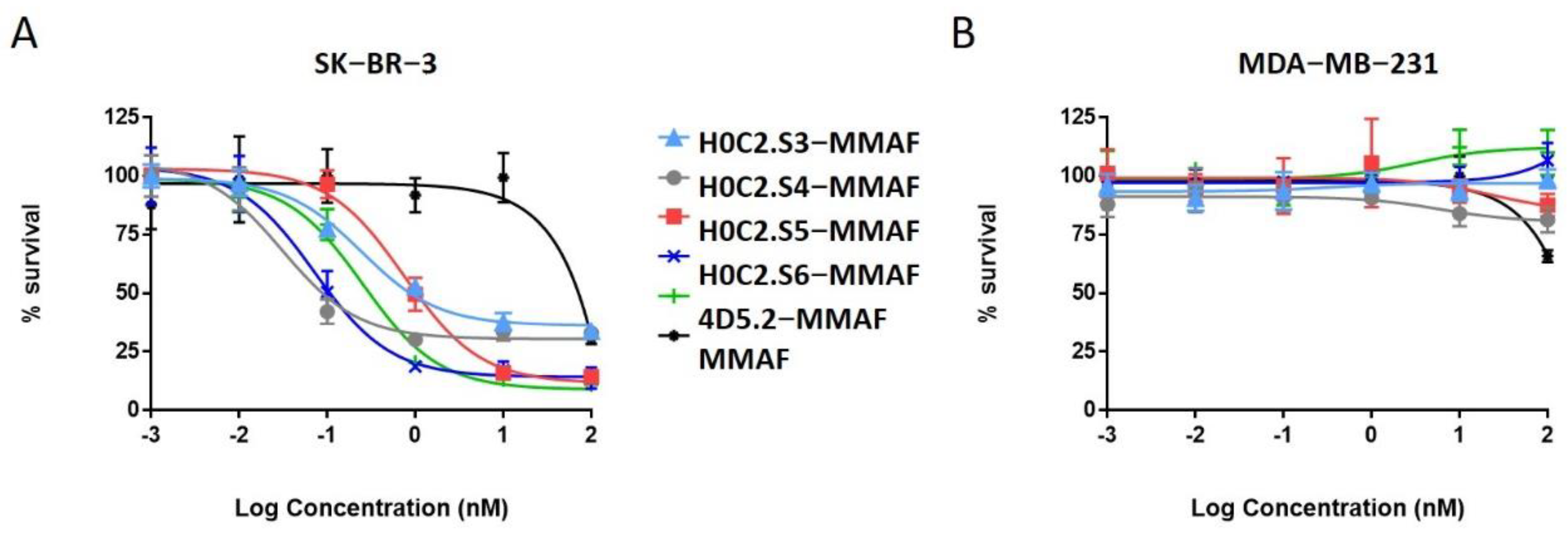
No cytotoxic activity was observed with the native scFv, or the untargeted MMAF payload alone (due to a charged carboxylic acid at physiological pH), on both cell lines. Only FDCs H0C2.Sx-MMAF (x = S3 to S6) and 4D5.2-MMAF were cytotoxic on the HER2-positive SK-BR-3 cell line while sparing the HER2-low MDA-MB-231 cell line. This demonstrated that the therapeutic effect of FDCs H0C2.Sx-MMAF (x = S3 to S6) and 4D5.2-MMAF was due to HER2-targeted uptake of the FDCs rather than non-specific protein uptake, and was limited to the HER2-overexpressing cell line. The difference in cytotoxicity between FDCs H0C2.Sx-MMAF (x = S3 to S6) and 4D5.2-MMAF, and their native counterparts used as control, confirmed that cell killing was via payload-mediated cytotoxicity rather than a therapeutic effect from the antibody fragment alone.
The most homogeneous FDC was obtained for H0C2.S5-MMAF (without DAR 0), with the highest average DAR (1.08). However, H0C2.S5-MMAF was the least cytotoxic FDC, probably due to a conformation issue, observable in particular by a strong drop in Tm and a rather lower affinity to HER2. In contrast, with a lower DAR (0.73) and the presence of competitive inhibitor DAR 0 (11% monomer and 20% dimer), with a similar affinity to HER2, FDC H0C2.S3-MMAF exhibited higher cytotoxicity than FDC H0C2.S5-MMAF and similar to reference 4D5.2-MMAF. The only factor that could explain this result and be in favor of FDC H0C2.S3-MMAF would be the position of the linker on the VL. Indeed, the results for FDCs H0C2.S4-MMAF and H0C2.S6-MMAF confirmed this statement: these two conjugated fragments exhibited the best toxicity and was similar to H0C2.S3-MMAF from the grafting of linker-MMAF 1 onto a basal position in the VL domain.
The more homogeneous DAR for H0C2.S4-MMAF, compared to H0C2.S6-MMAF, could explain its better cytotoxicity on SK-BR-3 cell line, reaching quite an impressive EC
50 of 31 ± 5 pM for an FDC with an average DAR of only 1, equivalent to an EC
50 observable for an ADC of DAR 4 [
26].
5. Conclusions and Prospects
We developed eight original scFvs (new IDC strategy), where no flag peptide was needed, and two residues at key positions were mutated to cysteine in order to have a linear or conformational “C.C” bioconjugation motif. Our results showed no proteolysis issue and satisfying production yields in CHO cells for new scFvs H0C2.S1 to H0C2.S8. In addition, we managed to demonstrate that the reaction of linker-MMAF 1 was more or less difficult depending on the location and accessibility of the bioconjugation site onto the scFvs. Indeed, among the eight proposed scFvs, only four scFvs (H0C2.S3 to H0C.S6) displayed excellent bioconjugation ability, leading to the corresponding FDCs H0C2.Sx-MMAF (x = 3 to 6) with an average DAR close to 1.0.
Another objective was to determine if the localization of the linker could affect the efficiency of the FDCs. Two FDCs, H0C2.S4-MMAF and H0C2.S6-MMAF, exhibited a significantly higher EC
50 than our previous reference FDC 4D5.2-MMAF [
18] on the SK-BR-3 cell line (HER2 high expression). The location of linker-MMAF
1 onto a basal position in the VL domain thus seems to be an important criterion to optimize FDCs. More precisely, in the case of H0C2.S4-MMAF, the two cysteines were located on the FR4 of the VL, a sequence that remains relatively constant. It is very likely that the results obtained on the scFv fragment of herceptin can easily be transposed to other antibodies. The characteristics of scFv H0C2.S4 or FDC H0C2.S4-MMAF were relatively similar to those of scFv 4D5.2 or FDC 4D5.2-MMAF, respectively, although H0C2.S4 (native and conjugated forms) exhibited neither proteolysis nor degradation of the conjugated linker. Therefore, the localization of linker-MMAF
1 onto a basal position in the VL domain (IDC strategy) was better than on the tag (ITC strategy) to produce FDCs with optimized properties and increased efficacy.
Finally, the scFv format is associated with a too fast clearance to be effective in vivo. Nevertheless, the scFv is very interesting as the backbone from which many other antibody formats are built. Thus, this original IDC strategy is an innovative tool to create new conjugation sites on more complex antibody formats (e.g., minibody, diabody, scFv-Fc). These larger antibody formats should have better pharmacokinetic properties and should be better suited against solid tumors. We believe that this new advancement in antibody technology could be implemented to produce novel and more effective antibody–drug conjugates as targeted cancer therapies, to explore the full potential of our IDC strategy.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





