
A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease

Abstract
1. Introduction
2. Materials and Methods
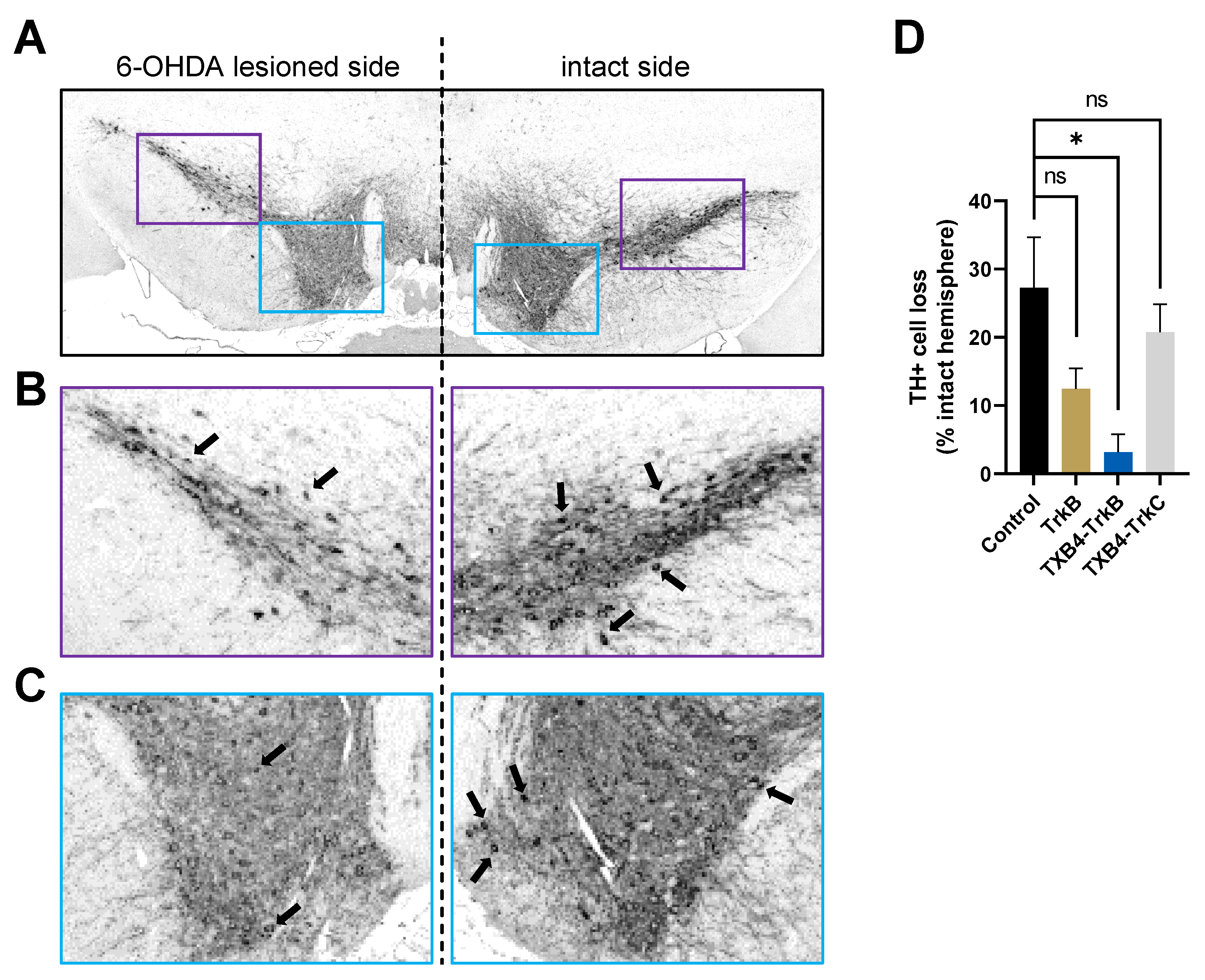
3. Results
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic Potential of Neurotrophins for Repair After Brain Injury: A Helping Hand From Biomaterials. Front. Neurosci. 2019, 13, 790. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A. Modulating Neurotrophin Receptor Signaling as a Therapeutic Strategy for Huntington’s Disease. J. Huntingt. Dis. 2017, 6, 303–325. [Google Scholar] [CrossRef]
- Ding, Y.X.; Xia, Y.; Jiao, X.Y.; Duan, L.; Yu, J.; Wang, X.; Chen, L.W. The TrkB-positive dopaminergic neurons are less sensitive to MPTP insult in the substantia nigra of adult C57/BL mice. Neurochem. Res. 2011, 36, 1759–1766. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, Z.; Yu, Z.; Yang, Z.; Zhao, H.; Liu, L.; Zhao, J. rAAV-mediated delivery of brain-derived neurotrophic factor promotes neurite outgrowth and protects neurodegeneration in focal ischemic model. Int. J. Clin. Exp. Pathol. 2011, 4, 496–504. [Google Scholar]
- Takeshima, Y.; Nakamura, M.; Miyake, H.; Tamaki, R.; Inui, T.; Horiuchi, K.; Wajima, D.; Nakase, H. Neuroprotection with intraventricular brain-derived neurotrophic factor in rat venous occlusion model. Neurosurgery 2011, 68, 1334–1341. [Google Scholar] [CrossRef]
- Kiprianova, I.; Freiman, T.M.; Desiderato, S.; Schwab, S.; Galmbacher, R.; Gillardon, F.; Spranger, M. Brain-derived neurotrophic factor prevents neuronal death and glial activation after global ischemia in the rat. J. Neurosci. Res. 1999, 56, 21–27. [Google Scholar] [CrossRef]
- Bejot, Y.; Mossiat, C.; Giroud, M.; Prigent-Tessier, A.; Marie, C. Circulating and brain BDNF levels in stroke rats. Relevance to clinical studies. PLoS ONE 2011, 6, e29405. [Google Scholar] [CrossRef]
- Kobayashi, N.R.; Fan, D.P.; Giehl, K.M.; Bedard, A.M.; Wiegand, S.J.; Tetzlaff, W. BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J. Neurosci. 1997, 17, 9583–9595. [Google Scholar] [CrossRef]
- Tuinstra, H.M.; Aviles, M.O.; Shin, S.; Holland, S.J.; Zelivyanskaya, M.L.; Fast, A.G.; Ko, S.Y.; Margul, D.J.; Bartels, A.K.; Boehler, R.M.; et al. Multifunctional, multichannel bridges that deliver neurotrophin encoding lentivirus for regeneration following spinal cord injury. Biomaterials 2012, 33, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Fouad, K.; Vavrek, R.; Cho, S. A TrkB antibody agonist promotes plasticity following cervical spinal cord injury in adult rats. J. Neurotrauma 2021, 38, 1338–1348. [Google Scholar] [CrossRef]
- Vilar, M.; Mira, H. Regulation of Neurogenesis by Neurotrophins during Adulthood: Expected and Unexpected Roles. Front. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1999, 270, 45–48. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, C.; Yun, W. Peripheral BDNF/TrkB protein expression is decreased in Parkinson’s disease but not in Essential tremor. J. Clin. Neurosci. 2019, 63, 176–181. [Google Scholar] [CrossRef]
- Huang, Y.; Yun, W.; Zhang, M.; Luo, W.; Zhou, X. Serum concentration and clinical significance of brain-derived neurotrophic factor in patients with Parkinson’s disease or essential tremor. J. Int. Med. Res. 2018, 46, 1477–1485. [Google Scholar] [CrossRef]
- Tanila, H. The role of BDNF in Alzheimer’s disease. Neurobiol. Dis. 2017, 97, 114–118. [Google Scholar] [CrossRef]
- Park, H. Cortical Axonal Secretion of BDNF in the Striatum Is Disrupted in the Mutant-huntingtin Knock-in Mouse Model of Huntington’s Disease. Exp. Neurobiol. 2018, 27, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.Q.; Rymar, V.V.; Sadikot, A.F. Impaired TrkB Signaling Underlies Reduced BDNF-Mediated Trophic Support of Striatal Neurons in the R6/2 Mouse Model of Huntington’s Disease. Front. Cell. Neurosci. 2016, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Yang, J.; Li, T.; Milner, T.A.; Hempstead, B.L. Selective reduction of striatal mature BDNF without induction of proBDNF in the zQ175 mouse model of Huntington’s disease. Neurobiol. Dis. 2015, 82, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C. Brain-Derived Neurotrophic Factor, Depression, and Physical Activity: Making the Neuroplastic Connection. Neural Plast. 2017, 2017, 7260130. [Google Scholar] [CrossRef]
- Rantamaki, T. TrkB neurotrophin receptor at the core of antidepressant effects, but how? Cell Tissue Res. 2019, 377, 115–124. [Google Scholar] [CrossRef]
- Sakane, T.; Pardridge, W.M. Carboxyl-directed pegylation of brain-derived neurotrophic factor markedly reduces systemic clearance with minimal loss of biologic activity. Pharm. Res. 1997, 14, 1085–1091. [Google Scholar] [CrossRef]
- Croll, S.D.; Chesnutt, C.R.; Rudge, J.S.; Acheson, A.; Ryan, T.E.; Siuciak, J.A.; DiStefano, P.S.; Wiegand, S.J.; Lindsay, R.M. Co-infusion with a TrkB-Fc receptor body carrier enhances BDNF distribution in the adult rat brain. Exp. Neurol. 1998, 152, 20–33. [Google Scholar] [CrossRef]
- Perreault, M.; Feng, G.; Will, S.; Gareski, T.; Kubasiak, D.; Marquette, K.; Vugmeyster, Y.; Unger, T.J.; Jones, J.; Qadri, A.; et al. Activation of TrkB with TAM-163 results in opposite effects on body weight in rodents and non-human primates. PLoS ONE 2013, 8, e62616. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Cohen, S.B.; DiPetrillo, K. TrkB agonist antibody dose-dependently raises blood pressure in mice with diet-induced obesity. Am. J. Hypertens. 2010, 23, 732–736. [Google Scholar] [CrossRef][Green Version]
- Sahenk, Z.; Galloway, G.; Edwards, C.; Malik, V.; Kaspar, B.K.; Eagle, A.; Yetter, B.; Forgie, A.; Tsao, D.; Lin, J.C. TrkB and TrkC agonist antibodies improve function, electrophysiologic and pathologic features in Trembler J mice. Exp. Neurol. 2010, 224, 495–506. [Google Scholar] [CrossRef]
- Kim, G.S.; Cho, S.; Nelson, J.W.; Zipfel, G.J.; Han, B.H. TrkB agonist antibody pretreatment enhances neuronal survival and long-term sensory motor function following hypoxic ischemic injury in neonatal rats. PLoS ONE 2014, 9, e88962. [Google Scholar] [CrossRef] [PubMed]
- Todd, D.; Gowers, I.; Dowler, S.J.; Wall, M.D.; McAllister, G.; Fischer, D.F.; Dijkstra, S.; Fratantoni, S.A.; van de Bospoort, R.; Veenman-Koepke, J.; et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS ONE 2014, 9, e87923. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.D.; Zhang, J.; Tan, X.Y.; Wood, A.; Gill, D.; Cho, S. Novel agonist monoclonal antibodies activate TrkB receptors and demonstrate potent neurotrophic activities. J. Neurosci. 2006, 26, 9394–9403. [Google Scholar] [CrossRef] [PubMed]
- Merkouris, S.; Barde, Y.A.; Binley, K.E.; Allen, N.D.; Stepanov, A.V.; Wu, N.C.; Grande, G.; Lin, C.W.; Li, M.; Nan, X.; et al. Fully human agonist antibodies to TrkB using autocrine cell-based selection from a combinatorial antibody library. Proc. Natl. Acad. Sci. USA 2018, 115, E7023–E7032. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert. Opin. Drug. Deliv. 2015, 12, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Stocki, P.; Szary, J.; Rasmussen, C.L.M.; Demydchuk, M.; Northall, L.; Logan, D.B.; Gauhar, A.; Thei, L.; Moos, T.; Walsh, F.S.; et al. Blood-brain barrier transport using a high affinity, brain-selective VNAR antibody targeting transferrin receptor 1. FASEB J. 2021, 35, e21172. [Google Scholar] [CrossRef] [PubMed]
- Stocki, P.; Wicher, K.B.; Szary, J.; Rutkowski, J.L. Improved TfR-Selective Binding Peptides Capable of Crossing the Blood Brain Barrier. U.S. Patent WO2019089395A1, 9 May 2019. [Google Scholar]
- Devaux, B.; Hongo, J.A.; Presta, L.G.; Shelton, D.L. Methods for Treating Neuropathy by Agonist Anti-Trk-C Monoclonal Antibodies. US7615383B2, 10 November 2009. [Google Scholar]
- Strohl, W.R. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009, 20, 685–691. [Google Scholar] [CrossRef]
- Fletcher, E.J.R.; Moon, L.D.F.; Duty, S. Chondroitinase ABC reduces dopaminergic nigral cell death and striatal terminal loss in a 6-hydroxydopamine partial lesion mouse model of Parkinson’s disease. BMC Neurosci. 2019, 20, 61. [Google Scholar] [CrossRef]
- Sommerfeld, M.T.; Schweigreiter, R.; Barde, Y.A.; Hoppe, E. Down-regulation of the neurotrophin receptor TrkB following ligand binding. Evidence for an involvement of the proteasome and differential regulation of TrkA and TrkB. J. Biol. Chem. 2000, 275, 8982–8990. [Google Scholar] [CrossRef]
- Nie, S.; Xu, Y.; Chen, G.; Ma, K.; Han, C.; Guo, Z.; Zhang, Z.; Ye, K.; Cao, X. Small molecule TrkB agonist deoxygedunin protects nigrostriatal dopaminergic neurons from 6-OHDA and MPTP induced neurotoxicity in rodents. Neuropharmacology 2015, 99, 448–458. [Google Scholar] [CrossRef]
- Altar, C.A.; Boylan, C.B.; Jackson, C.; Hershenson, S.; Miller, J.; Wiegand, S.J.; Lindsay, R.M.; Hyman, C. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11347–11351. [Google Scholar] [CrossRef]
- Klein, R.L.; Lewis, M.H.; Muzyczka, N.; Meyer, E.M. Prevention of 6-hydroxydopamine-induced rotational behavior by BDNF somatic gene transfer. Brain Res. 1999, 847, 314–320. [Google Scholar] [CrossRef]
- Somoza, R.; Juri, C.; Baes, M.; Wyneken, U.; Rubio, F.J. Intranigral transplantation of epigenetically induced BDNF-secreting human mesenchymal stem cells: Implications for cell-based therapies in Parkinson’s disease. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2010, 16, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Soderquist, R.G.; Milligan, E.D.; Sloane, E.M.; Harrison, J.A.; Douvas, K.K.; Potter, J.M.; Hughes, T.S.; Chavez, R.A.; Johnson, K.; Watkins, L.R.; et al. PEGylation of brain-derived neurotrophic factor for preserved biological activity and enhanced spinal cord distribution. J. Biomed. Mater. Res. Part A 2009, 91, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Barker, P.A. p75NTR is positively promiscuous: Novel partners and new insights. Neuron 2004, 42, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Williams, E.J.; Maison, P.; Pangalos, M.N.; Walsh, F.S.; Doherty, P. Overcoming the inhibitors of myelin with a novel neurotrophin strategy. J. Biol. Chem. 2005, 280, 5862–5869. [Google Scholar] [CrossRef]
- Bai, Y.; Xu, J.; Brahimi, F.; Zhuo, Y.; Sarunic, M.V.; Saragovi, H.U. An agonistic TrkB mAb causes sustained TrkB activation, delays RGC death, and protects the retinal structure in optic nerve axotomy and in glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4722–4731. [Google Scholar] [CrossRef]
- Hu, Y.; Cho, S.; Goldberg, J.L. Neurotrophic effect of a novel TrkB agonist on retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1747–1754. [Google Scholar] [CrossRef]
- Friden, P.M.; Walus, L.R.; Musso, G.F.; Taylor, M.A.; Malfroy, B.; Starzyk, R.M. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1991, 88, 4771–4775. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Buciak, J.L.; Friden, P.M. Selective transport of an anti-transferrin receptor antibody through the blood-brain barrier in vivo. J. Pharmacol. Exp. Ther. 1991, 259, 66–70. [Google Scholar]
- Kordower, J.H.; Charles, V.; Bayer, R.; Bartus, R.T.; Putney, S.; Walus, L.R.; Friden, P.M. Intravenous administration of a transferrin receptor antibody-nerve growth factor conjugate prevents the degeneration of cholinergic striatal neurons in a model of Huntington disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9077–9080. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Morgan, E.H. Restricted transport of anti-transferrin receptor antibody (OX26) through the blood-brain barrier in the rat. J. Neurochem. 2001, 79, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Manich, G.; Cabezon, I.; del Valle, J.; Duran-Vilaregut, J.; Camins, A.; Pallas, M.; Pelegri, C.; Vilaplana, J. Study of the transcytosis of an anti-transferrin receptor antibody with a Fab’ cargo across the blood-brain barrier in mice. Eur. J. Pharm. Sci. 2013, 49, 556–564. [Google Scholar] [CrossRef]
- Lesley, J.; Schulte, R.; Woods, J. Modulation of transferrin receptor expression and function by anti-transferrin receptor antibodies and antibody fragments. Exp. Cell Res. 1989, 182, 215–233. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J.; Patrick, D.J.; Ka-Wai Hui, E.; Lu, J.Z. Blood-Brain Barrier Transport, Plasma Pharmacokinetics, and Neuropathology Following Chronic Treatment of the Rhesus Monkey with a Brain Penetrating Humanized Monoclonal Antibody Against the Human Transferrin Receptor. Mol. Pharm. 2018, 15, 5207–5216. [Google Scholar] [CrossRef]
- Couch, J.A.; Yu, Y.J.; Zhang, Y.; Tarrant, J.M.; Fuji, R.N.; Meilandt, W.J.; Solanoy, H.; Tong, R.K.; Hoyte, K.; Luk, W.; et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 2013, 5, 183ra157. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/ENSG00000072274-TFRC/tissue (accessed on 20 April 2022).
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef]
- Webster, C.I.; Hatcher, J.; Burrell, M.; Thom, G.; Thornton, P.; Gurrell, I.; Chessell, I. Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti-transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. Pain 2017, 158, 660–668. [Google Scholar] [CrossRef]
- Thom, G.; Burrell, M.; Haqqani, A.S.; Yogi, A.; Lessard, E.; Brunette, E.; Delaney, C.; Baumann, E.; Callaghan, D.; Rodrigo, N.; et al. Enhanced Delivery of Galanin Conjugates to the Brain through Bioengineering of the Anti-Transferrin Receptor Antibody OX26. Mol. Pharm. 2018, 15, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juarez, K.; Urrutia, M.; Cauerhff, A.; Danquah, W.; Rissiek, B.; Scheuplein, F.; et al. Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Panaccio, M.; Zalcberg, J.R.; Thompson, C.H.; Leyden, M.J.; Sullivan, J.R.; Lichtenstein, M.; McKenzie, I.F. Heterogeneity of the human transferrin receptor and use of anti-transferrin receptor antibodies to detect tumours in vivo. Immunol. Cell Biol. 1987, 65 Pt 6, 461–472. [Google Scholar] [CrossRef]
- Rutkowski, J.L.; Walsh, F.S.; Sinclair, E.H.; Stocki, P. BBB-Shuttling-VNARs Conjugated to Neurotrophic Agonist Antibodies to Treat Neurodegenerative Diseases and Conditions. WO2021102276A1, 27 May 2021. [Google Scholar]
- Sehlin, D.; Stocki, P.; Gustavsson, T.; Hultqvist, G.; Walsh, F.S.; Rutkowski, J.L.; Syvanen, S. Brain delivery of biologics using a cross-species reactive transferrin receptor 1 VNAR shuttle. FASEB J. 2020, 34, 13272–13283. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Nagappan, G.; Lu, B. Differential effects of transient and sustained activation of BDNF-TrkB signaling. Dev. Neurobiol. 2018, 78, 647–659. [Google Scholar] [CrossRef]
- Glinka, Y.; Gassen, M.; Youdim, M.B. Mechanism of 6-hydroxydopamine neurotoxicity. J. Neural Transmission. Suppl. 1997, 50, 55–66. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef]
- Olmedo-Diaz, S.; Estevez-Silva, H.; Oradd, G.; Af Bjerken, S.; Marcellino, D.; Virel, A. An altered blood-brain barrier contributes to brain iron accumulation and neuroinflammation in the 6-OHDA rat model of Parkinson’s disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef]
- Han, F.; Guan, X.; Guo, W.; Lu, B. Therapeutic potential of a TrkB agonistic antibody for ischemic brain injury. Neurobiol. Dis. 2019, 127, 570–581. [Google Scholar] [CrossRef]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
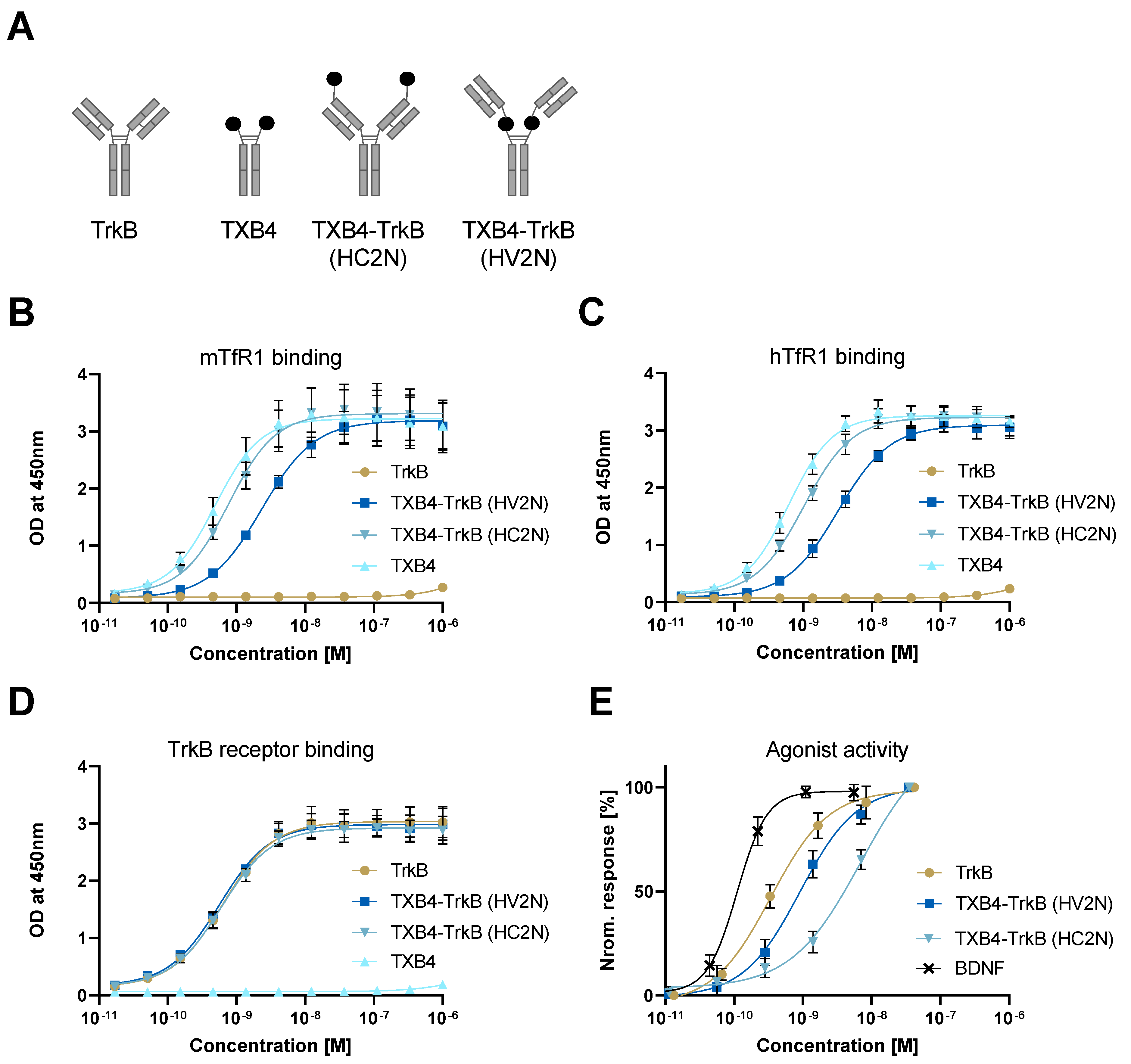
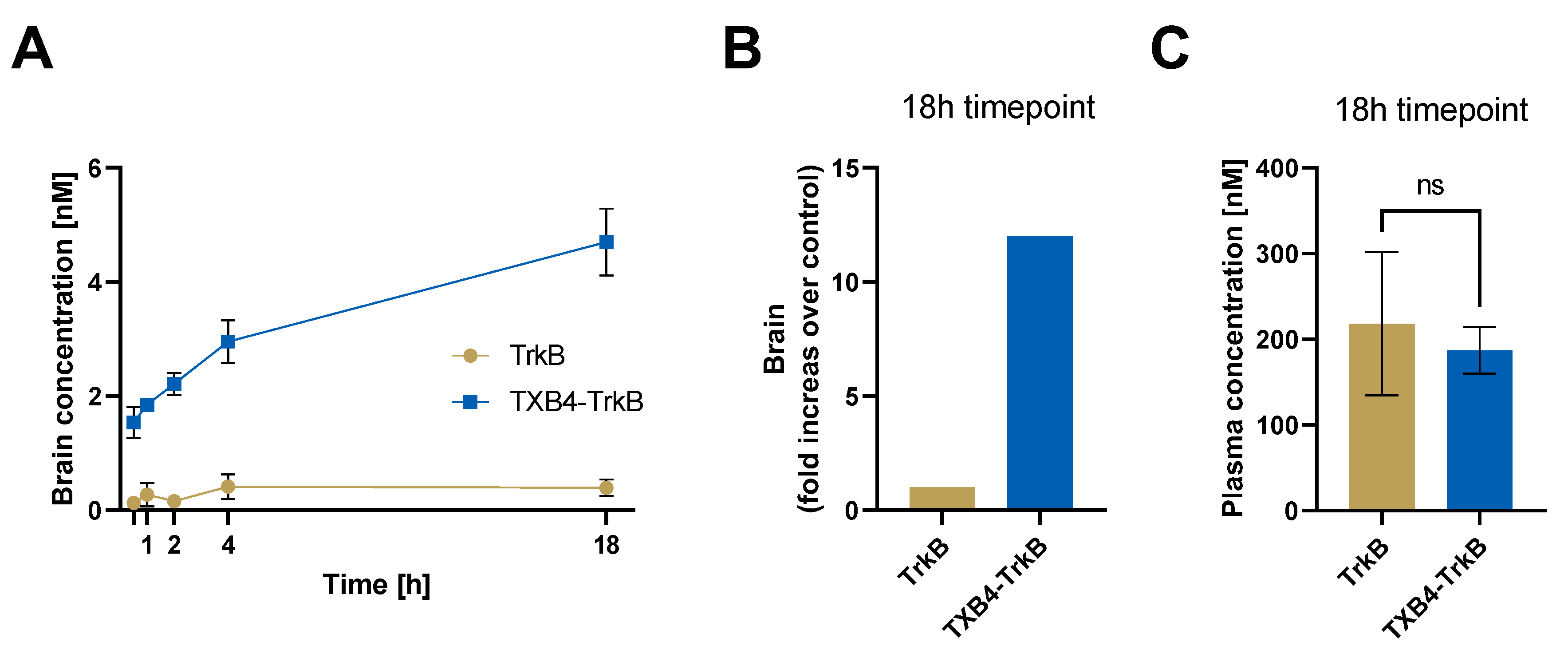
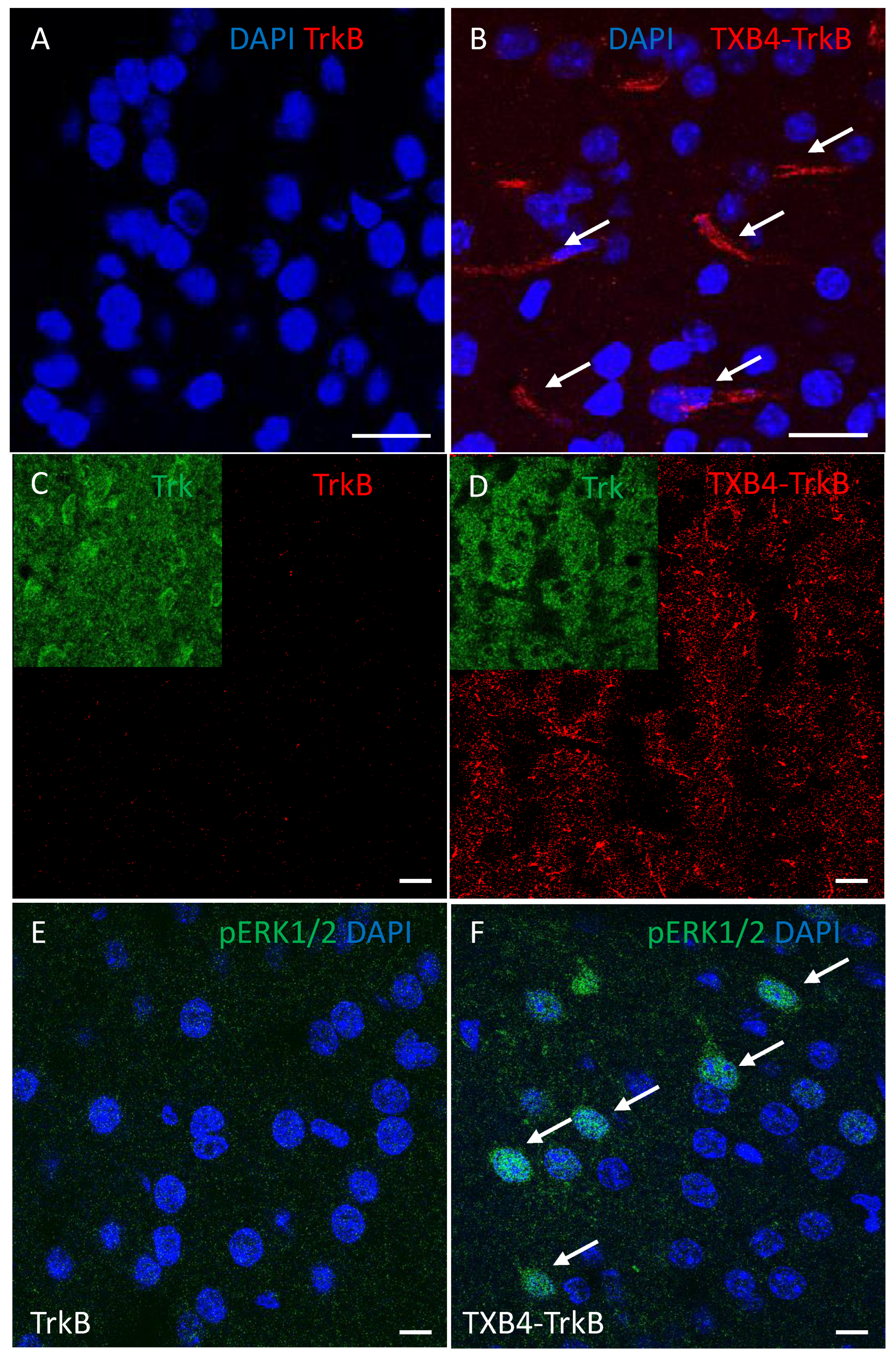
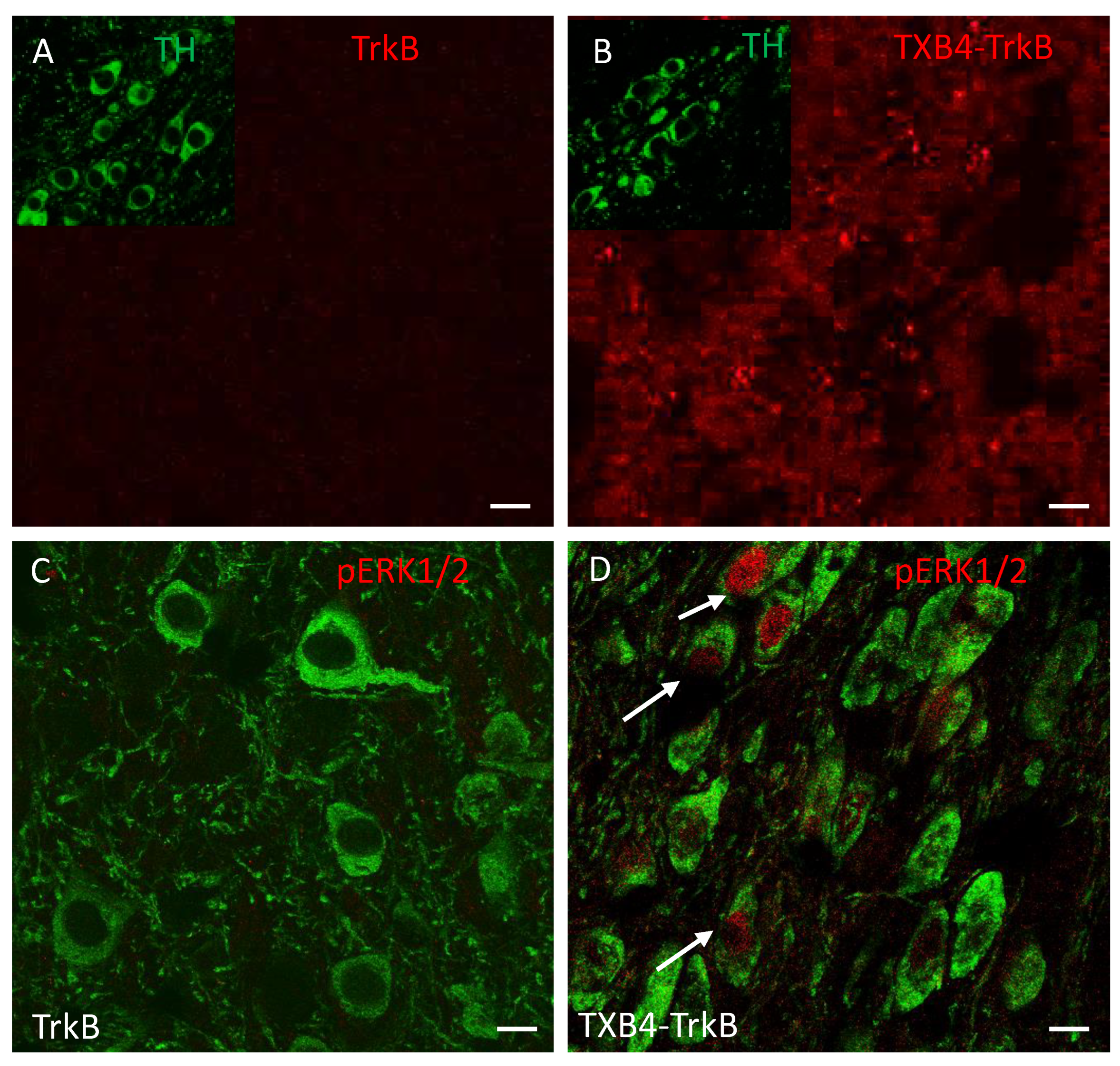
| TrkB Receptor | |||
|---|---|---|---|
| ka 1/[Ms] | kd [1/s] | KD [M] | |
| TrkB antibody | 2.18 × 105 | 3 × 10−4 | 1.37 × 10−9 |
| TXB4-TrkB (HV2N) | 2.32 × 105 | 3.02 × 10−4 | 1.3 × 10−9 |
| TXB4-TrkB (HC2N) | 2.20 × 105 | 9.77 × 10−5 | 4.44 × 10−10 |
| Agonist Activity EC50 [M] | ||||
|---|---|---|---|---|
| TrkB Antibody | TXB4-TrkB (HV2N) | TXB4-TrkB (HC2N) | TXB4 | BDNF |
| 3.4 × 10−10 | 9.1 × 10−10 | 6.9 × 10−9 | NA | 1.1 × 10−10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke, E.; Stocki, P.; Sinclair, E.H.; Gauhar, A.; Fletcher, E.J.R.; Krawczun-Rygmaczewska, A.; Duty, S.; Walsh, F.S.; Doherty, P.; Rutkowski, J.L. A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease. Pharmaceutics 2022, 14, 1335. https://doi.org/10.3390/pharmaceutics14071335
Clarke E, Stocki P, Sinclair EH, Gauhar A, Fletcher EJR, Krawczun-Rygmaczewska A, Duty S, Walsh FS, Doherty P, Rutkowski JL. A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease. Pharmaceutics. 2022; 14(7):1335. https://doi.org/10.3390/pharmaceutics14071335
Chicago/Turabian StyleClarke, Emily, Pawel Stocki, Elizabeth H. Sinclair, Aziz Gauhar, Edward J. R. Fletcher, Alicja Krawczun-Rygmaczewska, Susan Duty, Frank S. Walsh, Patrick Doherty, and Julia Lynn Rutkowski. 2022. "A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease" Pharmaceutics 14, no. 7: 1335. https://doi.org/10.3390/pharmaceutics14071335
APA StyleClarke, E., Stocki, P., Sinclair, E. H., Gauhar, A., Fletcher, E. J. R., Krawczun-Rygmaczewska, A., Duty, S., Walsh, F. S., Doherty, P., & Rutkowski, J. L. (2022). A Single Domain Shark Antibody Targeting the Transferrin Receptor 1 Delivers a TrkB Agonist Antibody to the Brain and Provides Full Neuroprotection in a Mouse Model of Parkinson’s Disease. Pharmaceutics, 14(7), 1335. https://doi.org/10.3390/pharmaceutics14071335