
Synthesis and Biological Evaluation of Highly Active 7-Anilino Triazolopyrimidines as Potent Antimicrotubule Agents

 ,
,  , , , , , ,
, , , , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. Biological Assays and Computational Studies
2.2.1. Cell Growth Conditions and Antiproliferative Assay for Human Cancer Cell Lines
2.2.2. Cytotoxicity in Human PBLs
2.2.3. Effects on Tubulin Polymerization and on Colchicine Binding to Tubulin
2.2.4. Molecular Modeling
2.2.5. Cell Cycle Analysis
2.2.6. Measurement of Apoptosis by Flow Cytometry
2.2.7. Measurement of Mitochondrial Membrane Potential and Reactive Oxygen Species (ROS) Production
2.2.8. 5-Ethynyl-2′-deoxyuridine (EdU) Cell Proliferation Assay
2.2.9. Evaluation of Cellular Protein Expression with Western Blots
2.2.10. Scratch-Migration Assay
2.2.11. Immunofluorescence Labeling of Microtubules
2.3. In Vivo Experiments on Zebrafish Model
2.3.1. Husbandry and Maintenance
2.3.2. Acute Toxicity Assessment in Zebrafish Embryos
2.3.3. Xenograft Model: Injection and Treatment
2.3.4. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
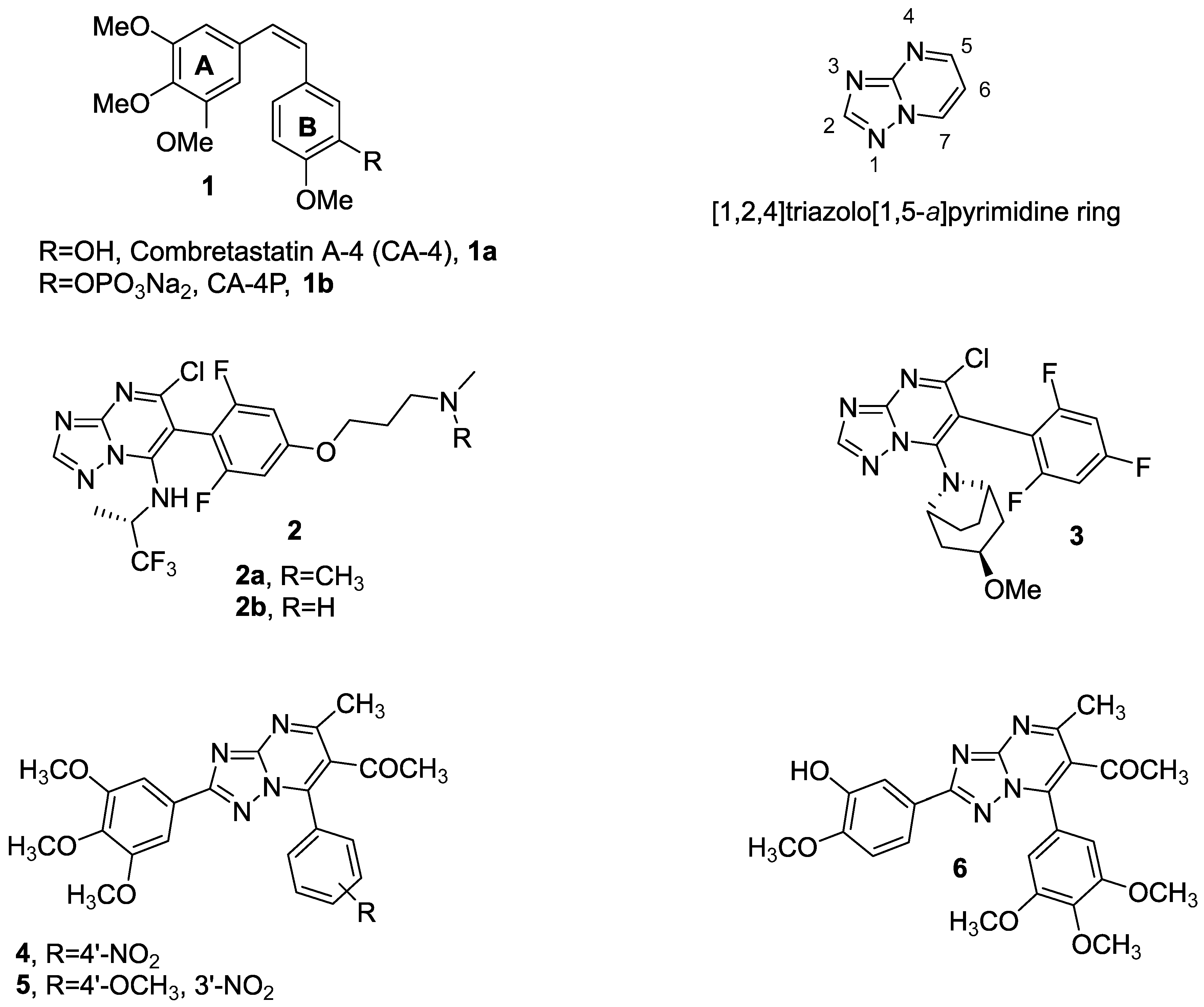
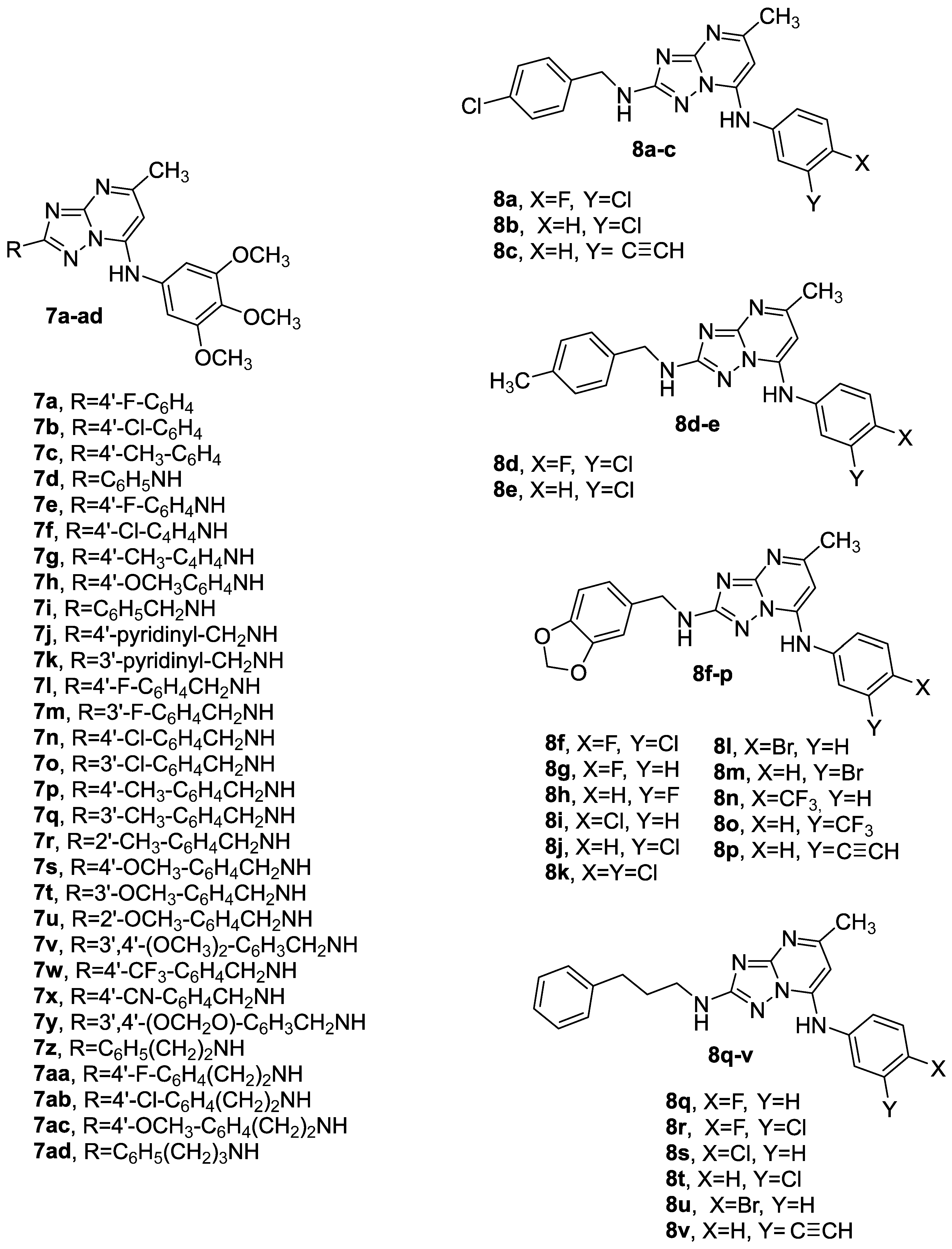
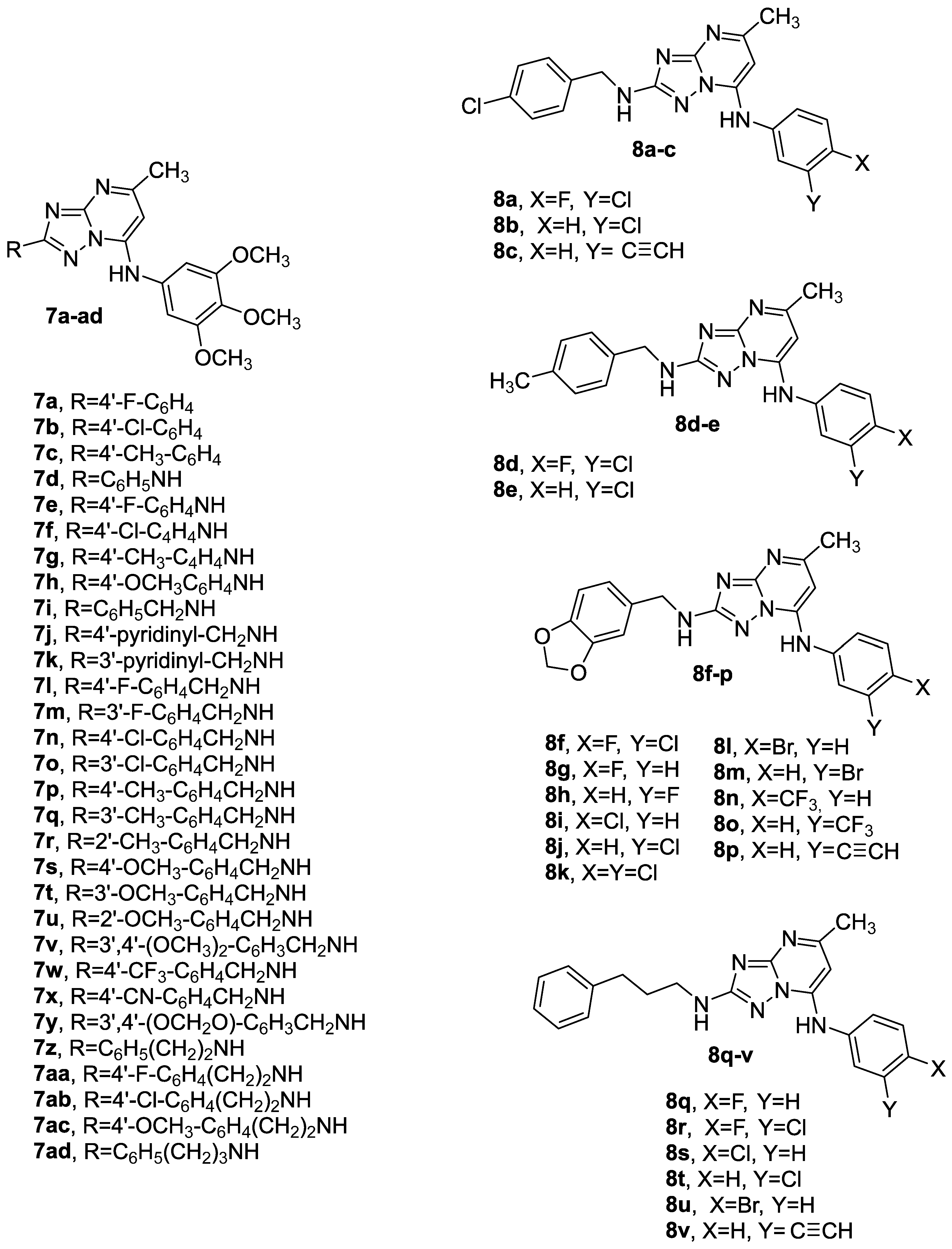
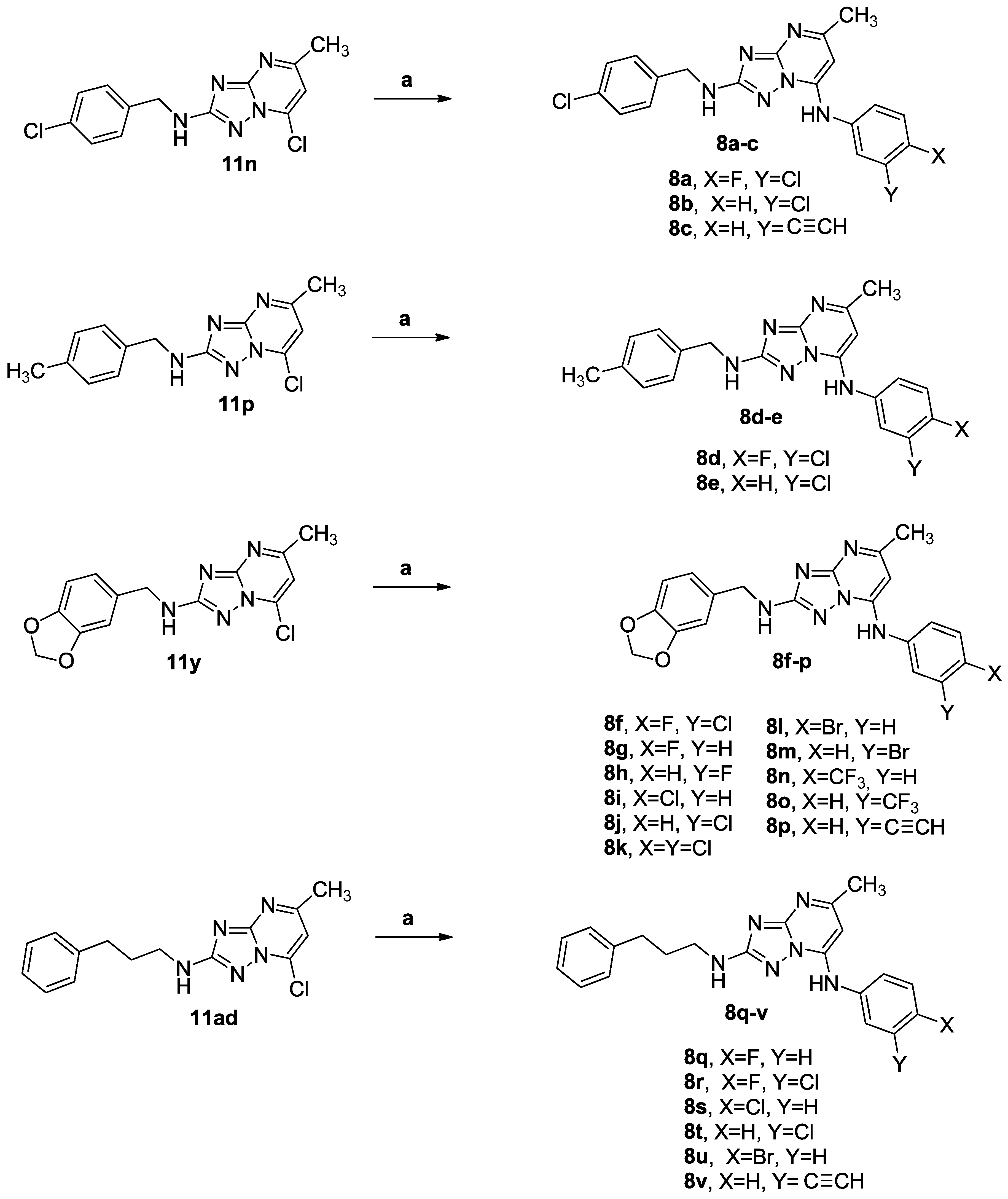
3.1.1. Synthetic Approach for the Preparation of 2,7-Disubstituted [1,2,4]triazolo [1,5-a]pyrimidines 7a–ad and 8a–v
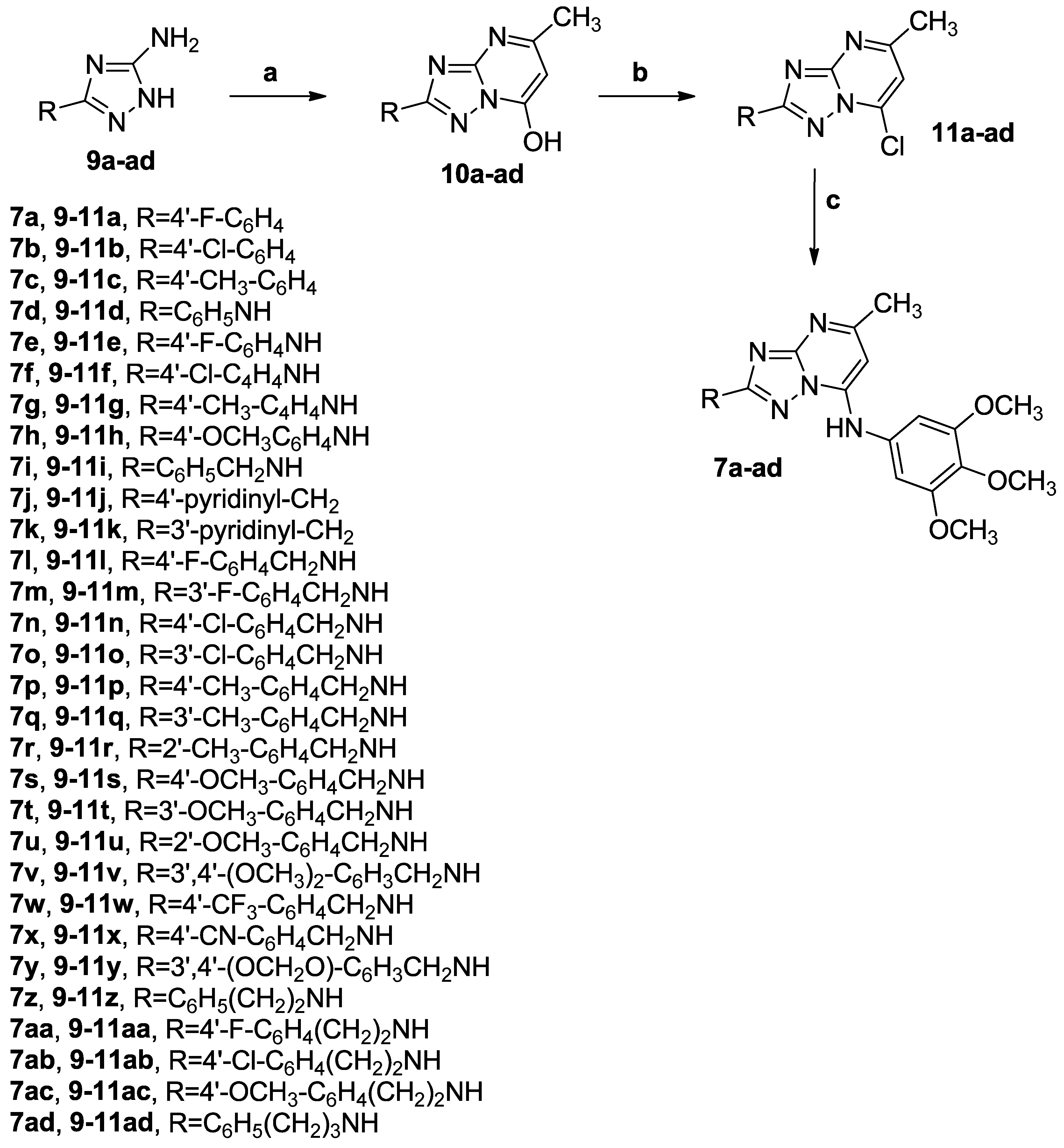
3.1.2. General Procedure A for the Synthesis of Compounds 10a–ad
3.1.3. General Procedure B for the Preparation of Compounds 11a–ad
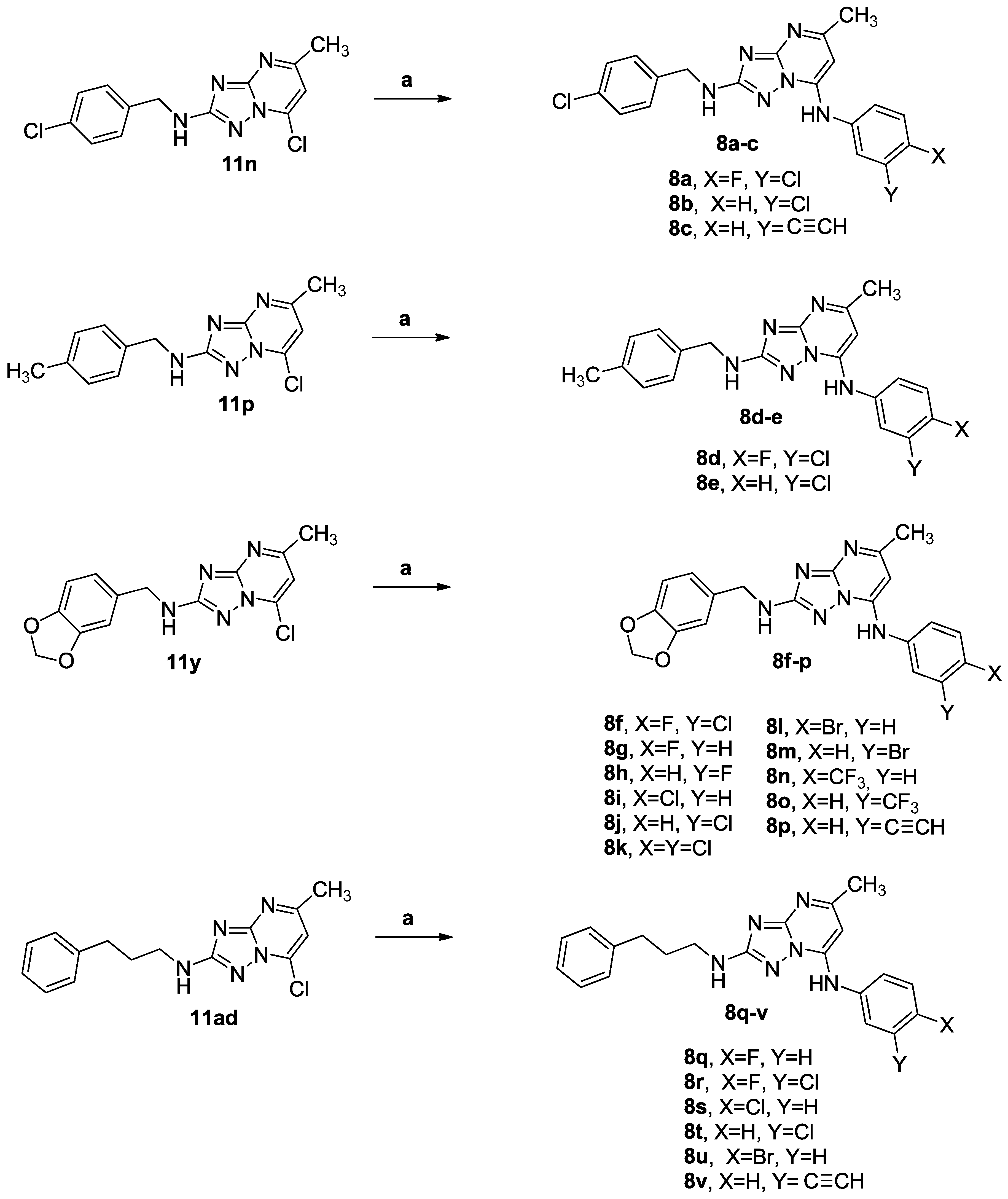
3.1.4. General Procedure C for the Synthesis of Compounds 7a–ad and 8a–v
3.2. Biological Activity
3.2.1. In Vitro Antiproliferative Activities
3.2.2. Effects of Compounds 7ad, 8q and 8r in Non-Tumor Cells
3.2.3. In Vitro Inhibition of Tubulin Polymerization and Colchicine Binding
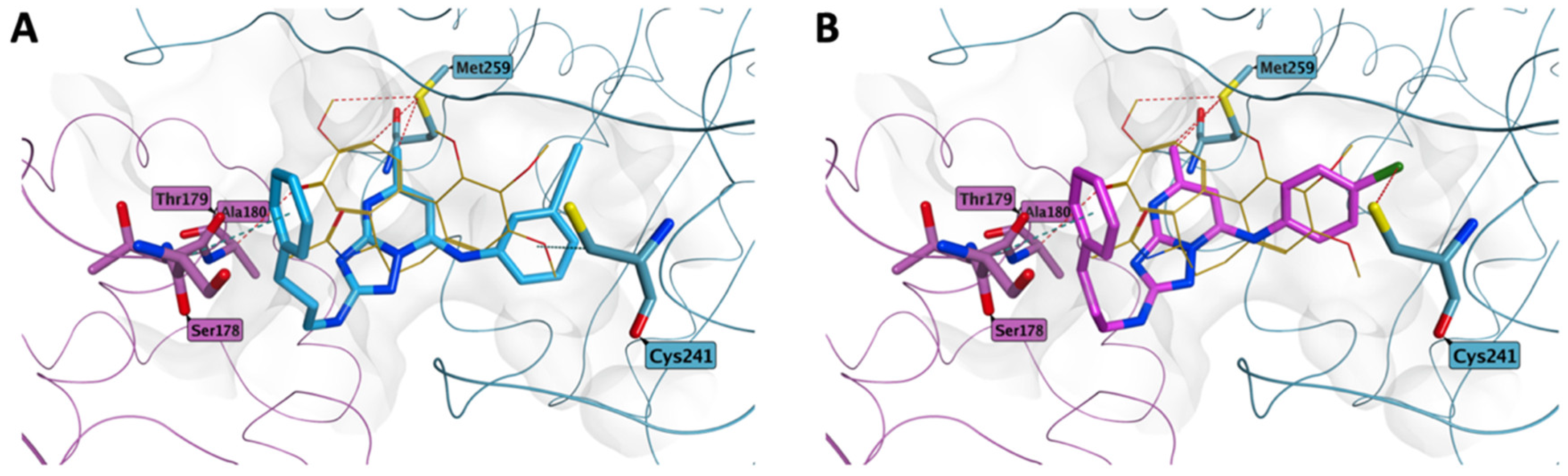
3.2.4. Molecular Modeling Studies
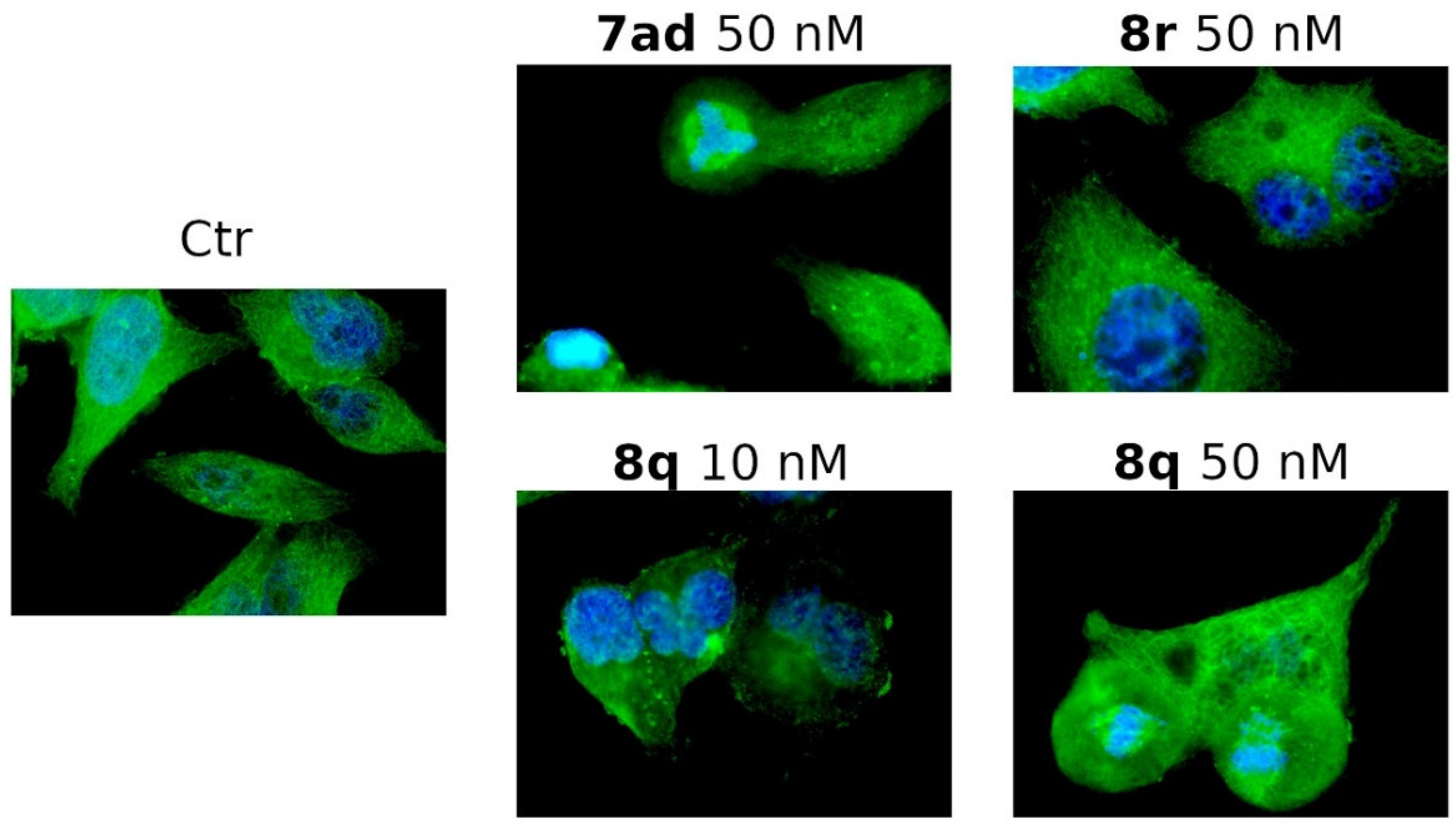
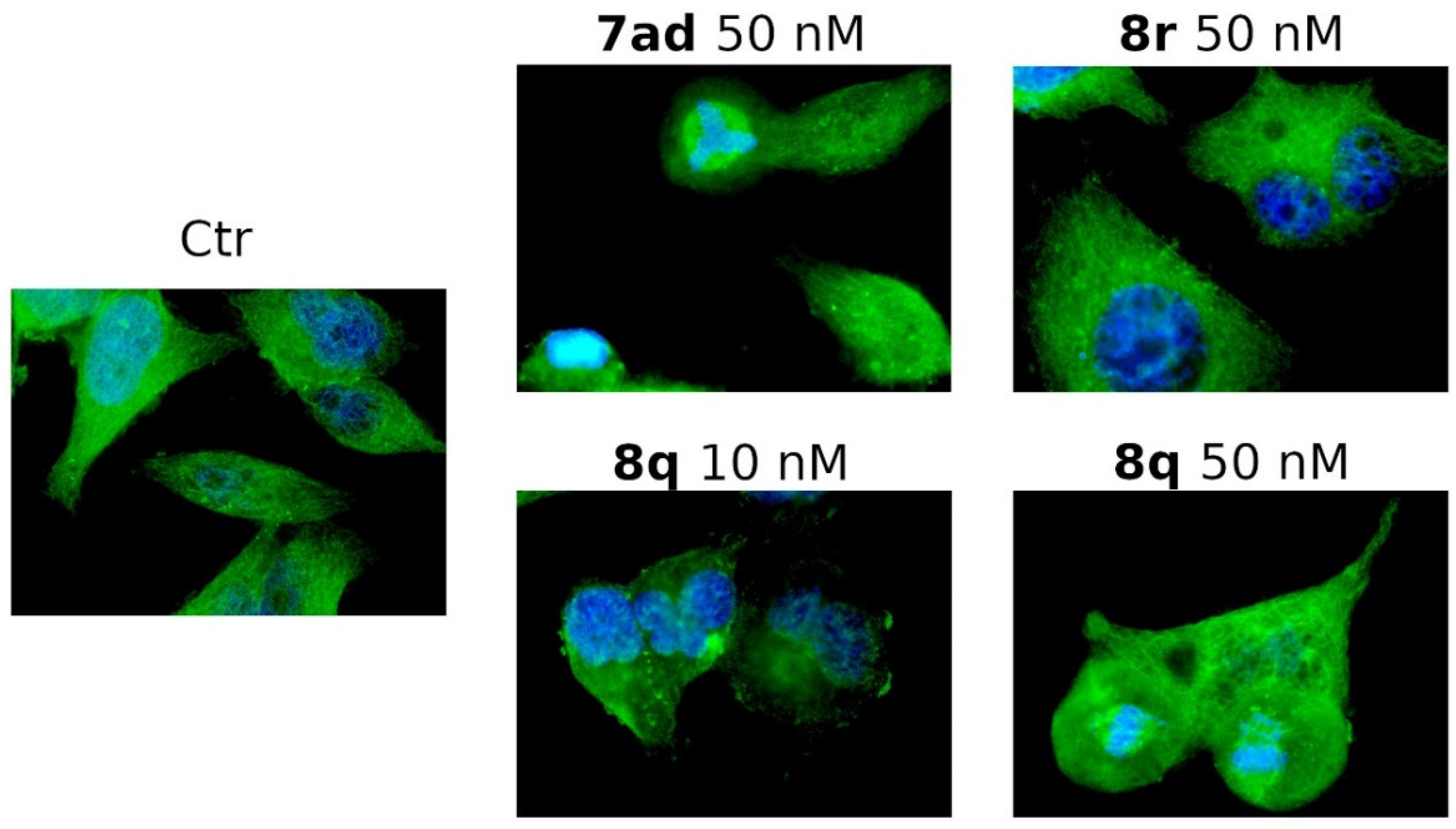
3.2.5. Compounds 7ad, 8q and 8r Induced Alterations of the Microtubule Network
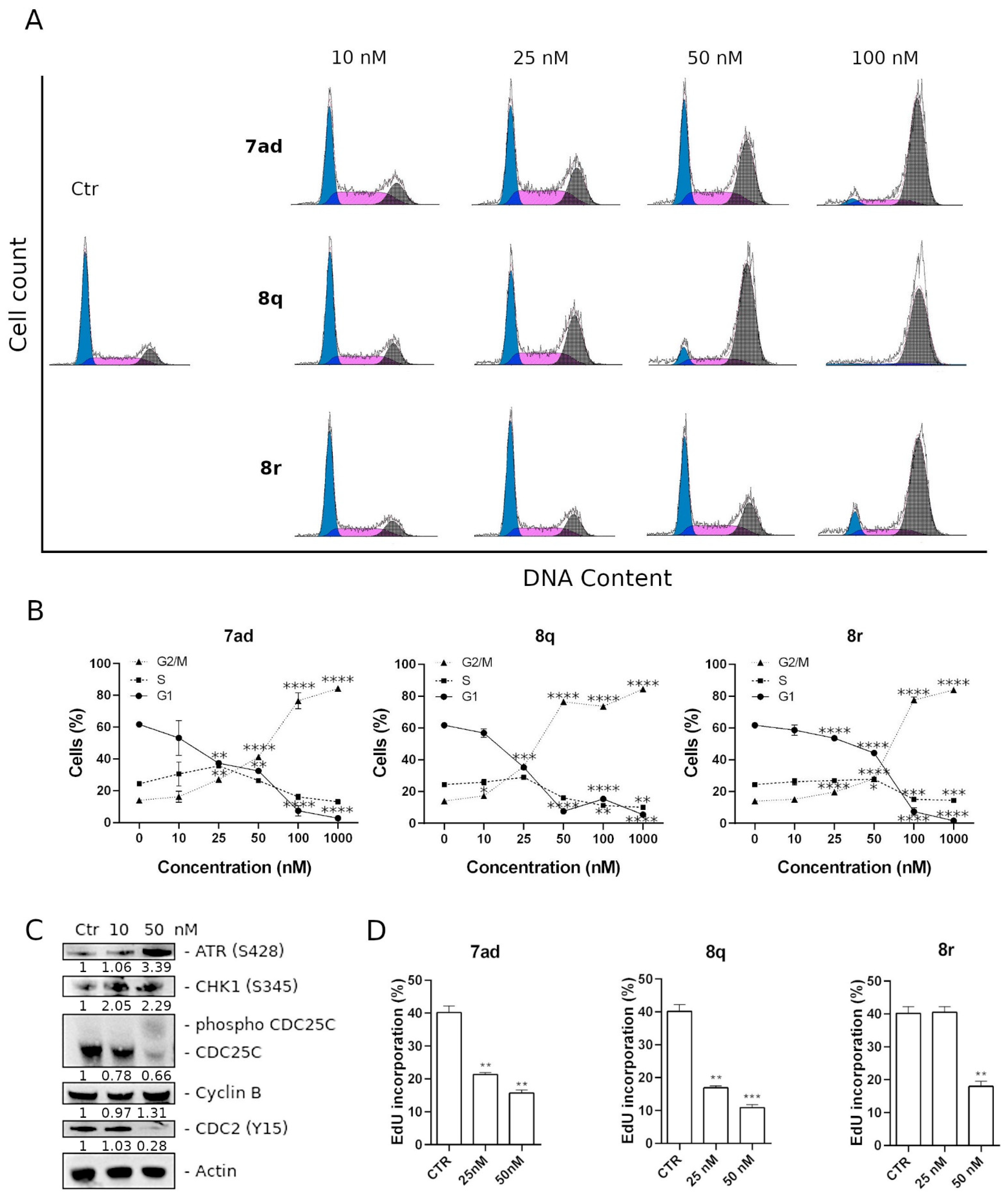
3.2.6. Compounds 7ad, 8q and 8r Induced Cell Cycle Arrest in G2/M along with Alteration of Cell Cycle and DNA Damage Checkpoint Proteins
3.2.7. Effects of 7ad, 8q and 8r Treatments on DNA Synthesis and Cell Proliferation
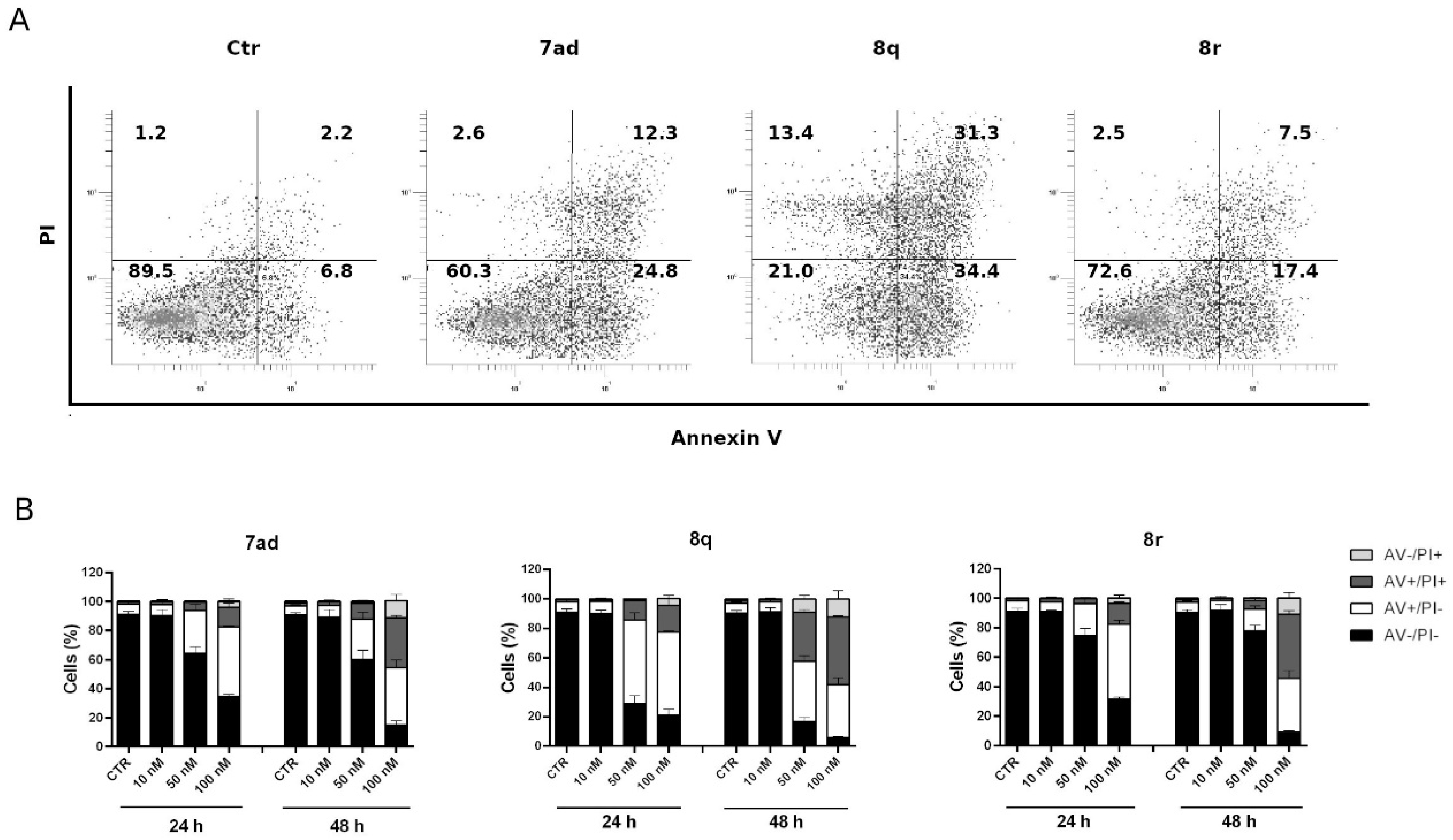
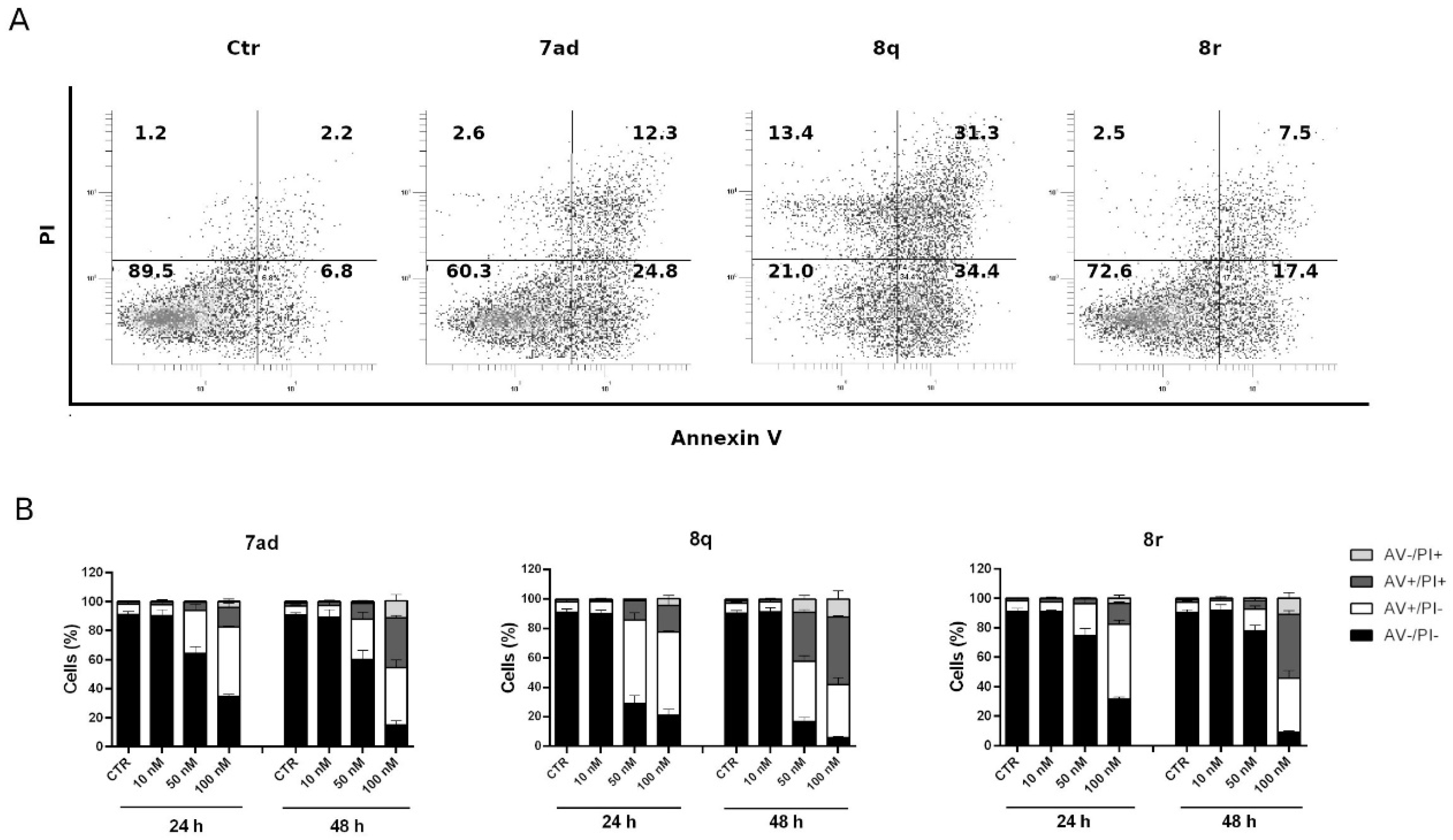
3.2.8. Compounds 7ad, 8q and 8r Induced Apoptosis in HeLa Cells
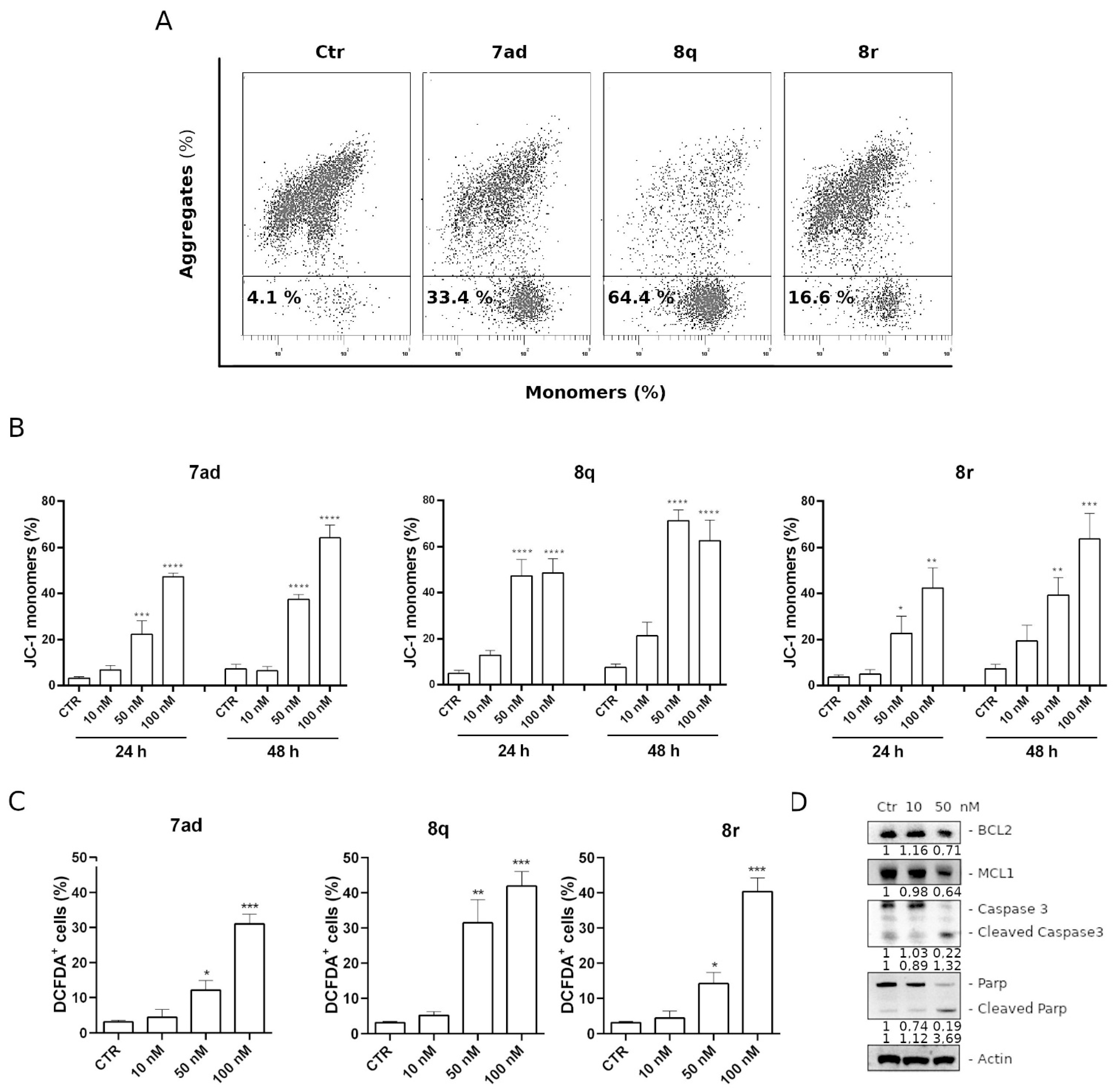
3.2.9. Apoptosis Induced by Compounds 7ad, 8q or 8r Follows the Mitochondrial Pathway
3.2.10. Compound 8q Induces Caspase-3 Activation and PARP Cleavage and Causes a Decrease in the Expression of Antiapoptotic Proteins
3.2.11. Compound 8q Impairs Cell Migration in HeLa Cells
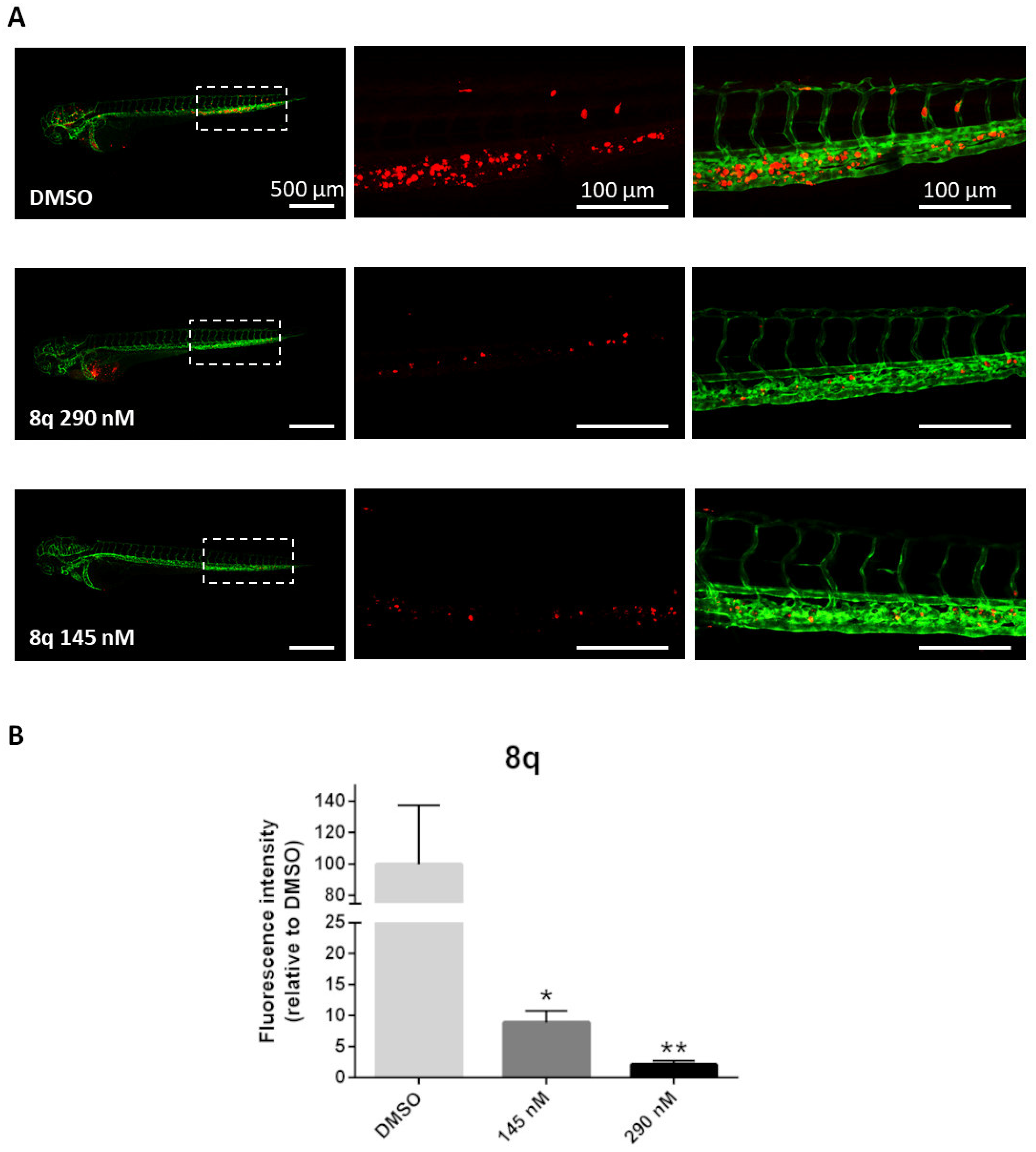
3.2.12. Effects of 8q Treatments on Zebrafish Embryos (Acute Toxicity Test)
3.2.13. In Vivo Antitumor Activity and Antimetastasis Effects of Compound 8q in a Zebrafish Xenograft
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Heron, M.; Anderson, R.N. Changes in the leading cause of death: Recent patterns in heart disease and cancer mortality. NCHS Data Brief 2016, 254, 1–8. [Google Scholar]
- Hulvat, M.C. Cancer incidence and trends. Surgical oncology for the general surgeon, an issue of surgical clinics. Surg. Clin. 2020, 100, 469. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.C.; Sloan, C.D.; Merril, R.M.; Harding, T.M.; Thacker, B.J.; Thacker, E.L. Transitions from heart disease to cancer as the leading cause of death in US states, 1999–2016. Prev. Chronic Dis. 2018, 15, E158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Gascoign, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [Green Version]
- Knossow, M.; Campanacci, V.; Khodja, L.A.; Gigant, B. The mechanism of tubulin assembly into microtubules: Insights from structural studies. iScience 2020, 23, 101511. [Google Scholar] [CrossRef]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Brouhard, G.J.; Rice, L.M. Microtubule dynamics: An interplay of biochemistry and mechanics. Nat. Rev. Mol. Cell Biol. 2018, 19, 451–463. [Google Scholar] [CrossRef]
- Cirillo, L.; Gotta, M.; Meraldi, P. The elephant in the room: The role of microtubules in cancer. Adv. Exp. Med. Biol. 2017, 1002, 93–124. [Google Scholar] [PubMed]
- Steinmetz, M.O.; Prota, A.E. Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Florian, S.; Mitchison, T.J. Anti-microtubule drugs. Methods Mol. Biol. 2016, 1413, 403–421. [Google Scholar] [PubMed]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calò, V.; Castiglia, M.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef] [Green Version]
- Mcloughlin, E.C.; O’Boyle, N.M. Colchicine-binding site inhibitors from chemistry to clinic: A review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Liu, F.; Zhou, H.; Zhai, S.; Yan, B. Novel natural product- and privileged scaffold-based tubulin inhibitors targeting the colchicine binding site. Molecules 2016, 21, 1375. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, R.; Medarde, M.; Pelaez, R. New ligands of the tubulin colchicine site based on X-ray structures. Curr. Top. Med. Chem. 2014, 14, 2231–2252. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, G.; Gill, R.K.; Soni, R.; Bariwal, J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendall, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experentia 1989, 45, 209–211. [Google Scholar] [CrossRef]
- Patil, P.O.; Patil, A.G.; Rane, R.A.; Patil, P.C.; Deshmukh, P.K.; Bari, S.B.; Patil, D.A.; Naphade, S.S. Recent advancement in discovery and development of natural product combretastatin-inspired anticancer agents. Anticancer Agents Med. Chem. 2015, 15, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.M.; Ho, H.H.; Pettit, G.R.; Hamel, E. The antimitotic natural products combretastatin A-4 and combretastatin A-2: Studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry 1989, 28, 6984–6991. [Google Scholar] [CrossRef] [PubMed]
- Hura, N.; Sawant, A.V.; Kumari, A.; Guchhait, S.K.; Panda, D. Combretastatin inspired heterocycles as antitubulin anticancer agents. ACS Omega 2018, 3, 9754–9769. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Sill, M.W.; Coleman, R.L.; Aghajanian, C.; Mannel, R.; DiSilvestro, P.A.; Powell, M.; Randall, L.M.; Farley, J.; Rubin, S.C.; et al. Bevacziumab plus fosbretabulin in recurrent ovarian cancer: Overall survival and exploratory analyses of a randomized Phase II NRG oncology/gynecologic oncology group study. Gynecol. Oncol. 2020, 159, 79–87. [Google Scholar] [CrossRef]
- Deng, C.; Zhao, J.; Zhou, S.; Dong, J.; Cao, J.; Gao, J.; Bai, Y.; Deng, H. The vascular disrupting agent CA4P improves the antitumor efficacy of CAR-T cells in preclinical models of solid human tumors. Mol. Ther. 2020, 28, 75–88. [Google Scholar] [CrossRef]
- Zhang, N.; Ayral-Kaloustian, S.; Nguyen, T.; Afragola, J.; Hernandez, R.; Lucas, J.; Gibbons, J.; Beyer, C. Synthesis and SAR of [1,2,4]triazolo [1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition. J. Med. Chem. 2007, 50, 319–327. [Google Scholar] [CrossRef]
- Beyer, C.F.; Zhang, N.; Hernandez, R.; Vitale, D.; Lucas, J.; Nguyen, T.; Discafani, C.; Ayral-Kaloustian, S.; Gibbons, J.J. TTI-237: A novel microtubule-active compound with in vivo antitumor activity. Cancer Res. 2008, 68, 2292–2300. [Google Scholar] [CrossRef] [Green Version]
- Wang-Gillam, A.; Arnold, S.M.; Bukowski, R.M.; Rothenberg, M.L.; Cooper, W.; Wang, K.K.; Gauthier, E.; Lockhart, A.C. A phase I dose escalation study of TTI-237 in patients with advanced malignant solid tumors. Investig. New Drugs 2012, 30, 266–272. [Google Scholar] [CrossRef]
- Sáez-Calvo, G.; Sharma, A.; Balaguer, F.A.; Barasoain, I.; Rodríguez-Salarichs, J.; Olieric, N.; Muñoz-Hernández, H.; Berbís, M.Á.; Wendeborn, S.; Peñalva, M.; et al. Triazolopyrimidines are microtubule-stabilizing agents that bind the Vinca inhibitor site of tubulin. Cell Chem. Biol. 2017, 24, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, L.Z.; Diao, P.C.; Jian, X.E.; Zhou, M.F.; Jiang, C.S.; You, W.W.; Ma, V.; Zhao, P.L. Novel [1,2,4]triazolo [1,5-a]pyrimidine derivatives as potent antitubulin agents: Design, multicomponent synthesis and antiproliferative activities. Bioorg. Chem. 2019, 92, 103260. [Google Scholar] [CrossRef]
- Huo, X.-S.; Jian, X.-E.; Ou-Yang, J.; Chen, L.; Yang, F.; Lv, D.-X.; You, W.-W.; Rao, J.-J.; Zhao, P.-L. Discovery of highly potent tubulin polymerization inhibitors: Design, synthesis, and structure-activity relationships of novel 2,7-diaryl-[1,2,4]triazolo [1,5-a]pyrimidines. Eur. J. Med. Chem. 2021, 220, 113449. [Google Scholar] [CrossRef] [PubMed]
- Gaukroger, K.; Hadfield, J.A.; Lawrence, N.J.; Nlan, S.; McGown, A.T. Structural requirements for the interaction of combretastatins with tubulin: How important is the trimethoxy unit? Org. Biomol. Chem. 2003, 1, 3033–3037. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E.; Lin, C.M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–21. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Lai, J.-Y.; Yoo, H.-D.; Yu, J.; Marquez, B.; Nagle, D.G.; Nambu, M.; White, J.D.; Falck, J.R.; Gerwick, W.H.; et al. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998, 53, 62–67. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE 2020.09); Chemical Computing Group, Inc.: Montreal, QC, Canada, 2020; Available online: http://www.chemcomp.com (accessed on 1 June 2022).
- Schrödinger Release 2021-1: Maestro; Schrödinger, LLC.: New York, NY, USA, 2021.
- Viola, G.; Vedaldi, D.; Dall’Acqua, F.; Fortunato, E.; Basso, G.; Bianchi, N.; Zuccato, C.; Borgatti, M.; Lampronti, I.; Gambari, R. Induction of γ-globin mRNA, erythroid differentiation and apoptosis in UVA-irradiated human erythroid cells in the presence of furocoumarine derivatives. Biochem. Pharm. 2008, 75, 810–825. [Google Scholar] [CrossRef]
- Hason, M.; Bartůnĕk, P. Zebrafish models of cancer-new insights on modeling human cancer in a non-mammalian vertebrate. Genes 2019, 10, 935. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.; Bongo, L.; Dyortsok, P. On triazoles XI. Structure elucidation of isomeric 1,2,4-triazolopyrimidinones. Tetrahedron 1987, 43, 2497–2504. [Google Scholar] [CrossRef]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Kimatrai Salvador, M.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilinoindoles as a new class of potent inhibitors of tubulin polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Prencipe, F.; Oliva, P.; Baraldi, S.; Tabrizi, M.A.; Lopez-Cara, L.C.; Ferla, S.; Brancale, A.; Hamel, E.; et al. Design and synthesis of potent in vitro and in vivo anticancer agents based on 1-(3′,4′,5′-trimethoxyphenyl)-2-aryl-1H-imidazole. Sci. Rep. 2016, 6, 26602. [Google Scholar] [PubMed] [Green Version]
- Jackson, J.R.; Patrick, D.R.; Dar, M.M.; Huang, P.S. Targeted anti-mitotic therapies: Can we improve on tubulin agents? Nat. Rev. Cancer 2007, 7, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Rovini, A.; Savry, A.; Braguer, D.; Carré, M. Microtubule-targeted agents: When mitochondria become essential to chemotherapy. Biochim. Biophys. Acta-Bioenergy 2011, 1807, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Lopez Cara, C.; Hamel, E.; Basso, G.; Bortolozzi, R.; Viola, G. Synthesis and biological evaluation of 2-(3′,4′,5′-trimethoxybenzoyl)-3-aryl/arylaminobenzo[b]thiophene derivatives as a novel class of antiproliferative agents. Eur. J. Med. Chem. 2010, 45, 5781–5791. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Tabrizi, M.A.; Brancale, A.; Fu, X.-H.; Li, J.; Zhang, S.-Z.; Hamel, E.; et al. Discovery and optimization of a series of 2-aryl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl)thiazoles as novel anticancer agents. J. Med. Chem. 2012, 55, 5433–5445. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Preti, D.; Aghazadeh Tabrizi, M.; Balzarini, J.; Bassetto, M.; Brancale, A.; Fu, X.-H.; Gao, Y.; et al. Concise synthesis and biological evaluation of 2-aroyl-5-amino benzo[b]thiophene derivatives as a novel class of potent antimitotic agents. J. Med. Chem. 2013, 56, 9296–9309. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [Green Version]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Decaudin, D.; Macho, A.; Hirsch, T.; Susin, S.A.; Petit, B.X.; Mignotte, B.; Kroemer, G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Aredia, F.; Scovassi, A.I. Poly(ADP-Ribose): A signaling molecule in different paradigms of cell death. Biochem. Pharmacol. 2014, 92, 157–163. [Google Scholar] [CrossRef]
- Widden, H.; Placzek, W.J. The multiple mechanisms of MCL1 in the regulation of cell fate. Commun. Biol. 2021, 4, 1029. [Google Scholar] [CrossRef]
- OECD. Test No. 236: Fish Embryo Acute Toxicity (FET) Test. In OECD Guidelines for the Testing of Chemicals; Section 2; OECD Publishing: Paris, France, 2016. [Google Scholar]
- Fohlen, A.; Bordji, K.; Assenat, E.; Gongora, C.; Bazille, C.; Boulonnais, J.; Naveau, M.; Breuil, C.; Pérès, E.A.; Bernaudin, M. Anticancer drugs for intra-arterial treatment of colorectal cancer liver metastases: In-vitro screening after short exposure time. Pharmaceuticals 2021, 14, 639. [Google Scholar] [CrossRef] [PubMed]
- Schobert, R.; Effenberger-Neidnicht, K.; Biersack, B. Stable combretastatin A-4 analogues with sub-nanomolar efficacy against chemoresistant HT-29 cells. Int. J. Clin. Pharmacol. Ther. 2011, 49, 71–72. [Google Scholar] [PubMed]
- Choi, J.-S.; Doh, K.-O.; Kim, B.-K.; Seu, Y.-B. Synthesis of cholesteryl doxorubicin and its anti-cancer activity. Bioorg. Med. Chem. Lett. 2017, 27, 723–728. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Bassetto, M.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Bortolozzi, R.; et al. Synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilino benzo[b]thiophenes and thieno [2,3-b]pyridines as new potent anticancer agents. J. Med. Chem. 2013, 56, 2606–2618. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Zhang, M.; Wang, Y.; Guo, J.; Wang, J.; Li, Y.; Ding, N. Synthesis and biological evaluation of novel cabazitaxel analogues. Bioorg. Med. Chem. 2021, 41, 116224. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | IC50 (μM) a | ||||
| A549 | MDA-MB-231 | HeLa | HT-29 | Jurkat | ||
| 7a | 4′-F-C6H4 | 2.71 ± 0.25 | 2.25 ± 0.19 | 3.76 ± 0.19 | >10 | 3.57 ± 0.45 |
| 7b | 4′-Cl-C6H4 | >10 | >10 | 5.79 ± 0.42 | >10 | >10 |
| 7c | 4′-CH3-C6H4 | >10 | 2.60 ± 0.27 | 2.08 ± 0.19 | >10 | 4.23 ± 1.15 |
| 7d | C6H5NH | 2.30 ± 0.12 | 1.43 ± 0.14 | 0.52 ± 0.06 | 0.82 ± 0.07 | 0.59 ± 0.01 |
| 7e | 4′-F-C6H4NH | 1.02 ± 0.08 | 0.71 ± 0.07 | 0.39 ± 0.02 | 0.69 ± 0.06 | 0.82 ± 0.001 |
| 7f | 4′-Cl-C6H4NH | 0.29 ± 0.03 | 0.47 ± 0.045 | 0.092 ± 0.01 | 0.32 ± 0.04 | >10 |
| 7g | 4′-CH3-C6H4NH | 1.55 ± 0.19 | 1.51 ± 0.14 | 0.50 ± 0.04 | 0.94 ± 0.11 | 0.85 ± 0.02 |
| 7h | 4′-OCH3-C6H4NH | 3.76 ± 0.14 | 1.17 ± 0.13 | 0.52 ± 0.03 | 0.92 ± 0.09 | 1.01 ± 0.13 |
| 7i | C6H5CH2NH | 3.75 ± 0.26 | 2.71 ± 0.31 | 3.20 ± 0.24 | 3.53 ± 0.14 | 4.40 ± 0.38 |
| 7j |  | >10 | 0.059 ± 0.01 | 3.01 ± 0.3 | 2.53 ± 0.25 | 9.64 ± 0.80 |
| 7k | 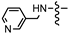 | 5.28 ± 0.65 | 0.56 ± 0.067 | 2.98 ± 0.32 | 3.57 ± 0.48 | 6.85 ± 0.63 |
| 7l | 4′-F-C6H4CH2NH | 5.89 ± 0.48 | 0.061 ± 0.009 | 1.86 ± 0.13 | 1.14 ± 0.09 | 4.68 ± 0.48 |
| 7m | 3′-F-C6H4CH2NH | 2.18 ± 0.31 | 0.077 ± 0.010 | 0.93 ± 0.02 | 1.53 ± 0.16 | 5.55 ± 0.48 |
| 7n | 4′-Cl-C6H4CH2NH | 0.11 ± 0.022 | 0.19 ± 0.02 | 0.031 ± 0.01 | 0.054 ± 0.01 | 0.32 ± 0.019 |
| 7o | 3′-Cl-C6H4CH2NH | 0.93 ± 0.11 | 0.060 ± 0.007 | 0.74 ± 0.08 | 0.79 ± 0.09 | 4.72 ± 0.36 |
| 7p | 4′-CH3-C6H4CH2NH | 0.42 ± 0.07 | 0.301 ± 0.04 | 0.060 ± 0.01 | 0.11 ± 0.02 | 0.43 ± 0.001 |
| 7q | 3′-CH3-C6H4CH2NH | 3.88 ± 0.28 | 1.68 ± 0.18 | 1.61 ± 0.13 | 3.46 ± 0.37 | 7.02 ± 0.62 |
| 7r | 2′-CH3-C6H4CH2NH | 1.92 ± 0.09 | 2.99 ± 0.18 | >10 | >10 | 4.44 ± 0.47 |
| 7s | 4′-OCH3-C6H4CH2NH | 0.81 ± 0.08 | 1.27 ± 0.12 | 0.22 ± 0.03 | 0.49 ± 0.06 | 0.90 ± 0.09 |
| 7t | 3′-OCH3-C6H4CH2NH | 1.65 ± 0.14 | 1.78 ± 0.15 | 0.88 ± 0.09 | 1.04 ± 0.08 | 1.65 ± 0.11 |
| 7u | 2′-OCH3-C6H4CH2NH | >10 | 4.40 ± 0.26 | >10 | >10 | >10 |
| 7v | 3′,4′-(OCH3)2-C6H3CH2NH | 1.00 ± 0.09 | 1.22 ± 0.15 | 0.56 ± 0.07 | >10 | 1.22 ± 0.09 |
| 7w | 4′-CF3-C6H4CH2NH | 1.28 ± 0.13 | 0.56 ± 0.03 | 0.52 ± 0.02 | 0.59 ± 0.26 | 0.96 ± 0.06 |
| 7x | 4′-CN-C6H4CH2NH | >10 | 1.55 ± 0.11 | 9.01 ± 0.21 | >10 | >10 |
| 7y | 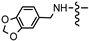 | 0.66 ± 0.03 | 0.30 ± 0.02 | 0.11 ± 0.01 | 0.17 ± 0.02 | 0.78 ± 0.09 |
| 7z | C6H5(CH2)2NH | 2.01 ± 0.16 | 0.94 ± 0.09 | 0.86 ± 0.10 | 1.89 ± 0.16 | 1.99 ± 0.24 |
| 7aa | 4′-F-C6H4(CH2)2NH | >10 | >10 | 2.50 ± 0.27 | 2.83 ± 0.22 | 9.61 ± 0.89 |
| 7ab | 4′-Cl-C6H4(CH2)2NH | >10 | >10 | 5.81 ± 0.48 | >10 | 5.94 ± 0.5 |
| 7ac | 4′-OCH3-C6H4(CH2)2NH | 8.88 ± 0.88 | 1.65 ± 0.19 | 5.05 ± 0.63 | 6.88 ± 0.49 | 9.59 ± 1.17 |
| 7ad | C6H5(CH2)3NH | 0.19 ± 0.02 | 0.091 ± 0.008 | 0.067 ± 0.007 | 0.14 ± 0.011 | 2.72 ± 0.13 |
| CA-4 | - | 0.18 ± 0.05 | 0.005 ± 0.002 | 0.004 ± 0.001 | 3.10 ± 0.03 | 0.001 ± 0.0002 |
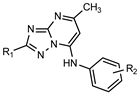 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) a | ||||
| A549 | MDA-MB-231 | HeLa | HT-29 | Jurkat | |||
| 8a | 4′-ClC6H4CH2NH | 3′-Cl, 4′-F | 0.32 ± 0.06 | 0.39 ± 0.04 | 0.19 ± 0.05 | 0.34 ± 0.02 | 0.50 ± 0.31 |
| 8b | 4′-ClC6H4CH2NH | 3′-Cl | 1.74 ± 0.13 | 1.88 ± 0.08 | 0.81 ± 0.15 | 1.64 ± 0.22 | 0.88 ± 0.50 |
| 8c | 4′-ClC6H4CH2NH | 3′-CCH | 0.61 ± 0.20 | 1.71 ± 0.06 | 0.42 ± 0.05 | 0.49 ± 0.06 | 0.50 ± 0.06 |
| 8d | 4′-CH3C6H4CH2NH | 3′-Cl, 4′-F | 0.30 ± 0.06 | 0.80 ± 0.26 | 0.19 ± 0.06 | 0.22 ± 0.04 | 0.29 ± 0.06 |
| 8e | 4′-CH3C6H4CH2NH | 3′-Cl | 1.64 ± 0.37 | 2.76 ± 0.55 | 0.68 ± 0.26 | 0.83 ± 0.003 | 0.63 ± 0.14 |
| 8f | 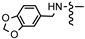 | 3′-Cl, 4′-F | 0.44 ± 0.05 | 0.70 ± 0.16 | 0.16 ± 0.09 | 0.30 ± 0.03 | 0.98 ± 0.06 |
| 8g |  | 4′-F | 0.65 ± 0.28 | 0.60 ± 0.21 | 0.16 ± 0.07 | 0.26 ± 0.10 | 0.20 ± 0.05 |
| 8h | | 3′-F | 2.44 ± 0.13 | 9.25 ± 0.12 | 5.88 ± 2.96 | 9.11 ± 0.93 | 5.98 ± 1.58 |
| 8i | | 4′-Cl | 0.81 ± 0.22 | 0.94 ± 0.34 | 0.19 ± 0.12 | 0.60 ± 0.16 | 0.28 ± 0.05 |
| 8j | | 3′-Cl | 2.17 ± 0.01 | 4.82 ± 1.91 | 0.96 ± 0.46 | 2.66 ± 0.66 | 1.66 ± 0.48 |
| 8k | | 3′,4′-Cl2 | 2.36 ± 0.34 | 2.04 ± 1.03 | 0.72 ± 0.34 | 1.38 ± 0.06 | 0.95 ± 0.12 |
| 8l | | 4′-Br | 0.43 ± 0.18 | 1.27 ± 0.45 | 0.22 ± 0.04 | 0.42 ± 0.02 | 0.26 ± 0.05 |
| 8m | | 3′-Br | 2.32 ± 0.23 | 3.99 ± 0.06 | 1.28 ± 0.30 | 2.24 ± 0.12 | 1.18 ± 0.11 |
| 8n | | 4′-CF3 | 3.27 ± 0.57 | 6.13 ± 1.46 | 3.64 ± 1.01 | 5.18 ± 0.27 | 1.60 ± 0.29 |
| 8o | | 3′-CF3 | 6.26 ± 2.02 | 9.82 ± 0.86 | 7.15 ± 0.60 | 7.13 ± 0.87 | 7.60 ± 0.06 |
| 8p | | 3′-CCH | 1.78 ± 0.51 | 3.09 ± 0.57 | 1.60 ± 0.07 | 2.08 ± 0.14 | 2.38 ± 0.18 |
| 8q | C6H5(CH2)3NH | 4′-F | 0.13 ± 0.09 | 0.062 ± 0.03 | 0.029 ± 0.000 | 0.049 ± 0.01 | 0.15 ± 0.02 |
| 8r | C6H5(CH2)3NH | 3′-Cl, 4′-F | 0.081 ± 0.002 | 0.098 ± 0.02 | 0.035 ± 0.000 | 0.058 ± 0.01 | 0.23 ± 0.02 |
| 8s | C6H5(CH2)3NH | 4′-Cl | 0.070 ± 0.01 | 0.20 ± 0.07 | 0.048 ± 0.001 | 0.053 ± 0.001 | 0.083 ± 0.02 |
| 8t | C6H5(CH2)3NH | 3′-Cl | 0.26 ± 0.07 | 1.32 ± 0.87 | 0.153 ± 0.01 | 0.38 ± 0.03 | 0.14 ± 0.07 |
| 8u | C6H5(CH2)3NH | 4′-Br | 0.048 ± 0.01 | 0.12 ± 0.04 | 0.022 ± 0.01 | 0.083 ± 0.02 | 0.15 ± 0.014 |
| 8v | C6H5(CH2)3NH | 3′-CCH | 0.11 ± 0.03 | 0.35 ± 0.01 | 0.069 ± 0.01 | 0.083 ± 0.02 | 0.16 ± 0.01 |
| CA-4 | - | - | 0.18 ± 0.05 | 0.005 ± 0.002 | 0.004 ± 0.001 | 3.10 ± 0.03 | 0.001 ± 0.00 |
| Compounds | IC50 (nM) a | |
|---|---|---|
| PBLresting b | PBLPHA c | |
| 7ad | >10,000 | >10,000 |
| 8q | >10,000 | >10,000 |
| 8r | >10,000 | >10,000 |
| CA-4 (1a) | >10,000 | 18.6 ± 3.8 |
| Compounds | Tubulin Assembly a IC50 ± SD (µM) | Colchicine Binding b % Inhibition ± SD |
|---|---|---|
| 7n | 0.39 ± 0.06 | 30 ± 0.3 |
| 7p | 0.66 ± 0.1 | 27 ± 3 |
| 7y | 1.0 ± 0.2 | 19 ± 05 |
| 7ad | 0.38 ± 0.06 | 55 ± 3 |
| 8q | 0.39 ± 0.02 | 88 ± 0.8 |
| 8r | 0.52 ± 0.06 | 87 ± 0.8 |
| 8s | 0.39 ± 0.01 | 88 ± 3 |
| 8u | 0.38 ± 0.02 | 84 ± 2 |
| 8v | 0.43 ±0.05 | 70 ± 3 |
| CA-4 | 0.75 ± 0.06 | 97 ± 0.1 |
| Condition | Number of Embryos | % Lethality | % Alterations | Results | ||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |||
| CTR | 16 | 0 | 0 | 0 | 0 | - |
| DMSO (vehicle-treated) | 21 | 0 | 0 | 0 | 0 | Not toxic |
| CA-4 (100 nM) | 21 | 100 | - | - | - | Toxic |
| 8q (100 nM) | 24 | 0 | 0 | 0 | 0 | Not toxic |
| 8q (1 μM) | 20 | 0 | 0 | 0 | 0 | Not toxic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, P.; Romagnoli, R.; Cacciari, B.; Manfredini, S.; Padroni, C.; Brancale, A.; Ferla, S.; Hamel, E.; Corallo, D.; Aveic, S.; et al. Synthesis and Biological Evaluation of Highly Active 7-Anilino Triazolopyrimidines as Potent Antimicrotubule Agents. Pharmaceutics 2022, 14, 1191. https://doi.org/10.3390/pharmaceutics14061191
Oliva P, Romagnoli R, Cacciari B, Manfredini S, Padroni C, Brancale A, Ferla S, Hamel E, Corallo D, Aveic S, et al. Synthesis and Biological Evaluation of Highly Active 7-Anilino Triazolopyrimidines as Potent Antimicrotubule Agents. Pharmaceutics. 2022; 14(6):1191. https://doi.org/10.3390/pharmaceutics14061191
Chicago/Turabian StyleOliva, Paola, Romeo Romagnoli, Barbara Cacciari, Stefano Manfredini, Chiara Padroni, Andrea Brancale, Salvatore Ferla, Ernest Hamel, Diana Corallo, Sanja Aveic, and et al. 2022. "Synthesis and Biological Evaluation of Highly Active 7-Anilino Triazolopyrimidines as Potent Antimicrotubule Agents" Pharmaceutics 14, no. 6: 1191. https://doi.org/10.3390/pharmaceutics14061191
APA StyleOliva, P., Romagnoli, R., Cacciari, B., Manfredini, S., Padroni, C., Brancale, A., Ferla, S., Hamel, E., Corallo, D., Aveic, S., Milan, N., Mariotto, E., Viola, G., & Bortolozzi, R. (2022). Synthesis and Biological Evaluation of Highly Active 7-Anilino Triazolopyrimidines as Potent Antimicrotubule Agents. Pharmaceutics, 14(6), 1191. https://doi.org/10.3390/pharmaceutics14061191