
Raman Mapping-Based Reverse Engineering Facilitates Development of Sustained-Release Nifedipine Tablet




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
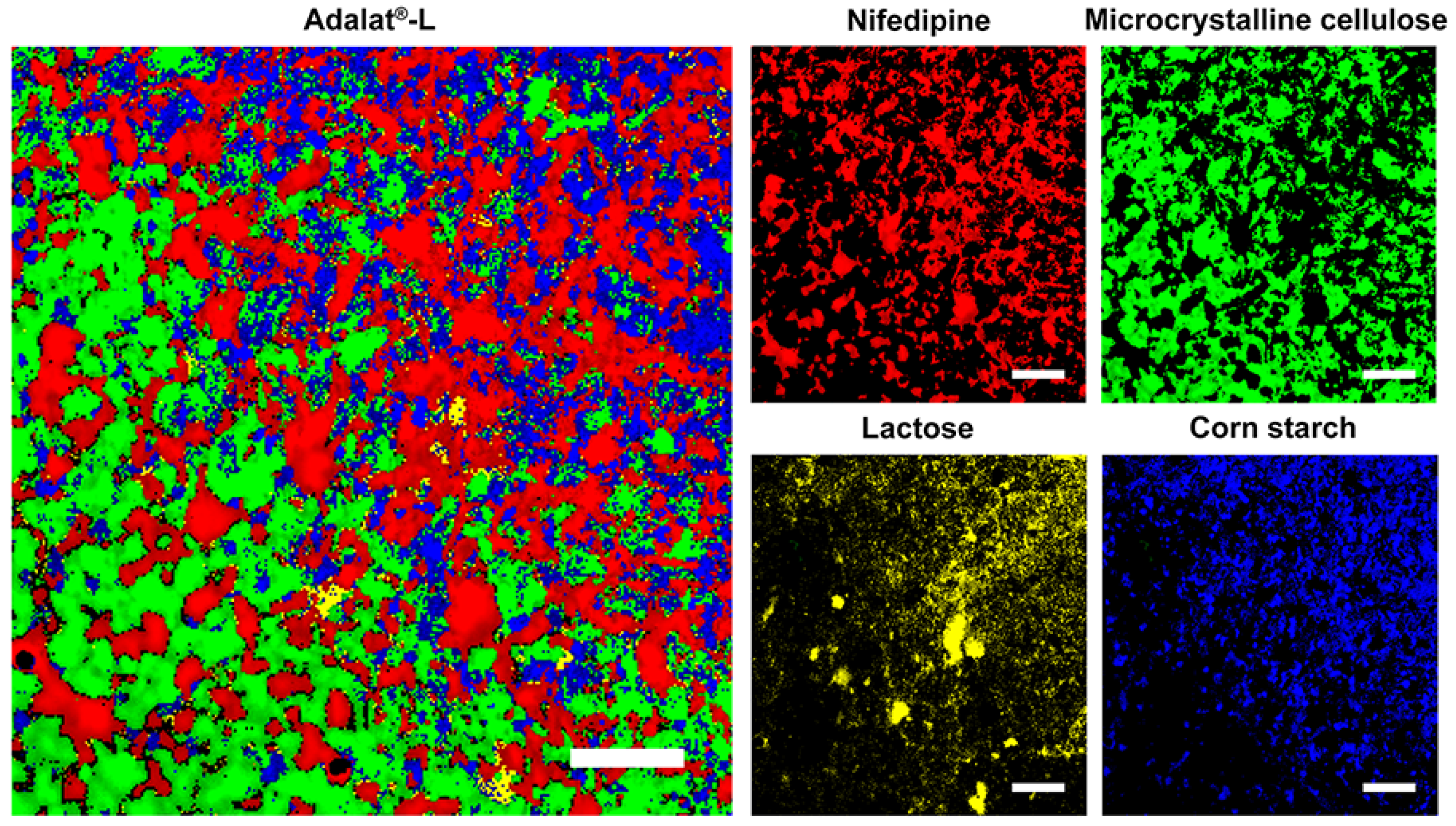
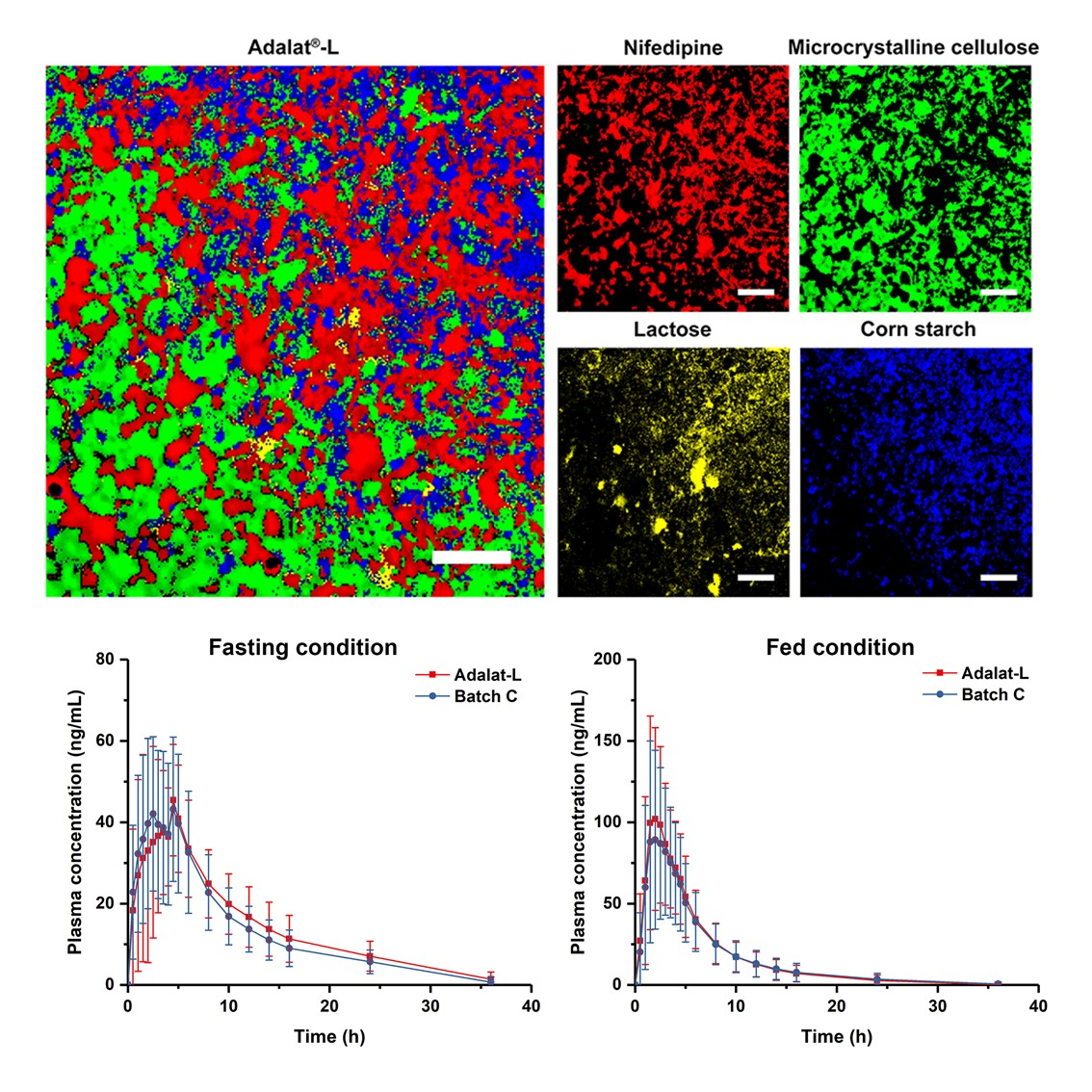
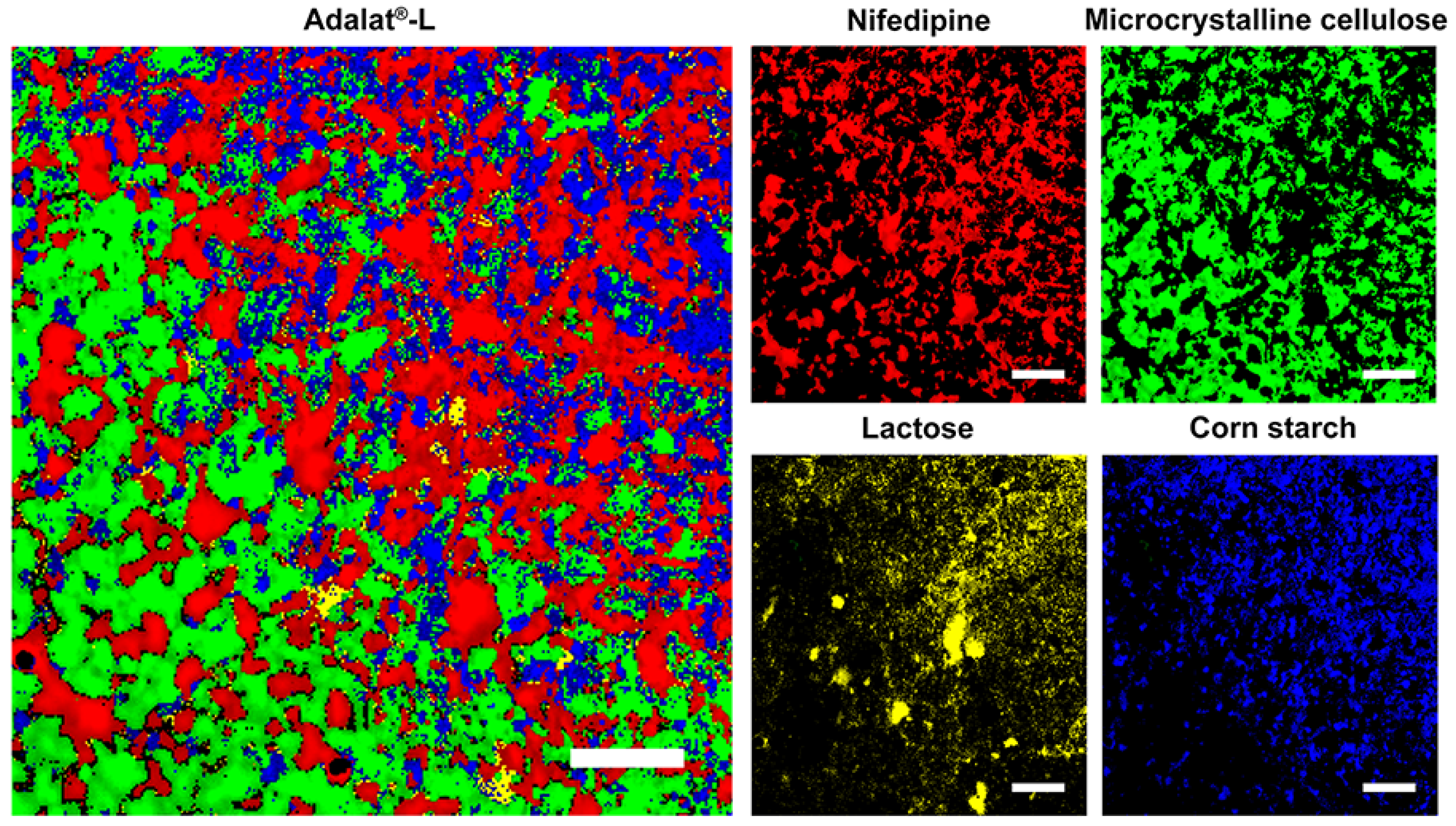
2.2. Raman Mapping of Adalat®-L
2.3. Preparation of Nifedipine Sustained-Release Tablets
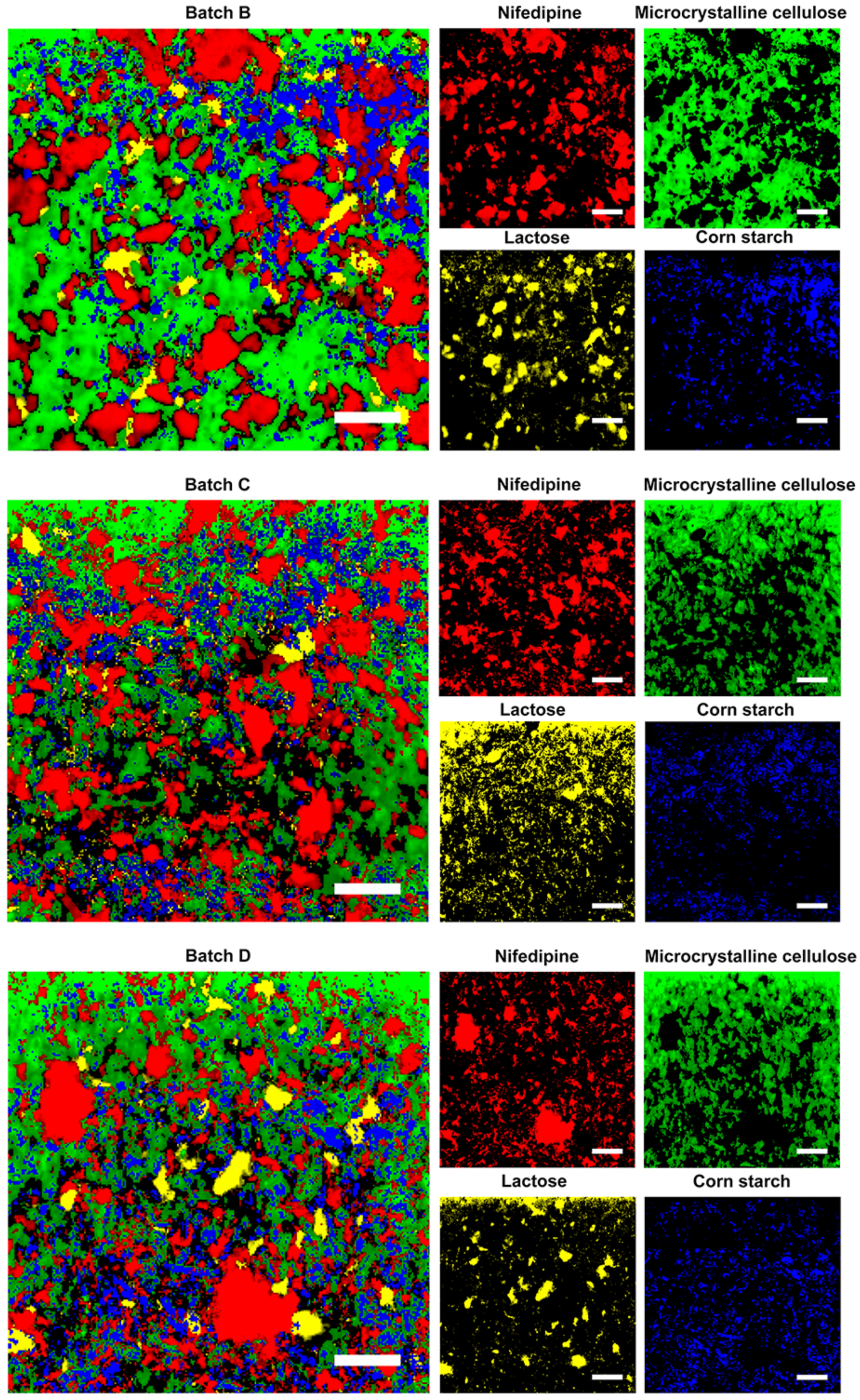
2.4. Validation of the Prepared Tablets by Raman Mapping
2.5. In Vitro Dissolution Studies
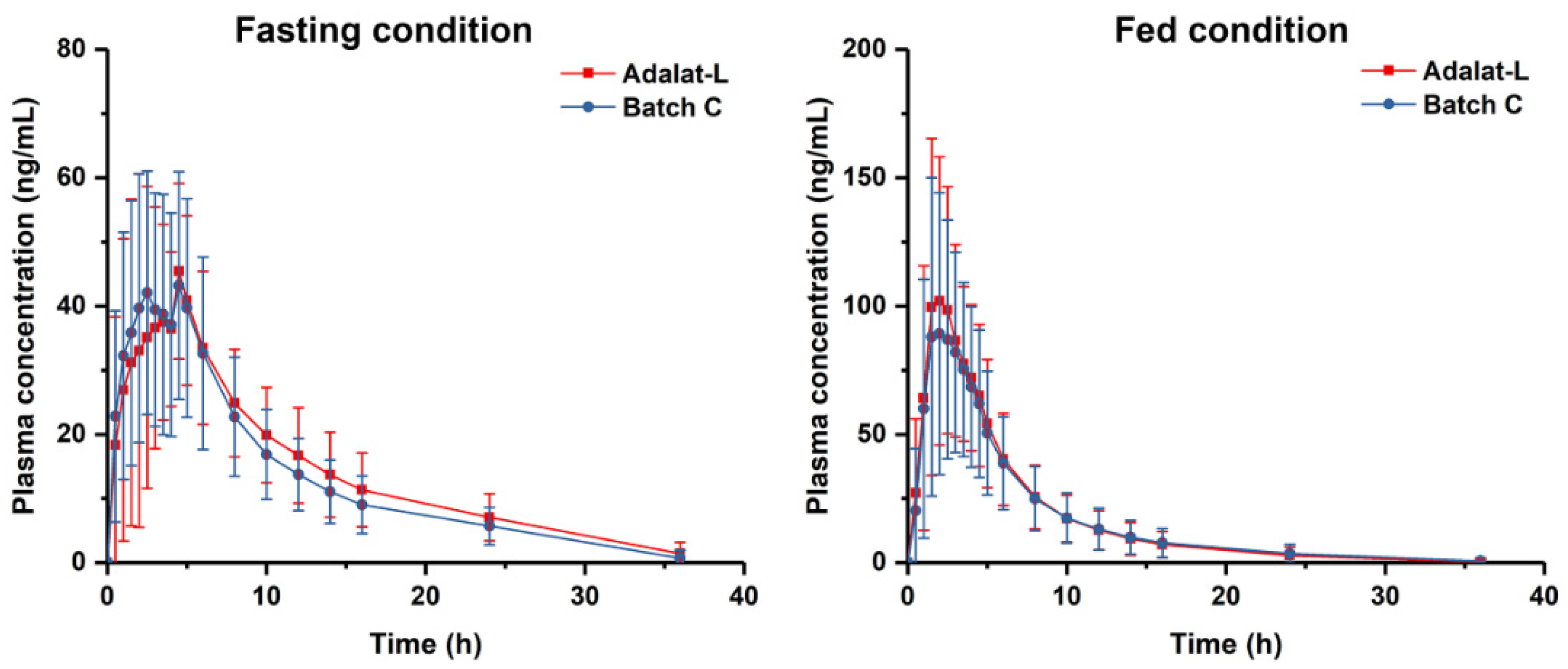
2.6. Bioequivalence Studies
3. Results and Discussion
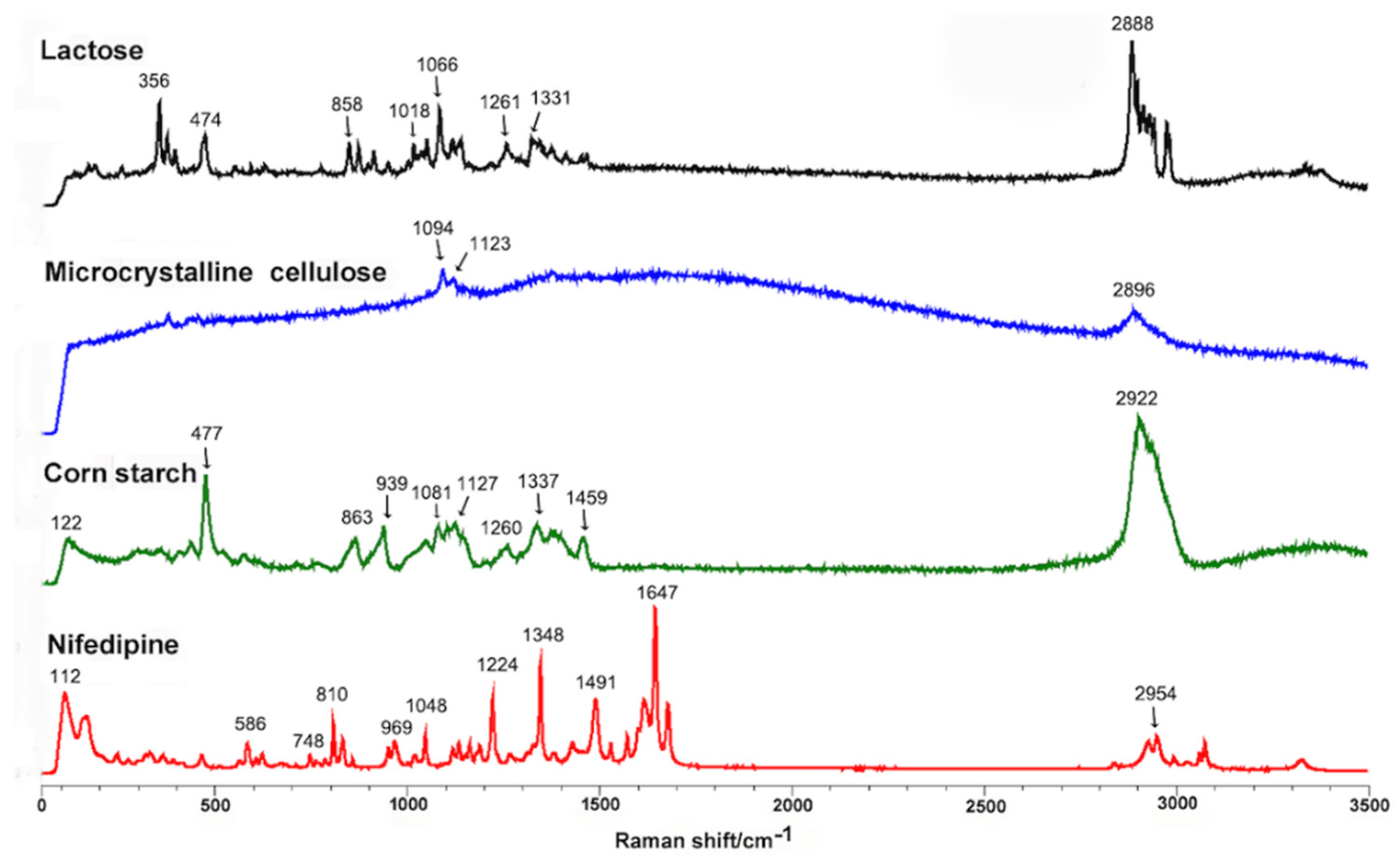
3.1. Analysis of Adalat®-L by Laser Raman Spectroscopy
3.2. Comminution and Particle Size Control of Nifedipine
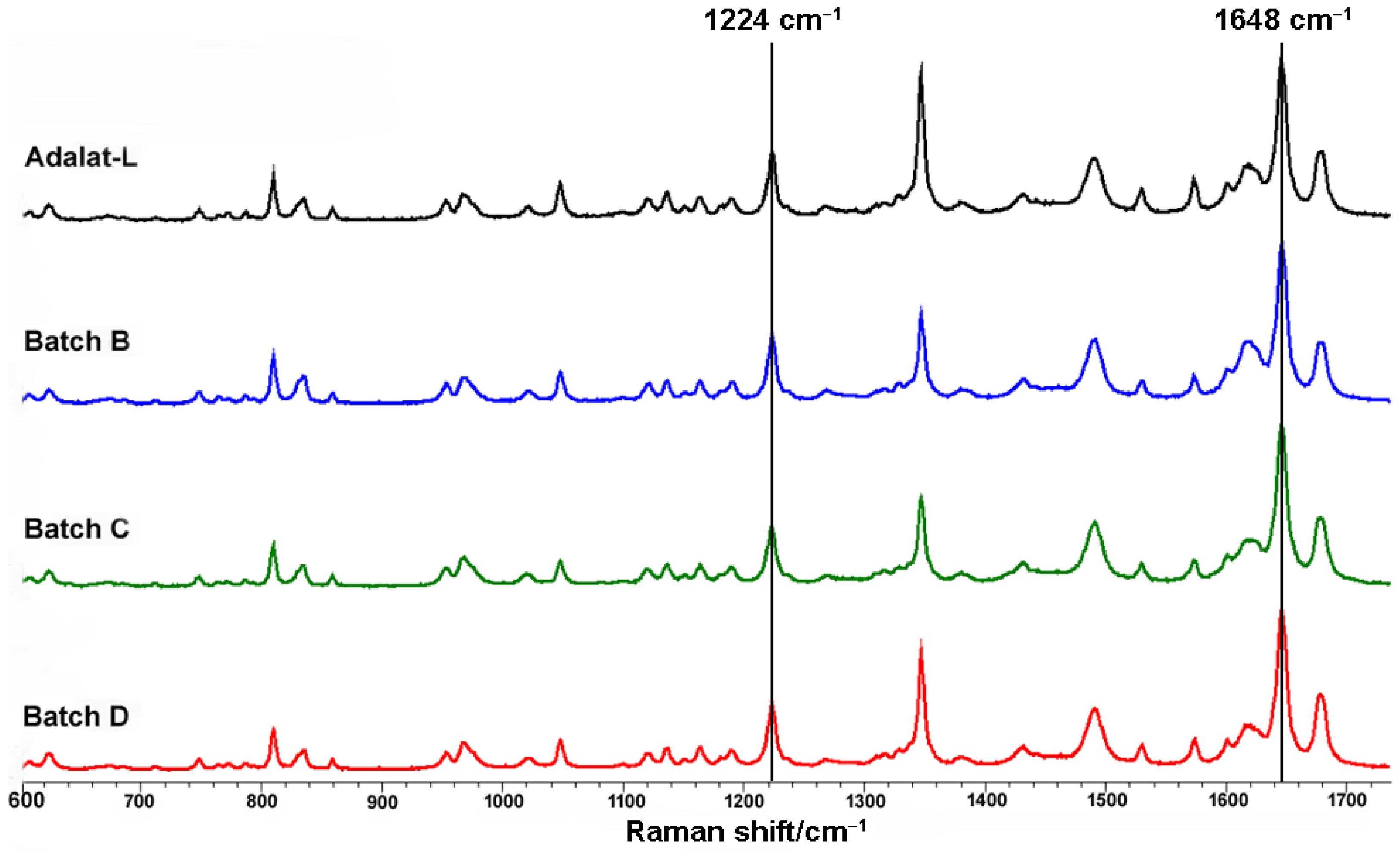
3.3. Validation by Raman Mapping of Prepared Tablets
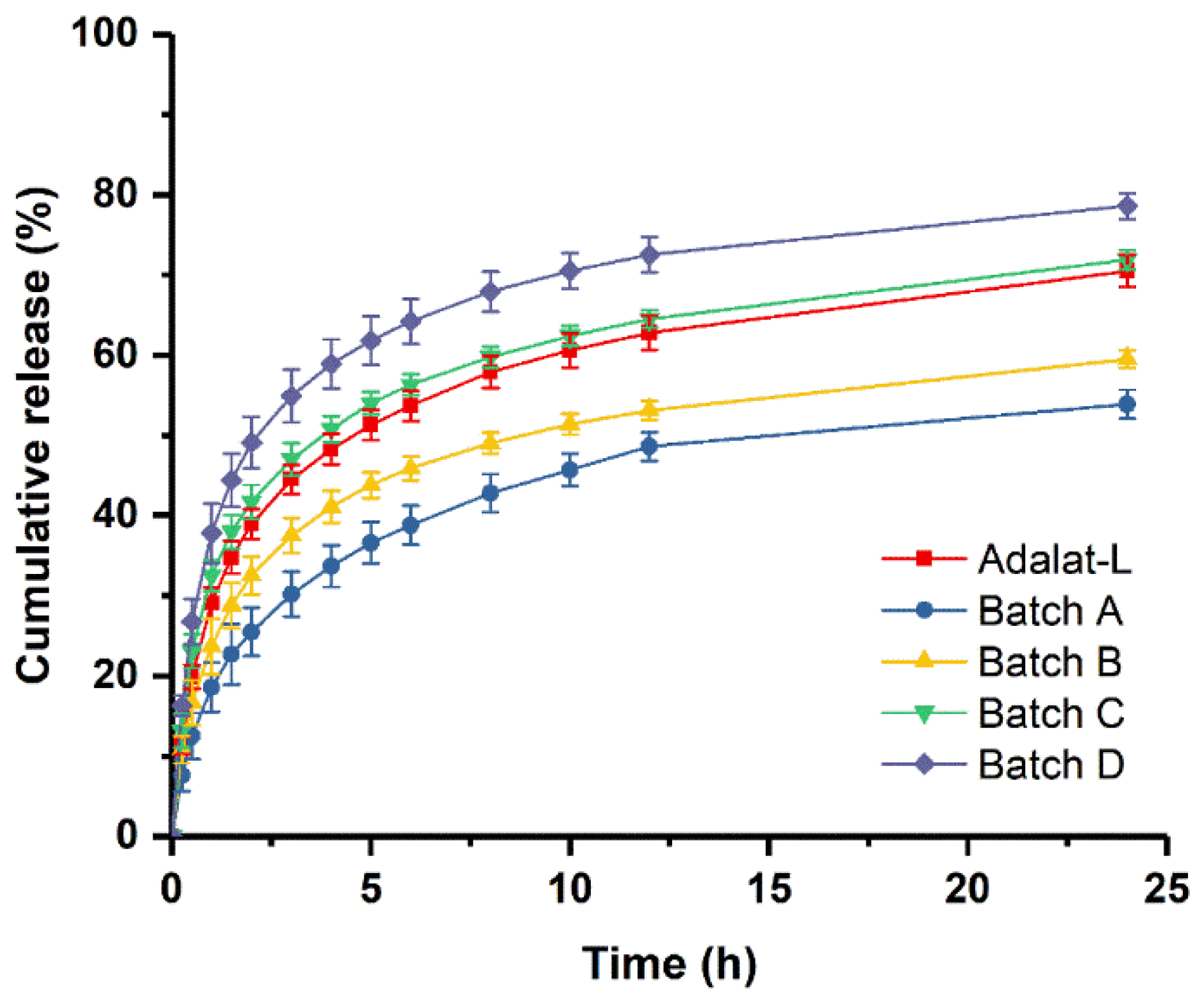
3.4. In Vitro Dissolution Studies
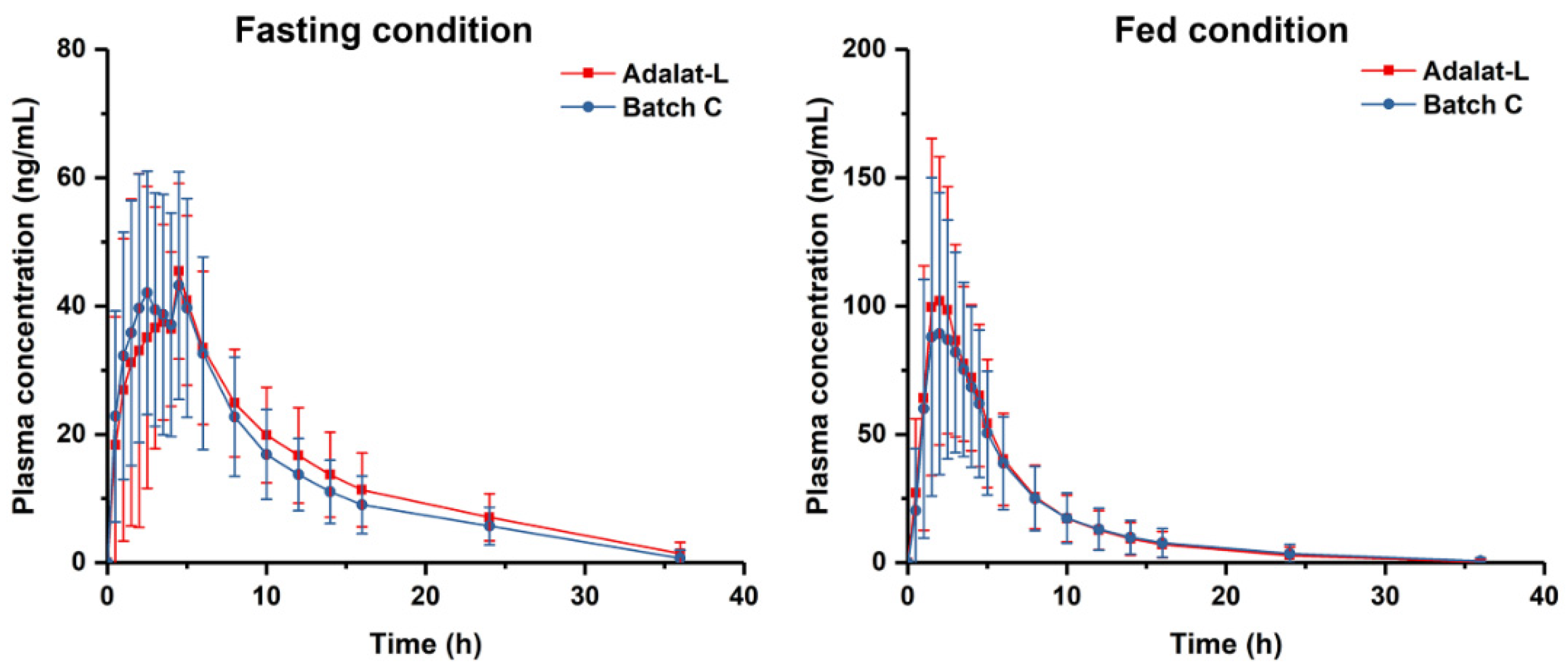
3.5. Bioequivalence Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wouters, O.J.; Kanavos, P.G.; McKee, M. Comparing generic drug markets in Europe and the United States: Prices, volumes, and spending. Milbank Q. 2017, 95, 554–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holman, A. Is Bioequivalence a Sufficient Measure of Equivalence? J. Leg. Med. 2019, 39, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Mishuk, A.U.; Qian, J.; Howard, J.N.; Harris, I.; Frank, G.; Kiptanui, Z.; Hansen, R. The association between patient sociodemographic characteristics and generic drug use: A systematic review and meta-analysis. J. Manag. Care Spec. Pharm. 2018, 24, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Meredith, P. Bioequivalence and other unresolved issues in generic drug substitution. Clin. Ther. 2003, 25, 2875–2890. [Google Scholar] [CrossRef]
- Yu, L.X. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2008, 25, 781–791. [Google Scholar] [CrossRef]
- Suarez-Sharp, S.; Li, M.; Duan, J.; Shah, H.; Seo, P. Regulatory experience with in vivo in vitro correlations (IVIVC) in new drug applications. AAPS J. 2016, 18, 1379–1390. [Google Scholar] [CrossRef]
- Lionberger, R.A.; Lee, S.L.; Lee, L.; Raw, A.; Yu, L.X. Quality by design: Concepts for ANDAs. AAPS J. 2008, 10, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Iacocca, R.G.; Burcham, C.L.; Hilden, L.R. Particle engineering: A strategy for establishing drug substance physical property specifications during small molecule development. J. Pharm. Sci. 2010, 99, 51–75. [Google Scholar] [CrossRef]
- Shekunov, B.Y.; Chattopadhyay, P.; Tong, H.H.; Chow, A.H. Particle size analysis in pharmaceutics: Principles, methods and applications. Pharm. Res. 2007, 24, 203–227. [Google Scholar] [CrossRef]
- Overhoff, K.A.; Johnston, K.P.; Williams III, R.O. Improvement of dissolution rate of poorly water soluble drugs using a new particle engineering process: Spray freezing into liquid. ACS Symp. Ser. 2006, 924, 305–319. [Google Scholar] [CrossRef]
- Bukovec, P.; Benkič, P.; Smrkolj, M.; Vrečzer, F. Effect of crystal habit on the dissolution behaviour of simvastatin crystals and its relationship to crystallization solvent properties. Die Pharm. Int. J. Pharm. Sci. 2016, 71, 263–268. [Google Scholar] [CrossRef]
- Hu, J.; Johnston, K.P.; Williams III, R.O. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev. Ind. Pharm. 2004, 30, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, F.; Garekani, H.A.; Goli, F. Tableting of Eudragit RS and propranolol hydrochloride solid dispersion: Effect of particle size, compaction force, and plasticizer addition on drug release. Drug Dev. Ind. Pharm. 2004, 30, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Eksi-Kocak, H.; Tamer, S.I.; Yilmaz, S.; Eryilmaz, M.; Boyaci, I.H.; Tamer, U. Quantification and spatial distribution of salicylic acid in film tablets using FT-Raman mapping with multivariate curve resolution. Asian J. Pharm. Sci. 2018, 13, 155–162. [Google Scholar] [CrossRef]
- Ibrahim, M.; Zhang, J.; Repka, M.; Chen, R. Characterization of the solid physical state of API and its distribution in pharmaceutical hot melt extrudates using terahertz Raman imaging. AAPS PharmSciTech 2019, 20, 62. [Google Scholar] [CrossRef]
- Sacré, P.-Y.; De Bleye, C.; Chavez, P.-F.; Netchacovitch, L.; Hubert, P.; Ziemons, E. Data processing of vibrational chemical imaging for pharmaceutical applications. J. Pharm. Biomed. Anal. 2014, 101, 123–140. [Google Scholar] [CrossRef]
- Ewing, A.V.; Kazarian, S.G. Recent advances in the applications of vibrational spectroscopic imaging and mapping to pharmaceutical formulations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 197, 10–29. [Google Scholar] [CrossRef]
- Cailletaud, J.; De Bleye, C.; Dumont, E.; Sacré, P.-Y.; Netchacovitch, L.; Gut, Y.; Boiret, M.; Ginot, Y.-M.; Hubert, P.; Ziemons, E. Critical review of surface-enhanced Raman spectroscopy applications in the pharmaceutical field. J. Pharm. Biomed. Anal. 2018, 147, 458–472. [Google Scholar] [CrossRef]
- Wang, W.-t.; Zhang, H.; Yuan, Y.; Guo, Y.; He, S.-x. Research progress of Raman spectroscopy in drug analysis. AAPS PharmSciTech 2018, 19, 2921–2928. [Google Scholar] [CrossRef]
- Willett, D.R.; Rodriguez, J.D. Quantitative Raman assays for on-site analysis of stockpiled drugs. Anal. Chim. Acta. 2018, 1044, 131–137. [Google Scholar] [CrossRef]
- Matthews, T.E.; Coffman, C.; Kolwyck, D.; Hill, D.; Dickens, J.E. Enabling robust and rapid raw material identification and release by handheld raman spectroscopy. PDA J. Pharm. Sci. Technol. 2019, 73, 356–372. [Google Scholar] [CrossRef] [PubMed]
- Okotrub, K.A.; Zykova, V.A.; Adichtchev, S.V.; Surovtsev, N.V. Deciphering the orientation of lipid molecules by principal component analysis of Raman mapping data. Analyst 2020, 145, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Meek, C.; Hoe, J.; Evans, J.; Thurman, R.; Ashworth, L.; Leff, R. Raman Spectroscopy: A Sensitive and Specific Technique for Determining the Accuracy of Compounded Pharmaceutical Formulations. J. Pediatr. Pharmacol. Ther. 2016, 21, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Gallimore, P.J.; Davidson, N.M.; Kalberer, M.; Pope, F.D.; Ward, A.D. 1064 nm Dispersive Raman microspectroscopy and optical trapping of pharmaceutical aerosols. Anal. Chem. 2018, 90, 8838–8844. [Google Scholar] [CrossRef] [PubMed]
- Galata, D.L.; Zsiros, B.; Mészáros, L.A.; Nagy, B.; Szabó, E.; Farkas, A.; Nagy, Z.K. Raman mapping-based non-destructive dissolution prediction of sustained-release tablets. J. Pharm. Biomed. Anal. 2022, 212, 114661. [Google Scholar] [CrossRef]
- Navin, C.V.; Tondepu, C.; Toth, R.; Lawson, L.S.; Rodriguez, J.D. Quantitative determinations using portable Raman spectroscopy. J. Pharm. Biomed. Anal. 2017, 136, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Paiva, E.M.; da Silva, V.H.; Poppi, R.J.; Pereira, C.F.; Rohwedder, J.J. Comparison of macro and micro Raman measurement for reliable quantitative analysis of pharmaceutical polymorphs. J. Pharm. Biomed. Anal. 2018, 157, 107–115. [Google Scholar] [CrossRef]
- Ma, X.; Sun, X.; Wang, H.; Wang, Y.; Chen, D.; Li, Q. Raman spectroscopy for Pharmaceutical quantitative analysis by Low-Rank Estimation. Front. Chem. 2018, 6, 400. [Google Scholar] [CrossRef] [Green Version]
- Paiva, E.M.; Ribessi, R.L.; Pereira, C.F.; Rohwedder, J.J.R. Low-frequency Raman spectrophotometer with wide laser illumination on the sample: A tool for pharmaceutical analytical analysis. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 228, 117798. [Google Scholar] [CrossRef]
- Smith, M.; Logan, M.; Bazley, M.; Blanchfield, J.; Stokes, R.; Blanco, A.; McGee, R. A Semi-quantitative method for the detection of fentanyl using surface-enhanced Raman scattering (SERS) with a handheld Raman instrument. J. Forensic Sci. 2021, 66, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.A.; Sacré, P.-Y.; Coïc, L.; De Bleye, C.; Dumont, E.; Bouklouze, A.; Hubert, P.; Marini, R.; Ziemons, E. Quantitation of active pharmaceutical ingredient through the packaging using Raman handheld spectrophotometers: A comparison study. Talanta 2020, 207, 120306. [Google Scholar] [CrossRef] [PubMed]
- Čapková, T.; Pekárek, T.; Hanulíková, B.; Matějka, P. Application of reverse engineering in the field of pharmaceutical tablets using Raman mapping and chemometrics. J. Pharm. Biomed. Anal. 2022, 209, 114496. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Urasaki, T.; Hondo, S.; Murahashi, N.; Yonemochi, E.; Terada, K. Evaluation of the crystalline and amorphous states of drug products by nanothermal analysis and Raman imaging. J. Pharm. Biomed. Anal. 2013, 75, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Šašić, S.; Mehrens, S. Raman chemical mapping of low-content active pharmaceutical ingredient formulations. III. Statistically optimized sampling and detection of polymorphic forms in tablets on stability. Anal. Chem. 2012, 84, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Shao, Z.; Wang, Q.; Hu, X.; Yu, D. Quantitation of polymorphic impurity in entecavir polymorphic mixtures using powder X-ray diffractometry and Raman spectroscopy. J. Pharm. Biomed. Anal. 2018, 158, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Dadou, S.M.; El-Barghouthi, M.I.; Antonijevic, M.D.; Chowdhry, B.Z.; Badwan, A.A. Elucidation of the controlled-release behavior of metoprolol succinate from directly compressed xanthan gum/chitosan polymers: Computational and experimental studies. ACS Biomater. Sci. Eng. 2020, 6, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Čapková-Helešicová, T.; Pekárek, T.; Schöngut, M.; Matějka, P. New designed special cells for Raman mapping of the disintegration process of pharmaceutical tablets. J. Pharm. Biomed. Anal. 2019, 168, 113–123. [Google Scholar] [CrossRef]
- Esmonde-White, K.A.; Cuellar, M.; Uerpmann, C.; Lenain, B.; Lewis, I.R. Raman spectroscopy as a process analytical technology for pharmaceutical manufacturing and bioprocessing. Anal. Bioanal. Chem. 2017, 409, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Woo, Y.-A. Coating process optimization through in-line monitoring for coating weight gain using Raman spectroscopy and design of experiments. J. Pharm. Biomed. Anal. 2018, 154, 278–284. [Google Scholar] [CrossRef]
- Lim, Y.-I.; Han, J.; Woo, Y.-A.; Kim, J.; Kang, M.J. Rapid quantitation of atorvastatin in process pharmaceutical powder sample using Raman spectroscopy and evaluation of parameters related to accuracy of analysis. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 200, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhong, L.-m.; Cao, Y.-b.; Chen, H.; Lu, F. Quantitative analysis of excipient dominated drug formulations by Raman spectroscopy combined with deep learning. Anal. Methods 2021, 13, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.-Y.; Yang, X.; Mao, Z.-L.; Yao, X.-Y.; Chen, X.-B. Raman study of impurity influence on active center in artemisinin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 221, 117206. [Google Scholar] [CrossRef] [PubMed]
- Hegasy, A.; Ramsch, K.D. Solid Medicament Formulations Containing Nifedipine, and Processes for Their Preparation. U.S. Patent 5,264,446, 1 June 1992. [Google Scholar]
- Moore, J. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996, 20, 64–75. [Google Scholar]
- Ishida, H.; Wu, T.; Yu, L. Sudden rise of crystal growth rate of nifedipine near Tg without and with polyvinylpyrrolidone. J. Pharm. Sci. 2007, 96, 1131–1138. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Amount (mg) |
|---|---|
| Nifedipine | 20.0 |
| Polysorbate 80 | 0.4 |
| Microcrystalline cellulose | 26.0 |
| Lactose | 8.0 |
| Corn starch (for blending) | 12.4 |
| Corn starch (for starch slurry) | 12.4 |
| Magnesium stearate | 0.8 |
| SUM | 80.0 |
| D90 (μm) | D50 (μm) | D10 (μm) | |
|---|---|---|---|
| Nifedipine | 150 | 118 | 30.7 |
| Components | Nifedipine | Microcrystalline Cellulose | Lactose | Corn Starch |
|---|---|---|---|---|
| Proportion (%) | 33.94 | 33.02 | 1.40 | 31.65 |
| Batch | Particle Size | ||
|---|---|---|---|
| D (90) (μm) | D (50) (μm) | D (10) (μm) | |
| A | 145.2 ± 0.6 | 69.3 ± 0.6 | 12.1 ± 0.5 |
| B | 96.1 ± 0.7 | 38.2 ± 0.6 | 8.4 ± 0.6 |
| C | 62.0 ± 0.6 | 26.3 ± 0.2 | 5.5 ± 0.3 |
| D | 38.9 ± 0.5 | 18.8 ± 0.4 | 6.1 ± 0.4 |
| Particle Size | |||
|---|---|---|---|
| D (90) (μm) | D (50) (μm) | D (10) (μm) | |
| Adalat®-L | 150 | 118 | 30.7 |
| Batch B | 173 | 118 | 52.2 |
| Batch C | 147 | 121 | 47.5 |
| Batch D | 108 | 103 | 32.7 |
| Condition | Pharmacokinetic Parameters | Mean and Ratio | 90% Confidence Interval | ||
|---|---|---|---|---|---|
| Batch C (T) | Adalat®-L (R) | (T/R)% | |||
| Fasting (N = 28) | Cmax (ng/mL) | 56.4 ± 15.4 | 54.9 ± 16.3 | 102.75 | 92.15~114.56 |
| AUC0–t (ng/mL·h) | 443.4 ± 150.0 | 478.1 ± 156.2 | 92.73 | 86.67~99.21 | |
| AUC0–∞ (ng/mL·h) | 501.6 ± 147.8 | 536.6 ± 162.8 | 93.48 | 87.55~99.80 | |
| Fed (N = 28) | Cmax (ng/mL) | 107.5 ± 46.5 | 119.8 ± 44.1 | 89.67 | 81.95~98.11 |
| AUC0–t (ng/mL·h) | 539.6 ± 303.4 | 569.3 ± 283.0 | 94.78 | 88.39~101.63 | |
| AUC0–∞ (ng/mL·h) | 569.1 ± 314.9 | 597.3 ± 297.3 | 95.28 | 88.97~102.04 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, N.; Chang, L.; Lu, Y.; Wu, W. Raman Mapping-Based Reverse Engineering Facilitates Development of Sustained-Release Nifedipine Tablet. Pharmaceutics 2022, 14, 1052. https://doi.org/10.3390/pharmaceutics14051052
Sun N, Chang L, Lu Y, Wu W. Raman Mapping-Based Reverse Engineering Facilitates Development of Sustained-Release Nifedipine Tablet. Pharmaceutics. 2022; 14(5):1052. https://doi.org/10.3390/pharmaceutics14051052
Chicago/Turabian StyleSun, Ningyun, Liang Chang, Yi Lu, and Wei Wu. 2022. "Raman Mapping-Based Reverse Engineering Facilitates Development of Sustained-Release Nifedipine Tablet" Pharmaceutics 14, no. 5: 1052. https://doi.org/10.3390/pharmaceutics14051052
APA StyleSun, N., Chang, L., Lu, Y., & Wu, W. (2022). Raman Mapping-Based Reverse Engineering Facilitates Development of Sustained-Release Nifedipine Tablet. Pharmaceutics, 14(5), 1052. https://doi.org/10.3390/pharmaceutics14051052