2. Materials and Methods
2.1. Procedure for Glyconucleolipids Synthesis
All reactions were performed under an argon atmosphere. Unless otherwise stated, yields were determined on chromatographically and spectroscopically (nuclear magnetic resonance (1H-NMR)) homogeneous materials. All reagent-grade chemicals were purchased from commercial suppliers and used as received, unless otherwise stated. 1H-NMR and 13C-NMR spectra were acquired at 293 K (unless otherwise indicated) on a Bruker Avance 300 (1H: 300 MHz, 13C: 75.46 MHz) spectrometer with residual CHCl3 used as internal reference (7.26 ppm). The chemical shifts (δ) and coupling constants (J) were indicated in ppm and Hz, respectively. The following abbreviations were applied to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad. Fourier Transform InfraRed (FT-IR) spectra were acquired on a Perkin-Elmer spectrometer Spectrum two (UATR two). A Waters Micromass ZQ instrument equipped with an electrospray source (positive and/or negative mode) was used for the electrospray ionization high-resolution mass spectrometry (ESI HRMS) analyses. Matrix-assisted laser desorption ionization—time of flight (MALDI TOF) mass spectrometric analyses, using 3,4-dihydroxybenzoic acid as the matrix, were performed on a PerSeptive Biosystems Voyager-De Pro MALDI mass spectrometer in the Linear mode. Analytical thin-layer chromatography was performed using silica gel 60 F254 pre-coated plates (Merck Millipore, Saint Quentin Fallavier, France) with a revelation by UV light, potassium permanganate or sulfuric acid solutions. Silica gel (0.043–0.063 mm) was used for flash chromatography.
2.2. Synthesis of Compound 1: 5′-Azido-5′-deoxythymidine
Triphenylphosphine (1.2 equiv., 2.6 g, 9.91 mmol), sodium azide (5 equiv., 2.68 g, 41.3 mmol) and carbon tetrabromide (1.2 equiv., 3.29 g, 9.91 mmol) were sequentially added to a solution of thymidine (1 equiv., 2 g, 8.26 mmol), in anhydrous dimethylformamide (DMF) (40 mL) and under argon. The mixture was stirred for 24 h at room temperature until completion of the reaction. The reaction was then quenched by the addition of an aq. NaHCO
3 solution (50 mL) and the resulting mixture diluted with water (30 mL). Three extractions with dichloromethane (DCM; 3 × 30 mL) were applied to the aqueous phase, before drying the combined organic phases over Na
2SO
4 and concentrating to dryness under vacuum. A purification of the resulting crude by flash chromatography on a silica gel (DCM/MeOH, gradient 99:1 to 95:5) was required to obtain the expected compound
1 as a white powder (1.63 g, 74%). NMR data were consistent with the literature [
33].
Rf: 0.37 (DCM/MeOH, 95:5).
1H NMR (300 MHz, MeOD) δ (ppm): 1.90 (d,
J = 0.9 Hz, 3H, CH
3 thymidine), 2.25–2.33 (m, 2H, H
2′), 3.56 (dd,
J = 3.9 Hz, 13.2 Hz, 1H, H
5a′), 3.66 (dd,
J = 5.1 Hz, 13.5 Hz, 1H, H
5b′), 3.96 (q,
J = 3.9 Hz, 9 Hz, 2H, H
4′), 4.32–4.37 (m, 1H, H
3′), 6.27 (t,
J = 6.9 Hz, 1H, H
1′), 7.54 (s, 1H, H
6).
2.3. Synthesis of Compound 2: 3-(5′-Azido-5′-deoxythymidin-3-yl) Methyl Propanoate
DBU (diazabicycloundecene) (1.55 equiv., 433 µL, 2.90 mmol) and methyl acrylate (1.55 equiv., 261 µL, 2.90 mmol) were sequentially added to a solution of 1 (1 equiv., 0.500 g, 1.87 mmol) in anhydrous DMF (2 mL) and under argon. The mixture was stirred overnight at 60 °C until the completion of the reaction and followed by concentration to dryness under vacuum. The resulting crude was purified by flash chromatography on a silica gel (DCM/MeOH, gradient 98:2 to 96:4) to obtain the expected compound 2 as a colorless oil (0.436 g, 66%). Rf: 0.43 (DCM/MeOH, 95:5). 1H NMR (300 MHz, CDCl3) δ (ppm): 1.89 (d, J = 0.9 Hz, 3H, CH3 thymidine), 2.09–2.23 (m, 1H, H2′a), 2.28–2.41 (m, 1H, H2′b), 2.59 (t, J = 7.5 Hz, 2H, HB linker), 3.55 (dd, J = 3.6 Hz, 13.2 Hz, 1H, H5a′), 3.68 (dd, J = 3.0 Hz, 12.9 Hz, 1H, H5b′), 3.62 (s, 3H, HC linker), 4.01 (q, J = 3.6 Hz, 1H, H4′), 4.18 (t, J = 7.8 Hz, 2H, HA linker), 4.33–4.43 (m, 1H, H3′), 6.26 (t, J = 6.9 Hz, 1H, H1′), 7.34 (s, 1H, H6). 13C NMR (75.46 MHz, CDCl3) δ (ppm): 13.3 (CH3 thymidine), 32.1 (CH2-B linker), 37.2 (CH2-A linker), 40.2 (C2′), 51.9 (CH3-C linker), 52.2 (C5′), 71.4 (C3′), 84.5 (C4′), 85.5 (C1′), 110.4 (C5), 133.9 (C6), 150.6 (C=O thymidine), 163.2 (C=O thymidine), 171.9 (C=O linker).
2.4. Synthesis of Compound 3: 3-[3′-O-(tert-butyldimethylsilyl)-5′-azido-5′-deoxythymidin-3-yl] Methyl Propanoate
TBDMSCl (tert-Butyldimethylsilyl chloride) (1.2 equiv., 0.220 g, 1.46 mmol) and imidazole (1.2 equiv., 0.099 g, 1.46 mmol) were sequentially added to a solution of 2 (1 equiv., 0.430 mg, 1.22 mmol) in anhydrous DMF (2.6 mL) and under argon. The mixture was stirred overnight at room temperature until the completion of the reaction. DCM was added and the precipitate formed was filtered. The mixture was then diluted with toluene (100 mL), and DMF was co-evaporated under vacuum to obtain a yellow oil. The resulting crude was purified by flash chromatography on a silica gel (petroleum ether/EtOAc, 70:30) to obtain the expected compound 3 as a colorless oil (0.491 g, 86%). Rf: 0.63 (petroleum ether/EtOAc, 70:30). 1H NMR (300 MHz, CDCl3) δ (ppm): −0.05 (s, 6H, CH3-Si), 0.75 (s, 9H, tBu), 1.80 (s, 3H, CH3 thymidine), 1.97–2.24 (m, 2H, H2′), 2.49 (t, J = 7.5 Hz, 2H, HB linker), 3.38 (dd, J = 3.9 Hz, 13.5 Hz, 1H, H5a′), 3.48–3.59 (m, 4H, HC linker et H5b′), 3.78–3.85 (m, 1H, H4′), 4.09 (t, J = 7.8Hz, 2H, HA linker), 4.19–4.27 (m, 1H, H3′), 6.13 (t, J = 6.6 Hz, 1H, H1′), 7.20 (s, 1H, H6). 13C NMR (75.46 MHz, CDCl3) δ (ppm): −5.17 (CH3-Si), −4.9 (CH3-Si), 13.0 (CH3 thymidine), 17.6 (Cq tBu), 25.4 (CH3 tBu), 31.8 (CH2-B linker), 36.8 (CH2-A linker), 40.4 (C2′), 51.4 (CH3-C linker), 51.5 (C5′), 71.5 (C3′), 84.6 (C4′), 85.3 (C1′), 110.0 (C5), 133.4 (C6), 150.3 (C=O thymidine), 162.7 (C=O thymidine), 171.3 (C=O linker).
2.5. Synthesis of Compound 4: Propargyl N-dodecylcarbamate
Triethylamine (1.05 equiv., 0.229 g, 2.27 mmol) and drop by drop propargyl chloroformate (1 equiv., 0.256 g, 2.16 mmol) were sequentially added to a solution of dodecylamine (1 equiv., 0.400 g, 2.16 mmol) in anhydrous DCM (6 mL) and under argon. The mixture was stirred for 3 h at 0 °C until the completion of the reaction, followed by concentration to dryness under vacuum. The resulting crude was purified by flash chromatography on a silica gel (petroleum ether/EtOAc, 90:10) to obtain the expected compound 4 as a colorless oil (0.333 g, 97%). Rf: 0.61 (petroleum ether/EtOAc, 90:10). 1H NMR (300 MHz, CDCl3) δ (ppm): 0.72–0.85 (m, 3H, CH3 lipid), 1.19 (s, 18H, CH2 lipid), 1.37–1.52 (m, 2H, CH2-CH3 lipid), 2.40 (s, 1H, CH propargyl), 3.03–3.17 (m, 2H, CH2-NH lipid), 4.60 (s, 2H, CH2 propargyl), 4.99–5.24 (bs, 1H, NH lipid). 13C NMR (75.46 MHz, CDCl3) δ (ppm): 14.1 (CH3 lipid), 22.7 (CH2 lipid), 26.7 (CH2 lipid), 29.3 (CH2 lipid), 29.3 (CH2 lipid), 29.5 (CH2 lipid), 29.6 (CH2 lipid), 29.6 (CH2 lipid), 29.6 (CH2 lipid), 29.8 (CH2-CH3 lipid), 31.9 (CH2 lipid), 41.2 (CH2-NH lipid), 52.2 (CH2 propargyl), 74.4 (CH propargyl), 78.4 (C propargyl), 155.5 (C=O lipid).
2.6. Synthesis of Compound 5: 3′-[3′-O-(tert-butyldimethylsilyl)-5′-{4-[(dodecylcarbamoyloxy)methyl]-1H-1,2,3-triazol-1-yl}-5′-deoxythymidin-3-yl] Methyl Propanoate
Compound 4 (1 equiv., 0.181 g, 0.68 mmol), copper (II) sulfate pentahydrate (0.1 equiv., 0.011 g, 0.07 mmol) and sodium ascorbate (0.2 equiv., 0.027 g, 0.14 mmol) were sequentially added to a solution of 3 (1 equiv., 0.316 g, 0.68 mmol) in a mixture of H2O/THF (tetrahydrofuran) (1:1, 5 mL). The mixture was stirred overnight at 60 °C until the completion of the reaction, followed by concentration to dryness under vacuum to obtain a green product. This was washed several times with EDTA (Ethylenediaminetetraacetic acid) until colorless organic phases were obtained, followed by concentration to dryness under vacuum. The resulting crude was purified by flash chromatography on a silica gel (toluene/acetone, 80:20) to obtain the expected compound 5 as a white powder (0.433 g, 87%). Rf: 0.33 (toluene/acetone, 80:20). 1H NMR (300 MHz, CDCl3) δ (ppm): 0.04–0.07 (m, 6H, CH3-Si), 0.78–0.92 (m, 12H, CH3 lipid et tBu), 1.20 (m, 18H, CH2 lipid), 1.33–1.49 (m, 2H, CH2-CH3 lipid), 1.84 (s, 3H, CH3 thymidine), 2.00–2.77 (m, 2H, H2′), 2.58 (t, J = 7.2 Hz, 2H, HB linker), 3.03–3.14 (m, 2H, CH2-NH lipid), 3.62 (s, 3H, HC linker), 4.02–4.11 (m, 1H, H4′), 4.17 (t, J = 7.8 Hz, 2H, HA linker), 4.34–4.48 (m, 1H, H3′), 4.52–4.63 (m, 2H, H5′), 4.93–5.04 (m, 1H, NH lipid), 5.10 (s, 2H, O-CH2 triazole), 6.15 (t, J = 6.6 Hz, 1H, H1′), 6.60 (s, 1H, H6), 7.66 (s, 1H, CH triazole). 13C NMR (75.46 MHz, CDCl3) δ (ppm): −4.8 (CH3-Si), −4.6 (CH3-Si), 13.2 (CH3 thymidine), 14.1 (CH3 lipid), 17.8 (Cq tBu), 22.7 (CH2 lipid), 25.7 (CH3 tBu), 26.7 (CH2 lipid), 29.3 (CH2 lipid), 29.3 (CH2 lipid), 29.6 (CH2 lipid), 29.6 (CH2 lipid), 29.6 (CH2 lipid), 29.8 (CH2-CH3 lipid), 29.9 (CH2 lipid), 31.9 (CH2 lipid), 32.1 (CH2-B linker), 37.1 (CH2-A linker), 39.5 (C2′), 41.1 (CH2-NH lipid), 50.5 (C5′), 51.8 (CH3-C linker), 57.7 (O-CH2 triazole), 71.7 (C3′), 83.9 (C4′), 86.0 (C1′), 110.8 (C5), 125.6 (CH triazole), 133.8 (C6), 143.6 (C triazole), 150.5 (C=O thymidine), 156.2 (C=O lipid), 162.9 (C=O thymidine), 171.6 (C=O linker). HRMS (ESI) [M + H+]: calcd= 735.44712, found= 735.44576.
2.7. Synthesis of Compound 6: 3′-[3′-O-(tert-butyldimethylsilyl)-5′-{4-[(dodecylcarbamoyloxy) methyl]-1H-1,2,3-triazol-1-yl}-5′-deoxythymidin-3-yl] Propanoic Acid
Compound 5 (1 equiv., 0.431 g, 0.59 mmol) was dissolved in 1,4-dioxane (13 mL) before adding a solution of NaOH (1N) in MeOH (1.3 mL). The mixture was stirred overnight at room temperature until the completion of the reaction. The reaction mixture was treated with a solution of HCl (5%) to reach neutrality and then concentrated to dryness under vacuum. The resulting crude was purified by flash chromatography on a silica gel (DCM/MeOH, 90:10) to obtain the expected compound 6 (0.310 g, 73%). Rf: 0.26 (DCM/MeOH, 90:10). 1H NMR (300 MHz, CDCl3) δ (ppm): −0.05–0.05 (m, 6H, CH3-Si), 0.73–0.85 (m, 12H, CH3 lipid et tBu), 1.10–1.27 (m, 18H, CH2 lipid), 1.29–1.47 (m, 2H, CH2-CH3 lipid), 1.79 (s, 3H, CH3 thymidine), 2.07–2.21 (m, 2H, H2′), 2.37–2.51 (m, 2H, HB linker), 2.89–3.11 (m, 2H, CH2-NH lipid), 3.97–4.15 (m, 3H, H4′ et HA linker), 4.30–4.43 (m, 1H, H3′), 4.50–4.59 (m, 2H, H5′), 5.07 (s, 2H, O-CH2 triazole), 5.21–5.31 (m, 1H, NH lipid), 6.09 (t, J = 6.6 Hz, 1H, H1′), 6.61 (s, 1H, H6), 7.67 (s, 1H, CH triazole), 7.75–8.12 (bs, 1H, COOH linker). 13C NMR (75.46 MHz, CDCl3) δ (ppm): −4.9 (CH3-Si), −4.8 (CH3-Si), 13.1 (CH3 thymidine), 14.0 (CH3 lipid), 17.7 (Cq tBu), 22.6 (CH2 lipid), 25.5 (CH3 tBu), 26.7 (CH2 lipid), 29.2 (CH2 lipid), 29.2 (CH2 lipid), 29.3 (CH2 lipid), 29.4 (CH2 lipid), 29.5 (CH2 lipid), 29.5 (CH2-CH3 lipid), 29.8 (CH2 lipid), 31.8 (CH2 lipid), 33.6 (CH2-B linker), 38.0 (CH2-A linker), 39.2 (C2′), 41.0 (CH2-NH lipid), 50.6 (C5′), 57.6 (O-CH2 triazole), 71.8 (C3′), 84.0 (C4′), 86.3 (C1′), 110.5 (C5), 125.3 (CH triazole), 133.8 (C6), 143.5 (C triazole), 150.4 (C=O thymidine), 156.1 (C=O lipid), 162.9 (C=O thymidine), 175.9 (C=O linker). HRMS (ESI) [M + H+]: calcd = 721.43147, found = 721.43198.
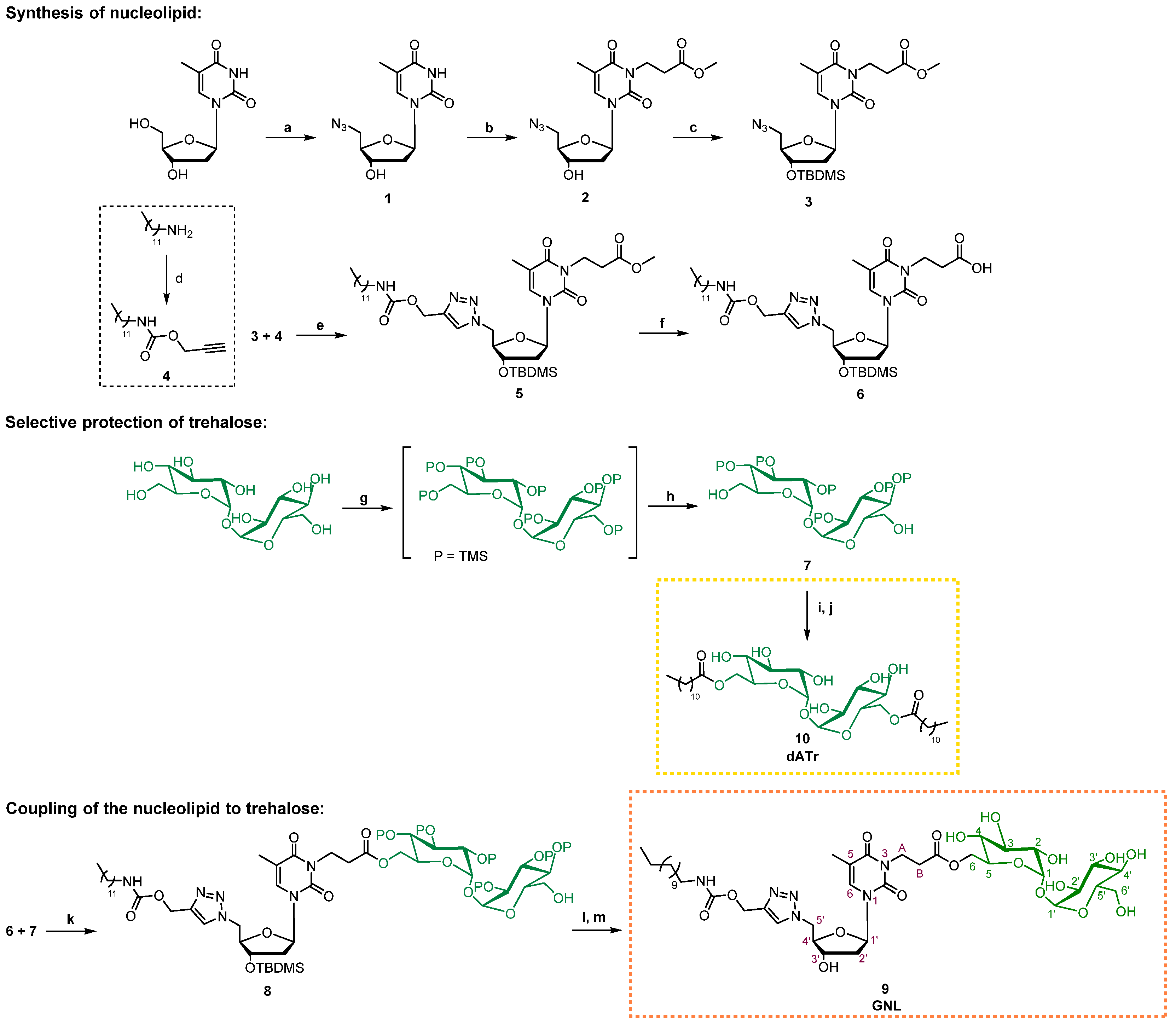
2.8. Synthesis of Compound 7: 2, 2′, 3, 3′, 4, 4′, 6′-Hepta-O- (trimethylsilyl)-α,α’-D-trehalose
D-(+)-trehalose dihydrate (1 equiv., 0.500 g, 1.32 mmol) was co-evaporated to dryness under vacuum with anhydrous DMF (3 × 10 mL). Following this, N,O-bistrimethylsilylacetamide (8.6 equiv., 2.78 mL, 11.37 mmol) and TBAF (Tetra-n-butylammonium fluoride) (0.06 equiv., 79 µL, 0.08 mmol) were sequentially added to a solution of the D-(+)-trehalose dihydrate dissolved in anhydrous DMF (4 mL). The completion of the reaction was reached by stirring the mixture overnight at room temperature. The mixture was then diluted in MeOH (60 mL) before the addition of K2CO3 (0.11 equiv., 0.020 g, 0.145 mmol). It was then stirred 4 h at 0 °C, before a quenching of the reaction by the addition of NaHCO3 (50 mL, aq.) and a dilution with water (30 mL). The aqueous phase was extracted 3 times with EtOAc (3 × 30 mL), prior to a drying over Na2SO4 of the combined organic phases, and a concentration to dryness under vacuum. The expected compound 7 was obtained as a white powder by a purification using flash chromatography on a silica gel (petroleum ether/EtOAc/NEt3, 80:19:1) (0.530 g, 52%). Rf: 0.42 (petroleum ether/EtOAc/NEt3, 80:19:1). 1H NMR (300 MHz, MeOD) δ (ppm): 0.12–0.20 (m, 54H, 6 TMS), 1.90 (bs, 2H, OH), 3.48 (dd, 2H, J = 3.0 Hz, 9.3 Hz, H2, H2′), 3.49–3.52 (dd, 2H, H4, H4′), 3.65–3.71 (m, 4H, H6, H6′), 3.83–3.93 (m, 4H, H3, H3′, H5, H5′), 4.96 (d, 2H, H1, H1′). 13C NMR (75.46 MHz, MeOD) δ (ppm): 0.3, 1.2, 1.6 (6 TMS), 61.7 (C6, C6′), 72.5, 74.4, 74.9 (C2, C2′, C3, C3′, C4, C4′), 74.0 (C5, C5′), 95.5 (C1, C1′).
2.9. Synthesis of Compound 8: 2,2′,3,3′,4,4′,6′-Hepta-O-(trimethylsilyl)-a,a’-D-trehalos-6-yl 3-[3′-O-(tert-butyldimethylsilyl)-5′-{4-[(dodecylcarbamoyloxy)methyl]-1H-1,2,3-triazol-1-yl}-5′-deoxythymidin-3-yl] Propanoate
Compound 7 (1 equiv., 0.126 g, 0.16 mmol), DCC (N,N′-Dicyclohexylcarbodiimide) (1.5 equiv., 0.050 g, 0.24 mmol) and DMAP (4-Dimethylaminopyridine) (0.5 equiv., 0.010 g, 0.08 mmol) were sequentially added to a solution of 6 (1.5 equiv., 0.175 g, 0.24 mmol) in anhydrous DCM (3 mL) and under argon. The mixture was stirred overnight at room temperature until the completion of the reaction. The precipitate was filtered and washed with DCM, followed by concentration to dryness of the filtrate under vacuum. The resulting crude was purified by flash chromatography on a silica gel (petroleum ether/EtOAc, 75:25) to obtain the expected compound 8 as a white powder (0.147 mg, 41%). Rf: 0.49 (petroleum ether/EtOAc, 75:25). 1H NMR (300 MHz, CDCl3) δ (ppm): −0.01–0.21 (m, 60H, CH3-Si et OTMS), 0.78–0.93 (m, 12H, CH3 lipid et tBu), 1.15–1.32 (m, 18H, CH2 lipid), 1.36–1.51 (m, 2H, CH2-CH3 lipid), 1.87 (s, 3H, CH3 thymidine), 1.96–2.13 (m, 1H, H2′), 2.17–2.31 (m, 1H, H2′), 2.60–2.75 (m, 2H, HB linker), 3.04–3.16 (m, 2H, CH2-NH lipid), 3.34–3.51 (m, 4H, HA linker et H2 trehalose, H2′ trehalose), 3.60–3.74 (m, 2H, H6′ trehalose), 3.77–4.33 (m, 10H, H4′, H6 trehalose, H3 trehalose, H3′ trehalose, H4 trehalose, H4′ trehalose, H5 trehalose, H5′ trehalose et OH trehalose), 4.39–4.47 (m, 1H, H3′), 4.55–4.61 (m, 2H, H5′), 4.83–4.91 (d, 2H, H1 trehalose, H1′ trehalose), 4.93–5.02 (m, 1H, NH lipid), 5.13 (s, 2H, O-CH2 triazole), 6.19 (t, J = 6.6 Hz, 1H, H1′), 6.63 (s, 1H, H6), 7.69 (s, 1H, CH triazole). 13C NMR (75.46 MHz, CDCl3) δ (ppm): −4.7 (CH3-Si), −4.5 (CH3-Si), 0.5 (OTMS), 1.2 (OTMS), 1.5 (OTMS), 13.3 (CH3 thymidine), 14.5 (CH3 lipid), 18.6 (Cq tBu), 23.6 (CH2 lipid), 25.9 (CH3 tBu), 26.2 (CH2 lipid), 26.6 (CH2 lipid), 27.7 (CH2 lipid), 30.2 (CH2 lipid), 30.3 (CH2 lipid), 30.6 (CH2 lipid), 30.7 (CH2 lipid), 32.9 (CH2-CH3 lipid), 33.1 (CH2-B linker), 34.6 (CH2 lipid), 38.2 (CH2-A linker), 40.2 (C2′), 41.7 (CH2-NH lipid), 52.0 (C5′), 58.2 (O-CH2 triazole), 61.6 (C6′ trehalose), 64.9 (C6 trehalose), 71.6, 72.4 (C2 trehalose, C2′ trehalose), 73.0 (C5′ trehalose), 73.5 (C3′), 73.7 (C5 trehalose), 73.8, 74.4, 74.6, 74.7 (C3 trehalose, C3′ trehalose, C4 trehalose, C4′ trehalose), 85.9 (C4′), 88.2 (C1′), 95.2, 95.4 (C1 trehalose, C1′ trehalose), 111.0 (C5), 126.7 (CH triazole), 136.5 (C6), 144.7 (C triazole), 151.6 (C=O thymidine), 158.1 (C=O lipid), 164.8 (C=O thymidine), 172.6 (C=O linker). HRMS (ESI) [M + H+]: calcd = 1477.77427, found = 1477.77674.
2.10. Synthesis of Compound 9 (GNL): 5′-{4-[(Dodecylcarbamoyloxy)methyl]-1H-1,2,3-triazol-1-yl}-5′-deoxythymidin-3-yl Propanoate
Dowex 50WX2-100 ion exchange resin (0.350 g) was added to a solution of 8 (1 equiv., 0.105 g, 0.07 mmol) in MeOH (20 mL). The mixture was stirred for 15 min at room temperature until the completion of the reaction. The mixture was then filtered and the filtrate was evaporated under vacuum. Purification of compound 9′ (0.062 g, 85%) was not necessary and it was directly engaged in the next reaction after drying. TBAF (1 equiv., 62 µL, 0.06 mmol) was added to a solution of 9′ (1 equiv., 0.062 g, 0.06 mmol) in anhydrous THF (3 mL) and under argon at 0 °C. The mixture was stirred for 2 h at 0 °C until the completion of the reaction. The reaction was then quenched by the addition of water (5 mL, aq.). The expected compound 9 (white powder) was obtained after three extractions of the aqueous phase with DCM (3 × 10 mL), drying of the combined organic phases over Na2SO4, a concentration to dryness under vacuum and a purification of the resulting crude by flash chromatography on a silica gel (DCM/MeOH/Water, 80:18.5:1.5) (0.029 g, 52%). Rf: 0.31 (petroleum ether/EtOAc/NEt3, 80:18.5:1.5). 1H NMR (300 MHz, MeOD) δ (ppm): 0.84–0.98 (m, 3H, CH3 lipid), 1.18–1.40 (m, 18H, CH2 lipid), 1.41–1.55 (m, 2H, CH2-CH3 lipid), 1.92 (s, 3H, CH3 thymidine), 2.21–2.39 (m, 2H, H2′), 2.65 (t, J = 7.2 Hz, 2H, HB linker), 2.99–3.09 (m, 2H, CH2-NH lipid), 3.25–3.37 (m, 6H, H2 trehalose, H2′ trehalose, H3 trehalose, H3′ trehalose, H4 trehalose et H4′ trehalose,), 3.45 (q, J = 3.9 Hz, 9.9 Hz, 2H, H6′ trehalose), 3.70–3.80 (m, 1H, H5′ trehalose), 3.97–4.05 (m, 1H, H5 trehalose), 4.11–4.36 (m, 5H, H4′, HA linker, H6 trehalose), 4.36–4.44 (m, 1H, H3′), 4.65–4.79 (m, 2H, H5′), 4.83–5.02 (d, 2H, H1 trehalose, H1′ trehalose), 5.10 (s, 2H, O-CH2 triazole), 6.15 (t, J = 6.9 Hz, 1H, H1′), 7.23 (s, 1H, H6), 8.00 (s, 1H, CH triazole). 13C NMR (75.46 MHz, MeOD) δ (ppm): 13.1 (CH3 thymidine), 14.3 (CH3 lipid), 23.2 (CH2 lipid), 25.4 (CH3 tBu), 29.6 (CH2 lipid), 29.6 (CH2 lipid), 29.9 (CH2 lipid), 30.0 (CH2 lipid), 30.2 (CH2 lipid), 30.2 (CH2 lipid), 30.2 (CH2 lipid), 32.4 (CH2-CH3 lipid), 32.8 (CH2-B linker), 34.6 (CH2 lipid), 34.8 (CH2-A linker), 37.8 (C2′), 39.4 (CH2-NH lipid), 51.9 (C5′), 62.2 (O-CH2 triazole), 63.2 (C6′ trehalose), 64.9 (C6 trehalose), 69.2, 70.6 (C2 trehalose, C2′ trehalose), 70.9 (C5′ trehalose), 71.2 (C3′), 71.5 (C5 trehalose), 72.3, 73.0, 73.6, 73.8 (C3 trehalose, C3′ trehalose, C4 trehalose, C4′ trehalose), 84.6 (C4′), 87.3 (C1′), 94.3, 95.5 (C1 trehalose, C1′ trehalose), 110.8 (C5), 125.3 (CH triazole), 136.9 (C6), 145.1 (C triazole), 151.2 (C=O thymidine), 155.6 (C=O lipid), 164.2 (C=O thymidine), 172.3 (C=O linker). HRMS (ESI) [M + H+]: calcd = 931.45356, found = 931.64586.
2.11. Synthesis of Compound 10 (dATr): 6,6′-Bis-O-dodecanoyl-α,α’-D-trehalose
Compound 7 (1 equiv., 0.200 g, 0.26 mmol), DCC (2 equiv., 0.106 g, 0.52 mmol) and DMAP (0.5 equiv., 0.016 g, 0.13 mmol) were sequentially added to a solution of lauric acid (2 equiv., 0.104 g, 0.52 mmol) in anhydrous DCM (3 mL) and under argon. The mixture was stirred overnight at room temperature until the completion of the reaction. The precipitate was filtered and washed with DCM, followed by concentration to dryness of the filtrate under vacuum. The resulting crude was purified by flash chromatography on a silica gel (petroleum ether/EtOAc, 75:25) to obtain the expected compound 7′ as a white powder (0.254 mg, 86%). Next, Dowex 50WX2-100 ion exchange resin (0.700 g) was added to a solution of 8′ (1 equiv., 0.254 g, 0.22 mmol) in MeOH (40 mL). The mixture was stirred for 15 min at room temperature until the completion of the reaction. The mixture was then filtered and the filtrate was evaporated under vacuum. Purification of compound 10 (0.143 g, 91%) was not necessary and it was directly engaged in the next reaction after drying. Rf: 0.49 (petroleum ether/EtOAc, 75:25). 1H NMR (300 MHz, MeOD) δ (ppm): 0.70–0.82 (m, 3H, CH3 lipid), 1.03–1.25 (m, 32H, CH2 lipid), 1.28–1.42 (m, 4H, CH2-CH3 lipid), 3.15–3.26 (m, 4H, CH2-C=O lipid), 3.50 (dd, 2H, J = 3.1 Hz, 9.2 Hz, H2, H2′), 3.49–3.58 (m, 2H, H4, H4′), 3.63–3.70 (m, 4H, H6, H6′), 3.83–3.90 (m, 2H, H3, H3′), 3.92–4.02 (m, 2H, H5, H5′), 4.96 (d, 2H, H1, H1′). 13C NMR (75.46 MHz, MeOD) δ (ppm): 13.8 (CH3 lipid), 20.5 (CH2 lipid), 24.7 (CH2 lipid), 30.1 (CH2 lipid), 30.5 (CH2 lipid), 30.6 (CH2 lipid), 30.6 (CH2 lipid), 30.9 (CH2 lipid), 30.9 (CH2-CH3 lipid), 31.1 (CH2 lipid), 31.5 (CH2 lipid), 61.7, 61.8 (C6, C6′), 71.3, 72.5, 74.4 (C2, C2′, C3, C3′, C4, C4′), 74.9 (C5, C5′), 95.5 (C1, C1′).
2.12. General Procedure for Nanoparticles and Solid Lipid Nanoparticles Preparation
The PLGA was purchased from Sigma-Aldrich (Resomer
® RG 503H PLGA; lactide-glycoside ratio of 50:50; molecular weight from 24,000 to 38,000). The nanoprecipitation protocol was adapted from a previous study to prepare NP suspensions [
19,
34]. Briefly, 31 mg of Resomer
® RG 503H PLGA was dissolved in 3.1 mL of tetrahydrofuran (THF; Sigma-Aldrich). Under sonication (37Hz, 100%, 3 min), 200 µL of the previous solution was quickly added to 20 mL of deionized water. The resulting NP suspension was then slowly concentrated by centrifugation using 10 kDa centrifugal filters (Merck Millipore, Saint Quentin Fallavier, France, Amicon Ultra centrifugal Filter) for 21 min at 5000 RPM (Centrifuge 5804 R, Eppendorf). Once the supernatant had been collected, the resulting suspension was ready for use (typical concentration of NPs ~ 0.2 mg·mL
−1). A rapid mixing protocol was adapted from the literature to prepare the NP suspension. 500 μL of the previously prepared stock solution was simultaneously injected (3 mL·min
−1) with 1.5 mL of deionized water (9 mL·min
−1) using syringe pumps (kdScientific, Holliston, MA, USA). The obtained PLGA NP suspension was concentrated to a typical concentration of 2.5 mg·mL
−1. For the control condition, PLGA NPs loaded with Nile Red fluorophore (Sigma-Aldrich, St. Louis, MO, USA 19123) were formulated as previously described, except that 0.9 mg of Nile Red was added to the polymer stock solution.
F2 and
F3 were formulated similarly, except that 15% (
w/
w) of
dATr or
GNL were introduced into the stock solution (typical final concentration in trehalose 0.4 µM). For all experiments, PLGA NP suspensions were prepared extemporaneously.
2.13. Nanoparticle and Solid Lipid Nanoparticle Characterization and Stability Evaluation
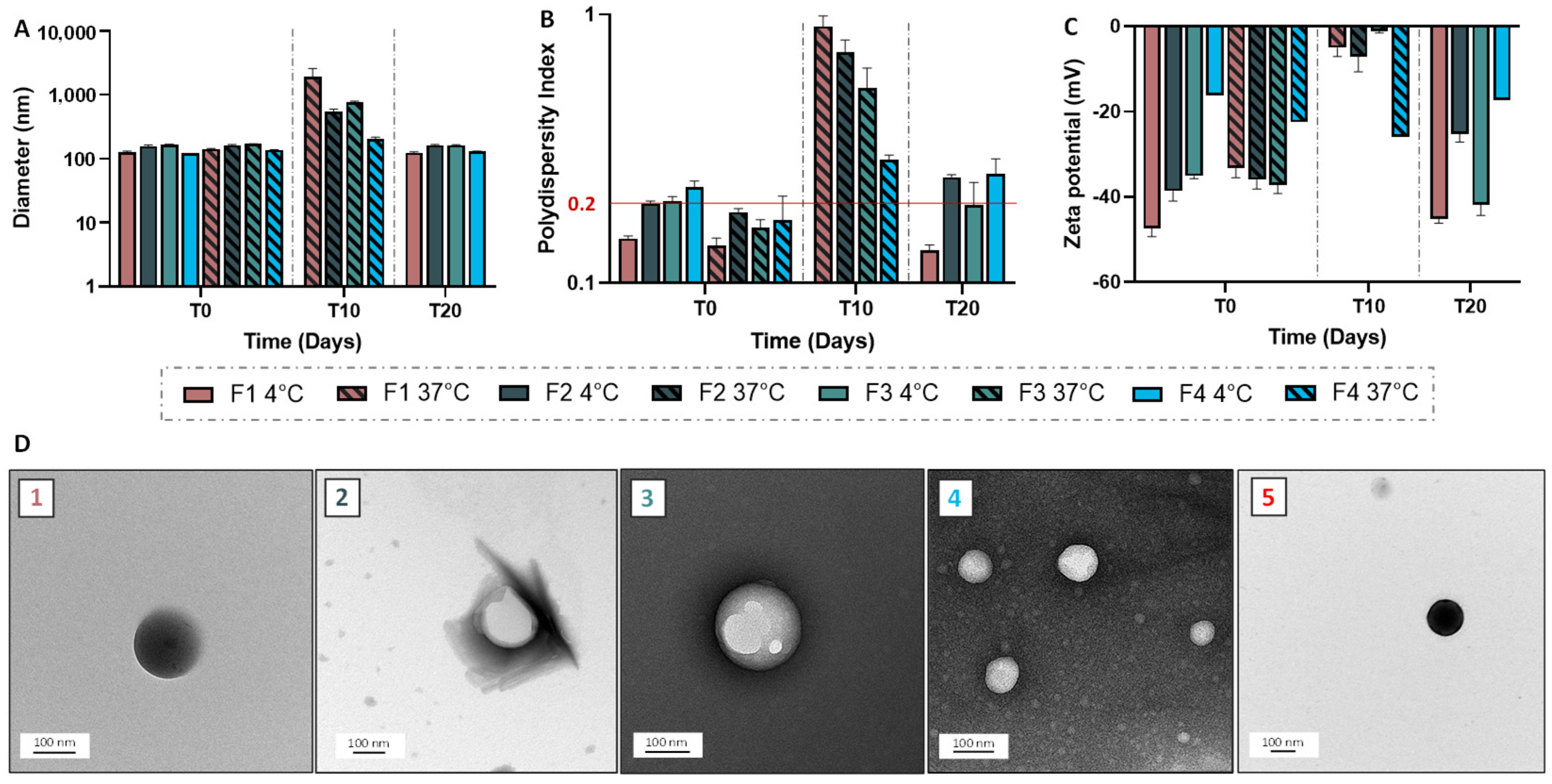
Granulometric profile of NPs and SLNs were acquired by Dynamic Light Scattering (DLS), using Malvern Instruments (Zetasizer Nano ZS, Palaiseau, France). NPs/SLNs were diluted at 1:1000 (v/v), then the average size and the polydispersity index were obtained in triplicate at 25 °C. To determine the Zeta potential, NPs/SLNs, at the same dilution, were analyzed using a Zetasizer Nano ZS coupled with Folded Capillary Cell (DTS1060). Short-term stability assessment was performed for a period of one month by visually checking the lack of creaming or phase separation and monitoring the size and zeta potential. Transmission electron microscopy was carried out using a Hitachi (Tokyo, Japan) coupled to an ORIUS SC1000 11MPX (Gatan, Pleasanton, CA, USA). All samples were deposited on carbon-coated grids (Delta Microscopies, Mauressac, France) and dried after 3 min of contact. A staining procedure using uranyless (Delta Microscopies, Mauressac, France) or nanotungstene was used.
2.14. Quantification of the Free GNL by Enzymatic Assay
Quantification of the free GNL in the suspensions of trehalose derivative-loaded NPs was performed using the Megazyme microplate assay procedure (Trehalose Assay Kit, Megazyne, Bray, Ireland) [
34]. Briefly, trehalose standard solutions (concentration between 25 and 100 µM) were prepared and 20 µL of each was used for the calibration curve. To these 20 µL were added 200 µL of deionized water, 20 µL of solution
1 (buffer), 10 µL of solution
2 (containing both nicotinamide adenine dinucleotide phosphate (NADP
+) and adenosine 5′ triphosphate) and 2 µL of solution
3 (containing the hexokinase and glucose-6-phosphate dehydrogenase). The solutions were mixed and the absorbance was measured at 340 nm (abs
0) before addition to the trehalase medium. The next step was the addition of 2 µL of suspension
4, containing trehalase, to the wells. The 96-well plate was shaken and incubated for 8 min at room temperature before the absorbance of the different solutions at 340 nm (abs
1) was read. The calibration curve was thus obtained by representing the background-corrected absorbance (abs
1-abs
0) as a function of the trehalose concentration. For the quantification of the NP samples, the same protocol was used except for the direct use of 220 µL of suspension instead of 20 µL of standard and 200 µL of deionized water, to ensure absorbances included in the calibration range.
2.15. Encapsulation Efficiency and Drug Loading
For the determination of the encapsulation efficiency (EE) and the drug loading (DL) of PLGA NPs,
GNL quantification was achieved by UV-Vis spectroscopy (Jasco V-630, France) using a standard calibration curve of
GNL dissolved in methanol (between 5 and 40 µg·mL
−1; absorbance at 266.5 nm).Equation (1) was used, where y is the absorbance value and x the
GNL concentration. The standard calibration curve was linear over the range of 5–40 µg·mL
−1 with r
2 = 0.9993. The amount of encapsulated
GNL in the PLGA NPs (also called DL) was calculated from the mass of the incorporated drug, using Equation (2):
The drug EE was defined as the ratio of the mass of the encapsulated drug over the mass of the drug used for the formulation, using the following equation:
The drug EE and the DL efficiency were calculated using Equations (1)–(3).
2.16. Infrared Analyses
Spectra of PLGA, GNL, F1 and F3 were acquired using an FT-IR spectrometer (FT-IR 4600, Jasco Inc., Tokyo, Japan) controlled by spectra manager software. Samples (3 mg) were mixed with 300 mg of KBr powder and then compressed with a mechanical die press to obtain translucent pellets. All spectra were recorded after averaging 16 accumulations with 8 cm−1 resolution between 4000 cm−1 and 450 cm−1. Background was performed on the atmosphere and subtracted to all acquisitions. All spectra were acquired in the transmittance mode. IR data were processed using spectra manager for IR spectra and IR derivatives (Savitzky–Golay: derivative order 1, polynome order 2 and window of 7 points).
2.17. Cell Culture and Cell Viability Assay
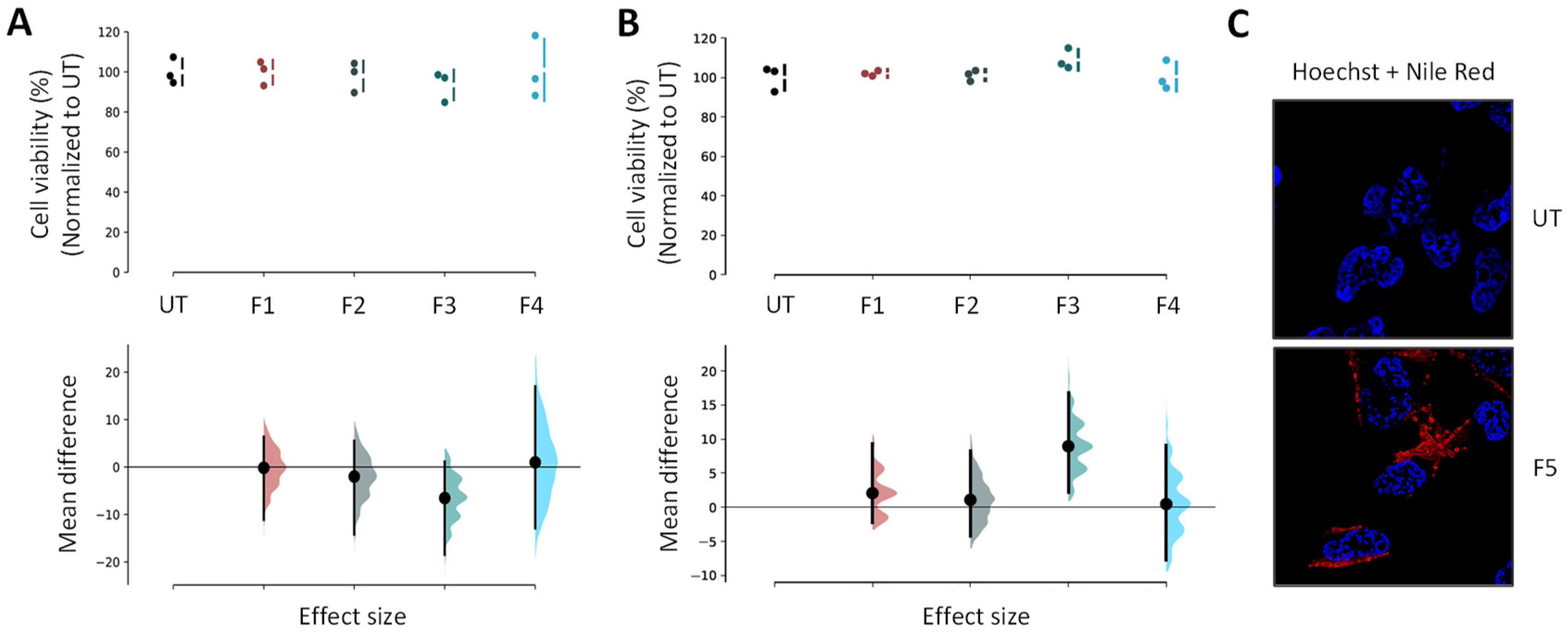
Human neuroblastoma cell lines (BE(2)-M17) from ATCC (CRL-2267) were grown in Opti-MEM (Life Technologies, Carlsbad, CA, USA; 31985-047) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin. All NP and SLN suspensions were used as freshly prepared on cells grown at 70% to 80% confluence. Cells were exposed at 0.5 µL of loaded NPs and SLNs for 24 h or 48 h, and each experiment was performed at least in triplicate. MTS assay (ATCC/LGC Promochem, Molsheim, France) estimated cell viability using the manufacturer instructions.
2.18. Western Blot Analysis
BE(2)-M17 cells were seeded on 6-well plates (NUNC) and grown to 70–80% confluence per well before being treated with F1, F2, F3, F4 and molecular trehalose as control (concentration of 0.4 µM). The cells were then transferred to Eppendorf tubes before centrifugation for 5 min at 3000 rpm and 4 °C. The supernatant was removed before the dropwise addition of 50 µL of P/X solution (P/X = 1.5 mL RB1X, 0.450 mL P4X, 0.050 mL dithiothreitol (DTT)) under mechanical agitation. The samples were then incubated for 5 min at 100 °C. Protein samples were loaded into 18% acrylamide gels and separated by SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis) before transfer to a nitrocellulose membrane (0.2 µm—Biorad—USA). The membranes were then blocked in 5% PBS (phosphate-buffered saline)-milk buffer before overnight incubation at 4 °C with rabbit anti-LC3 (light chain 3) primary antibody (1/1000°, Novus Biological, Littleton, CO, USA #NB100-2220). An incubation with mouse anti-actin antibody (1/5000°, Sigma) served as a loading control. The appropriate secondary antibodies were then used before revelation with the revelation kit (Super Signal West Pico Chemiluminescent kit—Immobilon Western, Chemiluminescent HRP substrate, Millipore). Chemiluminescence images were acquired using a ChemiDoc + XRS analysis system (BioRad, Hercules, CA, USA). The signal per well was quantified using ImageJ software and normalized by actin before statistical analysis.
2.19. Immunostaining and Imaging
Extemporaneous NP suspensions were used for all experiments. In the case of Nile Red-loaded NPs internalization and colocalization imaging assays, BE(2)-M17 cells were replated in 6-well plates (NUNC) with coverslips after trypsinization, and allowed growing at 70% to 80% confluence per well. They were then exposed to the tested formulations for 24 h at 37 °C. The final concentration of NPs in each well was set at ~3 µg·mL−1 for F5. The cell fixation was performed using 4% paraformaldehyde for 20 min at 4 °C, prior to the washing step with PBS 1X solution. A mixture, composed of 225 µL of triton, 450 µL of normal goat serum and 5 mL of PBS 1X, was added for cell permeabilization and blocking steps. The LAMP2 (Mx, H4B4) antibodies and their appropriate secondary antibodies conjugated with GAM 488 (Life Technologies) were used to mark the lysosomes overnight at 4 °C and for staining, respectively. The cells were incubated with 8 µM Hoechst dye (ThermoFisher Scientific, Waltham, MA, USA, #3342) (8 min; room temperature) to stain cytoplasm and nuclei, prior to mounting. After an air-drying step, slices were mounted on #1.5 coverslips using Dako fluorescent mounting medium, then left overnight in darkness to dry. A Leica TCS SP8 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) combined with a 63X Plan Apo CS oil immersion objective was used to acquire image stacks (pixel size ~100 nm; z-step 0.3 µm). Nile Red was observed in a detection window between 570 and 590 nm after an excitation at 568 nm (DPSS laser). For lysosomes, the excitation was performed at 488 nm (argon laser) and the detection within a window between 545 and 605 nm. In parallel, the untreated cells were required for autofluorescence controls using the previously described parameters. LAS AF v2.6 acquisition software implemented with HCS-A module (Leica Microsystems) was used for image analyses. The relative intensity of Nile Red to cell surface was determined using Definiens XD Developer v2.5 software (Definiens). The images were analyzed with the ImageJ (NIH, Bethesda, MD, USA; Fiji) software for colocalization, fluorescence profiles, orthogonal projections and maximal intensity projections.
2.20. GFP-LC3 Reporter Autophagy Assay
To assess autophagy activity, the green fluorescent protein (GFP)-LC3 reporter was used as previously described [
29,
35]. Briefly, a BE(2)-M17 cell line was plated on coverslips in 12-well plates which were transfected with mCherry-GFP-LC3 plasmid at 1.6 µg DNA using polyethylenimine (PEI)-mediated transfection. Cells were maintained for 24 h at 37 °C in 5% CO
2 before being treated with unloaded or loaded NPs or SLNs for 24 h at 1:1000. Cell coverslips were fixed at 4 °C for 20 min using 4% paraformaldehyde, followed by 3 washing steps of 5 min with PBS 1X. The final step consisted in a staining using DAPI solution (10 μM; Invitrogen, Waltham, MA, USA) for 8 min prior to long washes in PBS 1X solution. Mounting solution (Dako) was used to mount the coverslips onto slides, and images were obtained using a wide-field Olympus Epifluorescent Microscope (BX3-CBH) coupled with a Hamamatsu camera (ORCA-Flash 4.0 LT). Images were deconvolved using the cellSens Dimension software. Image analysis was performed using Fiji/ImageJ software.
2.21. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6 software. In the case of functional assays, a one-way analysis of variance (ANOVA) was used to evaluate the data statistical significance prior to a Tukey’s multiple comparison test. A significant difference was highlighted by p < 0.05. Data appear as estimation graphics called ‘Gardner–Altman plots’: in the top graph, the data from UT (untreated), F1, F2, F3 and F4 groups are presented as scatter plots showing the observed values, along with the above-defined descriptive statistics (mean ± standard deviation). Below each graph, a contrast graph using the difference axis to display an effect size, here the mean difference. Vertically aligned with the mean of the UT group, the mean difference is indicated by the black circle. The 95% confidence interval (CI) of the mean difference is illustrated by the black vertical line. The curve indicates the resampled distribution of the effect size, given the observed data.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}