
Emerging Albumin-Binding Anticancer Drugs for Tumor-Targeted Drug Delivery: Current Understandings and Clinical Translation


Abstract
1. Introduction
2. Properties of Albumin
2.1. Human Serum Albumin (HSA)
2.2. Other Types of Albumins
3. Albumin Binding of Anticancer Drugs
3.1. Albumin-Binding Methods
3.2. Albumin Binding and Anticancer Drug Half-Life
3.3. Albumin Binding and Cancer Targeting
4. Exogenous Albumin-Bound Anticancer Drug Formulations for Cancer Therapy
4.1. Developments in Exogenous Albumin-Bound Anticancer Drug Formulations
4.2. Methods for Albumin Nanoparticle Formation
4.3. Developments in Albumin-Bound Anticancer Drug Nanoformulations
4.4. Clinical Translation of Exogenous Albumin-Bound Anticancer Drug Formulations
5. Endogenous Albumin-Binding Anticancer Drugs for Cancer Therapy

{kind=link}
{kind=link}
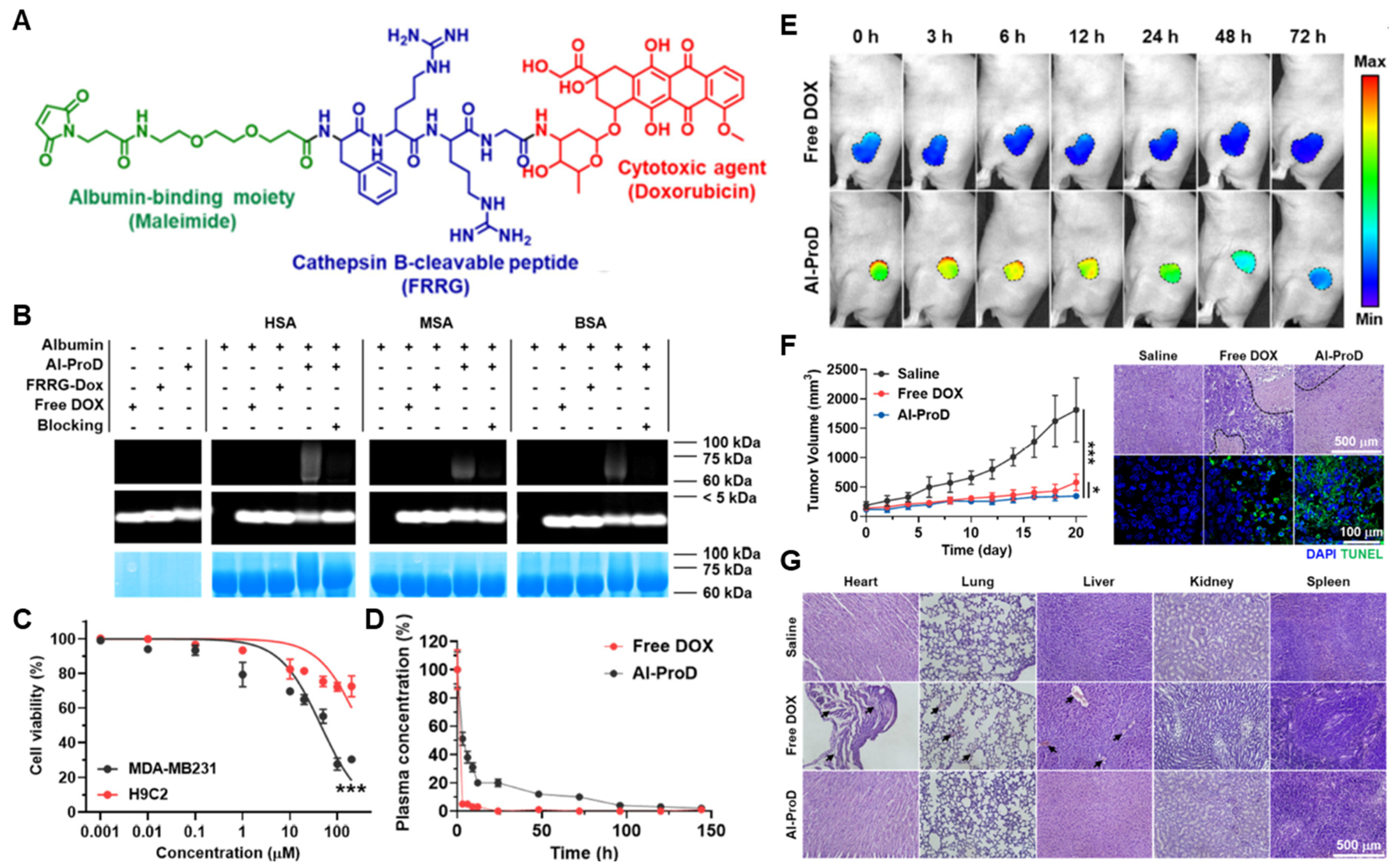{kind=link}
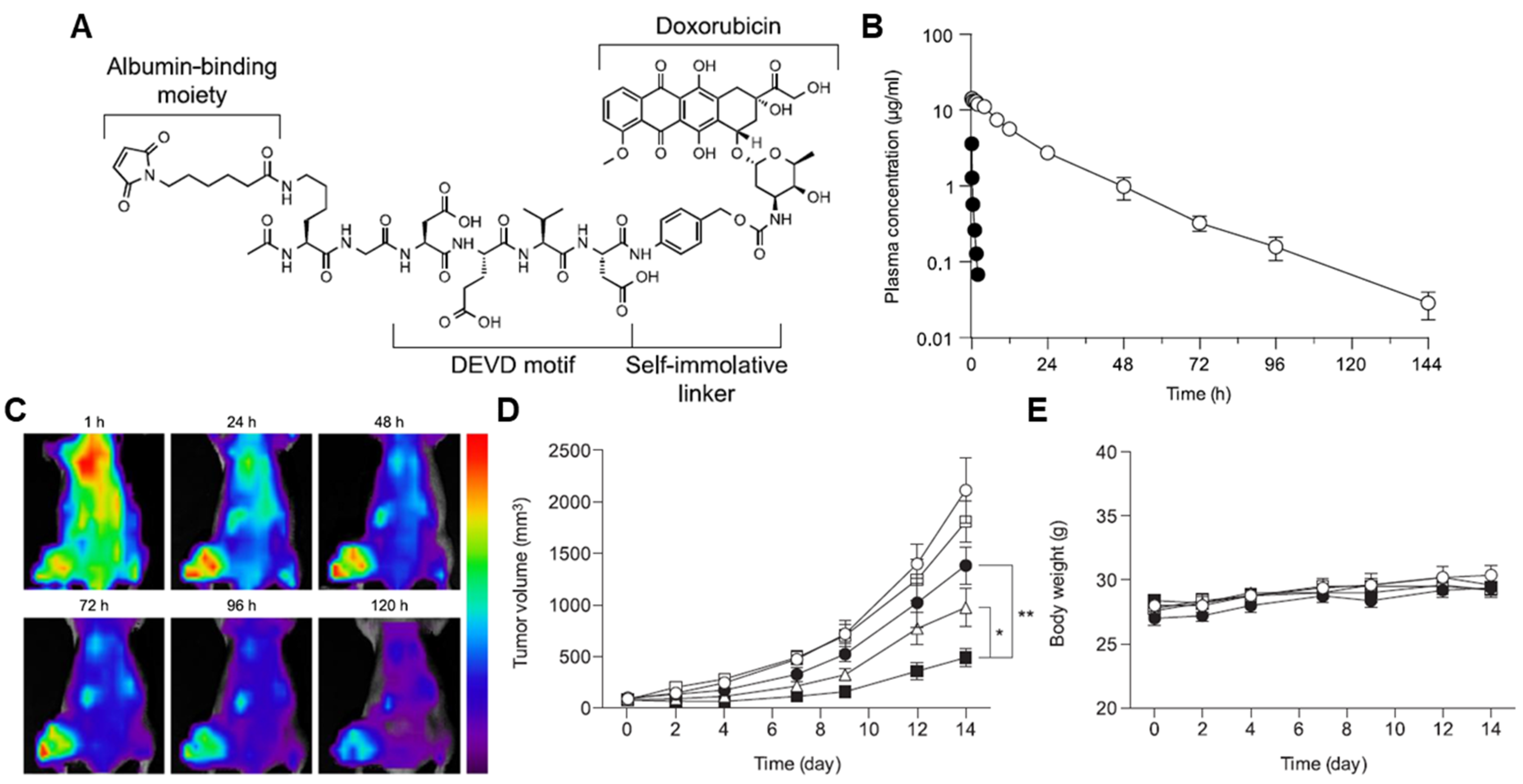{kind=link}
{kind=link}
{kind=link}
| Carrier Type | Trade Name | Therapeutic Agent | Target | Clinical Stage | Reference |
|---|---|---|---|---|---|
| Native albumin, exogenous | MTX-HSA | Methotrexate | Renal cell carcinoma, advanced or metastatic transitional cell carcinoma | Phase II | [174,245] |
| Recombinant albumin | MM-111 | HER2/HER3 antibory (anti-HER2/HER3) | Breast neoplasm, Her2-amplified solid tumors, metastatic breast cancer | Phase I/II | [246,247] NCT01097460, NCT00911898 |
| M0250 | Vascular endothelial growth fact of-A antibody (anti-VEGF-A), hepatocyte growth factor antibody (anti-HGF) | Advanced solid tumors | Phase I/II | [178,179] NCT02194426 | |
| Albumin nanoparticle | Abraxane® | Paclitaxel | Metastatic breast cancer, locally advanced or metastatic non-small cell lung cancer, Metastatic adenocarcinoma of the pancreas | Approved | [188] NCT01583426 |
| ABI-008 | Docetaxel | Hormone-refractory prostate cancer | Phase I/II | NCT00477529 | |
| ABI-009 | Rapamycin | Non-muscle invasive bladder cancer, solid tumors, PEComa, metastatic colorectal cancer, high grade recurrent glioma and newly diagnosed glioblastoma, soft tissue sarcoma | Phase I/II | NCT02009332, NCT00635284, NCT02494570, NCT03439642, NCT03463265, NCT03660930 | |
| ABI-010 | 17-Allylamino-17-demethoxygel danamycin | Solid tumors | Withdrawn (prior to Phase I) | NCT00820768 | |
| ABI-011 | Thiocolchicine dimer | Solid tumors and lymphoma | Phase I | NCT02582827 | |
| Native albumin, endogenous | Aldoxorubicin | Doxorubicin | Soft tissue sarcoma, glioblastoma, HIV positive Koposi’s sarcoma, pancreatic ductal adenocarcinoma | Phase III | [35] NCT02049905, NCT02014844, NCT02029430, NCT01580397 |
| Albumin-Binding Moiety | Cancer-Specific Cleavable Linker | Anticancer Drug | Reference |
|---|---|---|---|
| Maleimide | Cathepsin B-specific cleavable FRRG peptide | Doxorubicin | [216] |
| Maleimide | Cathepsin B-specific cleavable FL peptide | Doxorubicin | [217] |
| Maleimide | Caspase-3-specific cleavable KGDEVD peptide | Doxorubicin | [222] |
| Maleimide | Caspase-3-specific cleavable KGDEVD peptide | MMAE | [226] |
| Maleimide | MMP-specific cleavable GPLGIAGQ peptide | Doxorubicin | [232] |
| Maleimide | Prostate-specific antigen (PSA)-specific cleavable SSYYSG peptide | Doxorubicin | [234] |
| Maleimide | GSH-specific cleavable S-S linker | Gemcitabine | [237] |
| 2-acetylphenylboronic acid (APBA) | GSH-specific cleavable S-S linker | Camptothecin | [198] |
| Maleimide | Hypoxia-specific cleavable azo linker | Exatecan | [244] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Park, J.; Shim, M.K.; Um, W.; Yoon, H.Y.; Ryu, J.H.; Lim, D.-K.; Kim, K. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics 2019, 9, 7906–7923. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Choi, D.W.; Kim, H.N.; Park, C.G.; Lee, W.; Park, H.H. Protein-Based Nanoparticles as Drug Delivery Systems. Pharmaceutics 2020, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Huh, M.S.; Sun, I.-C.; Yuk, S.H.; Choi, K.; Kim, K.; Kwon, I.C. In Vivo Targeted Delivery of Nanoparticles for Theranosis. Acc. Chem. Res. 2011, 44, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.-H.; Shim, M.K.; Jung, B.; Jang, E.H.; Park, M.-J.; Kang, H.C.; Kim, J.-H. Heat shock responsive drug delivery system based on mesoporous silica nanoparticles coated with temperature sensitive gatekeeper. Microporous Mesoporous Mater. 2017, 253, 96–101. [Google Scholar] [CrossRef]
- Geng, W.; Jiang, N.; Qing, G.-Y.; Liu, X.; Wang, L.; Busscher, H.J.; Tian, G.; Sun, T.; Wang, L.-Y.; Montelongo, Y.; et al. Click Reaction for Reversible Encapsulation of Single Yeast Cells. ACS Nano 2019, 13, 14459–14467. [Google Scholar] [CrossRef] [PubMed]
- Geng, W.; Wang, L.; Jiang, N.; Cao, J.; Xiao, Y.-X.; Wei, H.; Yetisen, A.K.; Yang, X.-Y.; Su, B.-L. Single cells in nanoshells for the functionalization of living cells. Nanoscale 2018, 10, 3112–3129. [Google Scholar] [CrossRef]
- Shim, M.K.; Na, J.; Cho, I.K.; Jang, E.H.; Park, J.; Lee, S.; Kim, J.-H. Targeting of claudin-4 by Clostridium perfringens enterotoxin-conjugated polysialic acid nanoparticles for pancreatic cancer therapy. J. Control. Release 2021, 331, 434–442. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Kim, J.; Choi, Y.; Yang, S.; Lee, J.; Choi, J.; Moon, Y.; Kim, J.; Shim, N.; Cho, H.; Shim, M.K.; et al. Sustained and Long-Term Release of Doxorubicin from PLGA Nanoparticles for Eliciting Anti-Tumor Immune Responses. Pharmaceutics 2022, 14, 474. [Google Scholar] [CrossRef]
- Choi, Y.; Yoon, H.Y.; Kim, J.; Yang, S.; Lee, J.; Choi, J.W.; Moon, Y.; Kim, J.; Lim, S.; Shim, M.K.; et al. Doxorubicin-Loaded PLGA Nanoparticles for Cancer Therapy: Molecular Weight Effect of PLGA in Doxorubicin Release for Controlling Immunogenic Cell Death. Pharmaceutics 2020, 12, 1165. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Shim, M.-K.; Park, M.-J.; Jang, E.H.; Yoon, H.Y.; Kim, K.; Kim, J.-H. Hydrophobically modified polysaccharide-based on polysialic acid nanoparticles as carriers for anticancer drugs. Int. J. Pharm. 2017, 520, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Sun, I.-C.; Hwang, H.S.; Shim, M.K.; Yoon, H.Y.; Kim, K. Rediscovery of nanoparticle-based therapeutics: Boosting immunogenic cell death for potential application in cancer immunotherapy. J. Mater. Chem. B 2021, 9, 3983–4001. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef]
- Rahimizadeh, P.; Yang, S.; Lim, S.I. Albumin: An Emerging Opportunity in Drug Delivery. Biotechnol. Bioprocess Eng. 2020, 25, 985–995. [Google Scholar] [CrossRef]
- Sleep, D. Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 793–812. [Google Scholar] [CrossRef]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Control. Release 2012, 157, 4–28. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Z. Albumin Carriers for Cancer Theranostics: A Conventional Platform with New Promise. Adv. Mater. 2016, 28, 10557–10566. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Yhee, J.Y.; Jeon, S.; Yoon, H.Y.; Shim, M.K.; Ko, H.; Min, J.; Na, J.H.; Chang, H.; Han, H.; Kim, J.-H.; et al. Effects of tumor microenvironments on targeted delivery of glycol chitosan nanoparticles. J. Control. Release 2017, 267, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F. A clinical update of using albumin as a drug vehicl—A commentary. J. Control. Release 2014, 190, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Kyu Shim, M.; Yang, S.; Sun, I.-C.; Kim, K. Tumor-activated carrier-free prodrug nanoparticles for targeted cancer Immunotherapy: Preclinical evidence for safe and effective drug delivery. Adv. Drug Deliv. Rev. 2022, 183, 114177. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Zhou, Z.; Lu, Z.-R. Molecular imaging of the tumor microenvironment. Adv. Drug Deliv. Rev. 2017, 113, 24–48. [Google Scholar] [CrossRef]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.-H.; Kang, S.-W.; Seo, J.-W.; Hyun, S.-W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2019, 294, 376–389. [Google Scholar] [CrossRef]
- Shim, M.K.; Yoon, H.Y.; Lee, S.; Jo, M.K.; Park, J.; Kim, J.-H.; Jeong, S.Y.; Kwon, I.C.; Kim, K. Caspase-3/-7-Specific Metabolic Precursor for Bioorthogonal Tracking of Tumor Apoptosis. Sci. Rep. 2017, 7, 16635. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Shim, M.K.; Yang, S.; Hwang, H.S.; Cho, H.; Kim, J.; Yun, W.S.; Moon, Y.; Kim, J.; Yoon, H.Y.; et al. Visible-Light-Triggered Prodrug Nanoparticles Combine Chemotherapy and Photodynamic Therapy to Potentiate Checkpoint Blockade Cancer Immunotherapy. ACS Nano 2021, 15, 12086–12098. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.K.; Yoon, H.Y.; Ryu, J.H.; Koo, H.; Lee, S.; Park, J.H.; Kim, J.-H.; Lee, S.; Pomper, M.G.; Kwon, I.C.; et al. Cathepsin B-Specific Metabolic Precursor for In Vivo Tumor-Specific Fluorescence Imaging. Angew. Chem. Int. Ed. 2016, 55, 14698–14703. [Google Scholar] [CrossRef]
- Lee, S.; Jung, S.; Koo, H.; Na, J.H.; Yoon, H.Y.; Shim, M.K.; Park, J.; Kim, J.-H.; Lee, S.; Pomper, M.G.; et al. Nano-sized metabolic precursors for heterogeneous tumor-targeting strategy using bioorthogonal click chemistry in vivo. Biomaterials 2017, 148, 1–15. [Google Scholar] [CrossRef]
- Chawla, S.P.; Papai, Z.; Mukhametshina, G.; Sankhala, K.; Vasylyev, L.; Fedenko, A.; Khamly, K.; Ganjoo, K.; Nagarkar, R.; Wieland, S.; et al. First-Line Aldoxorubicin vs. Doxorubicin in Metastatic or Locally Advanced Unresectable Soft-Tissue Sarcoma: A Phase 2b Randomized Clinical Trial. JAMA Oncol. 2015, 1, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Board, S.A. Cross-Sectional Guidelines for Therapy with Blood Components and Plasma Derivatives: Chapter 5 Human Albumin–Revised. Transfus Med Hemother. 2016, 43, 223–232. [Google Scholar]
- Kobayashi, N.; Suzuki, Y.; Tsuge, T.; Okumura, K.; Ra, C.; Tomino, Y. FcRn-mediated transcytosis of immunoglobulin G in human renal proximal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2002, 282, F358–F365. [Google Scholar] [CrossRef]
- Fanali, G.; Di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Khorolskyi, O.V.; Malomuzh, N.P. Macromolecular sizes of serum albumins in its aqueous solutions. Proteins 2020, 2, 4. [Google Scholar] [CrossRef]
- Yang, X.; Bolsa-Ferruz, M.; Marichal, L.; Porcel, E.; Salado-Leza, D.; Lux, F.; Tillement, O.; Renault, J.-P.; Pin, S.; Wien, F. Human serum albumin in the presence of AGuIX nanoagents: Structure stabilisation without direct interaction. Int. J. Mol. Sci. 2020, 21, 4673. [Google Scholar] [CrossRef]
- Neumann, E.; Frei, E.; Funk, D.; Becker, M.D.; Schrenk, H.-H.; Müller-Ladner, U.; Fiehn, C. Native albumin for targeted drug delivery. Expert Opin. Drug Deliv. 2010, 7, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, I.; Saletsky, A. Study of the denaturation of human serum albumin by sodium dodecyl sulfate using the intrinsic fluorescence of albumin. J. Appl. Spectrosc. 2009, 76, 536–541. [Google Scholar] [CrossRef]
- Peters, T., Jr. All About Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Carter, D.C.; He, X.-m.; Munson, S.H.; Twigg, P.D.; Gernert, K.M.; Broom, M.B.; Miller, T.Y. Three-dimensional structure of human serum albumin. Science 1989, 244, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, J.; Sadler, P.J.; Tucker, A. 1H NMR of albumin in human blood plasma: Drug binding and redox reactions at Cys34. FEBS Lett. 1995, 376, 1–5. [Google Scholar] [CrossRef]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 1–12. [Google Scholar] [CrossRef]
- Hankins, J. The role of albumin in fluid and electrolyte balance. J. Infus. Nurs. 2006, 29, 260–265. [Google Scholar] [CrossRef]
- Rosenoer, V.M.; Oratz, M.; Rothschild, M.A. Albumin: Structure, Function and Uses; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Huntington, J.A.; Stein, P.E. Structure and properties of ovalbumin. J. Chromatogr. B: Biomed. Sci. Appl. 2001, 756, 189–198. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Yu, S.; Hu, J.; Pan, X.; Yao, P.; Jiang, M. Stable and pH-sensitive nanogels prepared by self-assembly of chitosan and ovalbumin. Langmuir 2006, 22, 2754–2759. [Google Scholar] [CrossRef]
- Van Der Maaden, K.; Varypataki, E.M.; Romeijn, S.; Ossendorp, F.; Jiskoot, W.; Bouwstra, J. Ovalbumin-coated pH-sensitive microneedle arrays effectively induce ovalbumin-specific antibody and T-cell responses in mice. Eur. J. Pharm. Biopharm. 2014, 88, 310–315. [Google Scholar] [CrossRef]
- Sassi, A.P.; Shaw, A.J.; Han, S.M.; Blanch, H.W.; Prausnitz, J.M. Partitioning of proteins and small biomolecules in temperature-and pH-sensitive hydrogels. Polymer 1996, 37, 2151–2164. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, A.; Ostadrahimi, A.; Jahanban-Esfahlan, R.; Roufegarinejad, L.; Tabibiazar, M.; Amarowicz, R. Recent developments in the detection of bovine serum albumin. Int. J. Biol. Macromol. 2019, 138, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Gelamo, E.; Tabak, M. Spectroscopic studies on the interaction of bovine (BSA) and human (HSA) serum albumins with ionic surfactants. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2000, 56, 2255–2271. [Google Scholar] [CrossRef]
- Bolel, P.; Mahapatra, N.; Halder, M. Optical spectroscopic exploration of binding of cochineal red A with two homologous serum albumins. J. Agric. Food Chem. 2012, 60, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.X.; Kim, H.-Y.; Dass, C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1237–1247. [Google Scholar] [CrossRef]
- Du, C.; Deng, D.; Shan, L.; Wan, S.; Cao, J.; Tian, J.; Achilefu, S.; Gu, Y. A pH-sensitive doxorubicin prodrug based on folate-conjugated BSA for tumor-targeted drug delivery. Biomaterials 2013, 34, 3087–3097. [Google Scholar] [CrossRef]
- Mondal, M.; Ramadas, K.; Natarajan, S. Molecular interaction of 2, 4-diacetylphloroglucinol (DAPG) with human serum albumin (HSA): The spectroscopic, calorimetric and computational investigation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 183, 90–102. [Google Scholar]
- Joseph, K.; Moser, A.C.; Basiaga, S.B.; Schiel, J.E.; Hage, D.S. Evaluation of alternatives to warfarin as probes for Sudlow site I of human serum albumin: Characterization by high-performance affinity chromatography. J. Chromatogr. A 2009, 1216, 3492–3500. [Google Scholar] [CrossRef]
- Moser, A.C.; Kingsbury, C.; Hage, D.S. Stability of warfarin solutions for drug–protein binding measurements: Spectroscopic and chromatographic studies. J. Pharm. Biomed. Anal. 2006, 41, 1101–1109. [Google Scholar] [CrossRef][Green Version]
- Fujiwara, S.-i.; Amisaki, T. Identification of high affinity fatty acid binding sites on human serum albumin by MM-PBSA method. Biophys. J. 2008, 94, 95–103. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Spectroscopic techniques in the study of protein binding. A fluorescence technique for the evaluation of the albumin binding and displacement of warfarin and warfarin-alcohol. Clin. Exp. Pharmacol. Physiol. 1975, 2, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Fehske, K.J.; Jähnchen, E.; Müller, W.E.; Stillbauer, A. Azapropazone binding to human serum albumin. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980, 313, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Chignell, C.F. Optical studies of drug-protein complexes: II. Interaction of phenylbutazone and its analogues with human serum albumin. Mol. Pharmacol. 1969, 5, 244–252. [Google Scholar] [PubMed]
- Isogai, H.; Hirayama, N. In silico prediction of interactions between site II on human serum albumin and profen drugs. Int. Sch. Res. Not. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Amézqueta, S.; Bolioli, A.M.; Beltrán, J.L.; Ràfols, C. Evaluation of the interactions between human serum albumin (HSA) and warfarin or diflunisal by using molecular fluorescence using two approaches. Admet Dmpk 2018, 6, 47–54. [Google Scholar] [CrossRef]
- Uddin, S.; Shilpi, J.; Murshid, G.; Rahman, A.; Sarder, M.; Alam, M. Determination of the binding sites of arsenic on bovine serum albumin using warfarin (site-I specific probe) and diazepam (site-II specific probe). J. Biol. Sci. 2004, 4, 609–612. [Google Scholar]
- Chuang, V.T.G.; Otagiri, M. Stereoselective binding of human serum albumin. Chirality Pharmacol. Biol. Chem. Conseq. Mol. Asymmetry 2006, 18, 159–166. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid–serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef]
- Karimi, M.; Bahrami, S.; Ravari, S.B.; Zangabad, P.S.; Mirshekari, H.; Bozorgomid, M.; Shahreza, S.; Sori, M.; Hamblin, M.R. Albumin nanostructures as advanced drug delivery systems. Expert Opin. Drug Deliv. 2016, 13, 1609–1623. [Google Scholar] [CrossRef]
- Stocker, R.; Glazer, A.N.; Ames, B.N. Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci. USA 1987, 84, 5918–5922. [Google Scholar] [CrossRef] [PubMed]
- Dockal, M.; Carter, D.C.; Rüker, F. The three recombinant domains of human serum albumin: Structural characterization and ligand binding properties. J. Biol. Chem. 1999, 274, 29303–29310. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Dou, W.-F.; Lei, J.-Y.; Zhang, L.-F.; Xu, Z.-H.; Chen, Y.; Jin, J. Expression, purification, and characterization of recombinant human serum albumin fusion protein with two human glucagon-like peptide-1 mutants in Pichia pastoris. Protein Expr. Purif. 2008, 61, 45–49. [Google Scholar] [CrossRef] [PubMed]
- De Bold, M.K.; Sheffield, W.P.; Martinuk, A.; Bhakta, V.; Eltringham-Smith, L.; Adolfo, J. Characterization of a long-acting recombinant human serum albumin-atrial natriuretic factor (ANF) expressed in Pichia pastoris. Regul. Pept. 2012, 175, 7–10. [Google Scholar] [CrossRef]
- Syed, S.; Schuyler, P.D.; Kulczycky, M.; Sheffield, W.P. Potent antithrombin activity and delayed clearance from the circulation characterize recombinant hirudin genetically fused to albumin. Blood J. Am. Soc. Hematol. 1997, 89, 3243–3252. [Google Scholar] [CrossRef]
- Duttaroy, A.; Kanakaraj, P.; Osborn, B.L.; Schneider, H.; Pickeral, O.K.; Chen, C.; Zhang, G.; Kaithamana, S.; Singh, M.; Schulingkamp, R. Development of a long-acting insulin analog using albumin fusion technology. Diabetes 2005, 54, 251–258. [Google Scholar] [CrossRef]
- Halpern, W.; Riccobene, T.A.; Agostini, H.; Baker, K.; Stolow, D.; Gu, M.-L.; Hirsch, J.; Mahoney, A.; Carrell, J.; Boyd, E. AlbugraninTM, a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharm. Res. 2002, 19, 1720–1729. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Baumann, A.; Tuerck, D.; Prabhu, S.; Dickmann, L.; Sims, J. Pharmacokinetics, metabolism and distribution of PEGs and PEGylated proteins: Quo vadis? Drug Discov. Today 2014, 19, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Garay, R.P.; Labaune, J.P. Immunogenicity of polyethylene glycol (PEG). In Proceedings of the Open Conf. Proc. J. 2011, 2, 104–107. [Google Scholar] [CrossRef]
- Kurtzhals, P.; Havelund, S.; Jonassen, I.; Kiehr, B.; Larsen, U.; Ribel, U.; Markussen, J. Albumin binding of insulins acylated with fatty acids: Characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem. J. 1995, 312, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Home, P.; Kurtzhals, P. Insulin detemir: From concept to clinical experience. Expert Opin. Pharmacother. 2006, 7, 325–343. [Google Scholar] [CrossRef]
- Dornhorst, A.; Lüddeke, H.J.; Sreenan, S.; Koenen, C.; Hansen, J.; Tsur, A.; Landstedt-Hallin, L.; Group, P.S. Safety and efficacy of insulin detemir in clinical practice: 14-week follow-up data from type 1 and type 2 diabetes patients in the predictivetm European cohort. Int. J. Clin. Pract. 2007, 61, 523–528. [Google Scholar] [CrossRef]
- Frei, E. Albumin binding ligands and albumin conjugate uptake by cancer cells. Diabetol. Metab. Syndr. 2011, 3, 1–4. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, Y.; Wu, A.; Rao, Y.; Huang, Y. Roles of Albumin-Binding Proteins in Cancer Progression and Biomimetic Targeted Drug Delivery. ChemBioChem 2018, 19, 1796–1805. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Malkinson, M. The transmission of passive immunity to Escherichia coli from mother to young in the domestic fowl (Gallus domesticus). Immunology 1965, 9, 311. [Google Scholar]
- Chaudhury, C.; Mehnaz, S.; Robinson, J.M.; Hayton, W.L.; Pearl, D.K.; Roopenian, D.C.; Anderson, C.L. The major histocompatibility complex–related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 2003, 197, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Sarav, M.; Wang, Y.; Hack, B.K.; Chang, A.; Jensen, M.; Bao, L.; Quigg, R.J. Renal FcRn reclaims albumin but facilitates elimination of IgG. J. Am. Soc. Nephrol. 2009, 20, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bronson, C.; Hayton, W.L.; Radmacher, M.D.; Roopenian, D.C.; Robinson, J.M.; Anderson, C.L. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G352–G360. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.; Sung, A.; Horvat, R.; Bravo, J. Preferential interaction of albumin-binding proteins, gp30 and gp18, with conformationally modified albumins. Presence in many cells and tissues with a possible role in catabolism. J. Biol. Chem. 1992, 267, 24544–24553. [Google Scholar] [CrossRef]
- Bito, R.; Hino, S.; Baba, A.; Tanaka, M.; Watabe, H.; Kawabata, H. Degradation of oxidative stress-induced denatured albumin in rat liver endothelial cells. Am. J. Physiol.-Cell Physiol. 2005, 289, C531–C542. [Google Scholar] [CrossRef]
- Schnitzer, J.; Bravo, J. High affinity binding, endocytosis, and degradation of conformationally modified albumins. Potential role of gp30 and gp18 as novel scavenger receptors. J. Biol. Chem. 1993, 268, 7562–7570. [Google Scholar] [CrossRef]
- Ottnad, E.; Via, D.; Frübis, J.; Sinn, H.; Friedrich, E.; Ziegler, R.; Dresel, H. Differentiation of binding sites on reconstituted hepatic scavenger receptors using oxidized low-density lipoprotein. Biochem. J. 1992, 281, 745–751. [Google Scholar] [CrossRef]
- Peters, T., Jr.; Brennan, S.O. All about albumin: Biochemistry, genetics and medical applications. Trends Biochem. Sci. 1996, 21, 451. [Google Scholar]
- Birn, H.; Fyfe, J.C.; Jacobsen, C.; Mounier, F.; Verroust, P.J.; Ørskov, H.; Willnow, T.E.; Moestrup, S.K.; Christensen, E.I. Cubilin is an albumin binding protein important for renal tubular albumin reabsorption. J. Clin. Investig. 2000, 105, 1353–1361. [Google Scholar] [CrossRef]
- Christensen, E.I.; Birn, H.; Storm, T.; Weyer, K.; Nielsen, R. Endocytic receptors in the renal proximal tubule. Physiology 2012, 27, 223–236. [Google Scholar] [CrossRef]
- Weyer, K.; Storm, T.; Shan, J.; Vainio, S.; Kozyraki, R.; Verroust, P.J.; Christensen, E.I.; Nielsen, R. Mouse model of proximal tubule endocytic dysfunction. Nephrol. Dial. Transplant. 2011, 26, 3446–3451. [Google Scholar] [CrossRef] [PubMed]
- Amsellem, S.; Gburek, J.; Hamard, G.; Nielsen, R.; Willnow, T.E.; Devuyst, O.; Nexo, E.; Verroust, P.J.; Christensen, E.I.; Kozyraki, R. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J. Am. Soc. Nephrol. 2010, 21, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Babson, A.L.; Winnick, T. Protein transfer in tumor-bearing rats. Cancer Res. 1954, 14, 606–611. [Google Scholar] [PubMed]
- Stehle, G.; Sinn, H.; Wunder, A.; Schrenk, H.H.; Stewart, J.C.M.; Hartung, G.; Maier-Borst, W.; Heene, D.L. Plasma protein (albumin) catabolism by the tumor itself—implications for tumor metabolism and the genesis of cachexia. Crit. Rev. Oncol./Hematol. 1997, 26, 77–100. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Stehle, G.; Wunder, A.; Schrenk, H.H.; Hartung, G.; Heene, D.L.; Sinn, H. Albumin-based drug carriers: Comparison between serum albumins of different species on pharmacokinetics and tumor uptake of the conjugate. Anti-Cancer Drugs 1999, 10, 785–790. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Wong, A.D.; Ye, M.; Ulmschneider, M.B.; Searson, P.C. Quantitative analysis of the enhanced permeation and retention (EPR) effect. PLoS ONE 2015, 10, e0123461. [Google Scholar] [CrossRef]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Desai, N.P.; Trieu, V.; Hwang, L.Y.; Wu, R.; Soon-Shiong, P.; Gradishar, W.J. Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anti-Cancer Drugs 2008, 19, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tang, L. SPARC in tumor pathophysiology and as a potential therapeutic target. Curr. Pharm. Des. 2014, 20, 6182–6190. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.E.; Oh, P. Albondin-mediated capillary permeability to albumin. Differential role of receptors in endothelial transcytosis and endocytosis of native and modified albumins. J. Biol. Chem. 1994, 269, 6072–6082. [Google Scholar] [CrossRef]
- Stehle, G.; Wunder, A.; Sinn, H.; Schrenk, H.H.; Schütt, S.; Frei, E.; Hartung, G.; Maier-Borst, W.; Heene, D.L. Pharmacokinetics of methotrexate-albumin conjugates in tumor-bearing rats. Anti-Cancer Drugs 1997, 8, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Wunder, A.; Stehle, G.; Schrenk, H.H.; Hartung, G.; Heene, D.L.; Maier-Borst, W.; Sinn, H. Antitumor activity of methotrexate-albumin conjugates in rats bearing a Walker-256 carcinoma. Int. J. Cancer 1998, 76, 884–890. [Google Scholar] [CrossRef]
- Burger, A.M.; Hartung, G.; Stehle, G.; Sinn, H.; Fiebig, H.H. Pre-clinical evaluation of a methotrexate–albumin conjugate (MTX-HSA) in human tumor xenografts in vivo. Int. J. Cancer 2001, 92, 718–724. [Google Scholar] [CrossRef]
- Sasaki, K.; Ishihara, J.; Ishihara, A.; Miura, R.; Mansurov, A.; Fukunaga, K.; Hubbell, J.A. Engineered collagen-binding serum albumin as a drug conjugate carrier for cancer therapy. Sci. Adv. 2019, 5, eaaw6081. [Google Scholar] [CrossRef]
- Gao, J.; Jiang, S.; Zhang, X.; Fu, Y.; Liu, Z. Preparation, characterization and in vitro activity of a docetaxel–albumin conjugate. Bioorganic Chem. 2019, 83, 154–160. [Google Scholar] [CrossRef]
- Yazaki, P.J.; Kassa, T.; Cheung, C.-W.; Crow, D.M.; Sherman, M.A.; Bading, J.R.; Anderson, A.-L.J.; Colcher, D.; Raubitschek, A. Biodistribution and tumor imaging of an anti-CEA single-chain antibody–albumin fusion protein. Nucl. Med. Biol. 2008, 35, 151–158. [Google Scholar] [CrossRef]
- Joshi, M.R.; Yao, N.; Myers, K.A.; Li, Z. Human serum albumin and p53-activating peptide fusion protein is able to promote apoptosis and deliver fatty acid-modified molecules. PLoS ONE 2013, 8, e80926. [Google Scholar] [CrossRef]
- Dong, D.; Xia, G.; Li, Z.; Li, Z. Human serum albumin and HER2-binding affibody fusion proteins for targeted delivery of fatty acid-modified molecules and therapy. Mol. Pharm. 2016, 13, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Bakker, T.R.; Phillips, D.J.; Cornelius, A.; Zitt, C.; Göttler, T.; Sigrist, G.; Fiedler, U.; Ekawardhani, S.; Dolado, I. Design and characterization of MP0250, A tri-specific anti-HGF/anti-VEGF DARPin® drug candidate. mAbs 2017, 9, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Coester, C.; Kreuter, J.; Langer, K. Desolvation process and surface characterisation of protein nanoparticles. Int. J. Pharm. 2000, 194, 91–102. [Google Scholar] [CrossRef]
- Langer, K.; Balthasar, S.; Vogel, V.; Dinauer, N.; Von Briesen, H.; Schubert, D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2003, 257, 169–180. [Google Scholar] [CrossRef]
- Rosenberger, I.; Schmithals, C.; Vandooren, J.; Bianchessi, S.; Milani, P.; Locatelli, E.; Israel, L.L.; Hübner, F.; Matteoli, M.; Lellouche, J.-P. Physico-chemical and toxicological characterization of iron-containing albumin nanoparticles as platforms for medical imaging. J. Control. Release 2014, 194, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Ma, K.; Kim, T.H.; Lee, E.S.; Oh, K.T.; Park, E.-S.; Lee, K.C.; Youn, Y.S. Doxorubicin-loaded human serum albumin nanoparticles surface-modified with TNF-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types. Biomaterials 2012, 33, 1536–1546. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, W.; Huang, Y.; Fu, Y.; Cheng, Y. Paclitaxel loaded human serum albumin nanoparticles stabilized with intermolecular disulfide bonds. MedChemComm 2014, 5, 1658–1663. [Google Scholar] [CrossRef]
- Khan, A.A.; Paul, A.; Abbasi, S.; Prakash, S. Mitotic and antiapoptotic effects of nanoparticles coencapsulating human VEGF and human angiopoietin-1 on vascular endothelial cells. Int. J. Nanomed. 2011, 6, 1069. [Google Scholar]
- Raoufinia, R.; Balkani, S.; Keyhanvar, N.; Mahdavipor, B.; Abdolalizadeh, J. Human albumin purification: A modified and concise method. J. Immunoass. Immunochem. 2018, 39, 687–695. [Google Scholar] [CrossRef]
- Jahanshahi, M.; Babaei, Z. Protein nanoparticle: A unique system as drug delivery vehicles. Afr. J. Biotechnol. 2008, 7, 25. [Google Scholar]
- Reis, C.P.; Neufeld, R.J.; Ribeiro, A.J.; Veiga, F. Nanoencapsulation, I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2006, 2, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cui, F.; Cun, D.; Tao, A.; Shi, K.; Lin, W. Preparation, characterization and biodistribution of the lactone form of 10-hydroxycamptothecin (HCPT)-loaded bovine serum albumin (BSA) nanoparticles. Int. J. Pharm. 2007, 340, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.A.; Patel, D.M.; Patel, J.K.; Patel, D.H. Solvent Emulsification Evaporation and Solvent Emulsification Diffusion Techniques for Nanoparticles. In Emerging Technologies for Nanoparticle Manufacturing; Springer: Berlin/Heidelberg, Germany, 2021; pp. 287–300. [Google Scholar]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Desai, N. Nanoparticle albumin-bound paclitaxel (Abraxane®). In Albumin in Medicine; Springer: Berlin/Heidelberg, Germany, 2016; pp. 101–119. [Google Scholar]
- Srivastava, A.; Prajapati, A. Albumin and functionalized albumin nanoparticles: Production strategies, characterization, and target indications. Asian Biomed. 2020, 14, 217–242. [Google Scholar] [CrossRef]
- Desai, N. Nanoparticle albumin bound (nab) technology: Targeting tumors through the endothelial gp60 receptor and SPARC. Nanomed. Nanotechnol. Biol. Med. 2007, 4, 339. [Google Scholar] [CrossRef]
- Karami, E.; Behdani, M.; Kazemi-Lomedasht, F. Albumin nanoparticles as nanocarriers for drug delivery: Focusing on antibody and nanobody delivery and albumin-based drugs. J. Drug Deliv. Sci. Technol. 2020, 55, 101471. [Google Scholar] [CrossRef]
- Boye, J.I.; Alli, I.; Ismail, A.A. Interactions involved in the gelation of bovine serum albumin. J. Agric. Food Chem. 1996, 44, 996–1004. [Google Scholar] [CrossRef]
- Yu, S.; Yao, P.; Jiang, M.; Zhang, G. Nanogels prepared by self-assembly of oppositely charged globular proteins. Biopolym. Orig. Res. Biomol. 2006, 83, 148–158. [Google Scholar] [CrossRef]
- Qi, J.; Yao, P.; He, F.; Yu, C.; Huang, C. Nanoparticles with dextran/chitosan shell and BSA/chitosan core—doxorubicin loading and delivery. Int. J. Pharm. 2010, 393, 177–185. [Google Scholar] [CrossRef]
- Ré, M.-I. Formulating drug delivery systems by spray drying. Dry. Technol. 2006, 24, 433–446. [Google Scholar] [CrossRef]
- Pedrozo, R.C.; Antônio, E.; Khalil, N.M.; Mainardes, R.M. Bovine serum albumin-based nanoparticles containing the flavonoid rutin produced by nano spray drying. Braz. J. Pharm. Sci. 2020, 56, e17692. [Google Scholar] [CrossRef]
- Lee, S.H.; Heng, D.; Ng, W.K.; Chan, H.-K.; Tan, R.B. Nano spray drying: A novel method for preparing protein nanoparticles for protein therapy. Int. J. Pharm. 2011, 403, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Meghani, N.; Loebenberg, R.; Cui, J.-H.; Cao, Q.-R.; Lee, B.-J. Fatty acid chain length impacts nanonizing capacity of albumin-fatty acid nanomicelles: Enhanced physicochemical property and cellular delivery of poorly water-soluble drug. Eur. J. Pharm. Biopharm. 2020, 152, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Fisher, M.; Juliano, R. Targeted albumin-based nanoparticles for delivery of amphipathic drugs. Bioconjugate Chem. 2011, 22, 870–878. [Google Scholar] [CrossRef]
- Dai, L.; Li, C.-X.; Liu, K.-F.; Su, H.-J.; Chen, B.-Q.; Zhang, G.-F.; He, J.; Lei, J.-D. Self-assembled serum albumin–poly (l-lactic acid) nanoparticles: A novel nanoparticle platform for drug delivery in cancer. RSC Adv. 2015, 5, 15612–15620. [Google Scholar] [CrossRef]
- Qu, N.; Lee, R.J.; Sun, Y.; Cai, G.; Wang, J.; Wang, M.; Lu, J.; Meng, Q.; Teng, L.; Wang, D. Cabazitaxel-loaded human serum albumin nanoparticles as a therapeutic agent against prostate cancer. Int. J. Nanomed. 2016, 11, 3451. [Google Scholar]
- Kudłacik-Kramarczyk, S.; Drabczyk, A.; Głąb, M.; Gajda, P.; Czopek, A.; Zagórska, A.; Jaromin, A.; Gubernator, J.; Makara, A.; Tyliszczak, B. The development of the innovative synthesis methodology of albumin nanoparticles supported by their physicochemical, cytotoxic and hemolytic evaluation. Materials 2021, 14, 4386. [Google Scholar] [CrossRef]
- Matei, I.; Buta, C.M.; Turcu, I.M.; Culita, D.; Munteanu, C.; Ionita, G. Formation and Stabilization of Gold Nanoparticles in Bovine Serum Albumin Solution. Molecules 2019, 24, 3395. [Google Scholar] [CrossRef]
- Khullar, P.; Singh, V.; Mahal, A.; Dave, P.N.; Thakur, S.; Kaur, G.; Singh, J.; Singh Kamboj, S.; Singh Bakshi, M. Bovine Serum Albumin Bioconjugated Gold Nanoparticles: Synthesis, Hemolysis, and Cytotoxicity toward Cancer Cell Lines. J. Phys. Chem. C 2012, 116, 8834–8843. [Google Scholar] [CrossRef]
- Boulos, S.P.; Davis, T.A.; Yang, J.A.; Lohse, S.E.; Alkilany, A.M.; Holland, L.A.; Murphy, C.J. Nanoparticle–Protein Interactions: A Thermodynamic and Kinetic Study of the Adsorption of Bovine Serum Albumin to Gold Nanoparticle Surfaces. Langmuir 2013, 29, 14984–14996. [Google Scholar] [CrossRef]
- Ruttala, H.B.; Ramasamy, T.; Poudel, B.K.; Ruttala, R.R.T.; Jin, S.G.; Choi, H.-G.; Ku, S.-K.; Yong, C.S.; Kim, J.O. Multi-responsive albumin-lonidamine conjugated hybridized gold nanoparticle as a combined photothermal-chemotherapy for synergistic tumor ablation. Acta Biomater. 2020, 101, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Kalidasan, V.; Liu, X.L.; Herng, T.S.; Yang, Y.; Ding, J. Bovine Serum Albumin-Conjugated Ferrimagnetic Iron Oxide Nanoparticles to Enhance the Biocompatibility and Magnetic Hyperthermia Performance. Nano-Micro Lett. 2016, 8, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Lian, X.; Dong, J.; Wan, Z.; Xia, C.; Song, X.; Fu, Y.; Gong, T.; Zhang, Z. Co-delivery of pirarubicin and paclitaxel by human serum albumin nanoparticles to enhance antitumor effect and reduce systemic toxicity in breast cancers. Mol. Pharm. 2015, 12, 4085–4098. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Sun, Y.; Li, Y.; Hao, F.; Qiu, P.; Teng, L.; Xie, J.; Gao, Y. Docetaxel-loaded human serum albumin (HSA) nanoparticles: Synthesis, characterization, and evaluation. Biomed. Eng. Online 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Huh, M.; Lee, S.J.; Koo, H.; Kwon, I.C.; Jeong, S.Y.; Kim, K. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics 2011, 1, 230. [Google Scholar] [CrossRef]
- Jiang, C.; Cheng, H.; Yuan, A.; Tang, X.; Wu, J.; Hu, Y. Hydrophobic IR780 encapsulated in biodegradable human serum albumin nanoparticles for photothermal and photodynamic therapy. Acta Biomater. 2015, 14, 61–69. [Google Scholar] [CrossRef]
- Son, S.; Song, S.; Lee, S.J.; Min, S.; Kim, S.A.; Yhee, J.Y.; Huh, M.S.; Kwon, I.C.; Jeong, S.Y.; Byun, Y. Self-crosslinked human serum albumin nanocarriers for systemic delivery of polymerized siRNA to tumors. Biomaterials 2013, 34, 9475–9485. [Google Scholar] [CrossRef]
- Kim, B.; Lee, C.; Lee, E.S.; Shin, B.S.; Youn, Y.S. Paclitaxel and curcumin co-bound albumin nanoparticles having antitumor potential to pancreatic cancer. Asian J. Pharm. Sci. 2016, 11, 708–714. [Google Scholar] [CrossRef]
- Kushwah, V.; Katiyar, S.S.; Dora, C.P.; Agrawal, A.K.; Lamprou, D.A.; Gupta, R.C.; Jain, S. Co-delivery of docetaxel and gemcitabine by anacardic acid modified self-assembled albumin nanoparticles for effective breast cancer management. Acta Biomater. 2018, 73, 424–436. [Google Scholar] [CrossRef]
- Huang, H.; Shi, H.; Liu, J.; Min, Y.; Wang, Y.; Wang, A.Z.; Wang, J.; Liu, Y. Co-delivery of all-trans-retinoic acid enhances the anti-metastasis effect of albumin-bound paclitaxel nanoparticles. Chem. Commun. 2017, 53, 212–215. [Google Scholar] [CrossRef]
- Yu, X.; Zhu, W.; Di, Y.; Gu, J.; Guo, Z.; Li, H.; Fu, D.; Jin, C. Triple-functional albumin-based nanoparticles for combined chemotherapy and photodynamic therapy of pancreatic cancer with lymphatic metastases. Int. J. Nanomed. 2017, 12, 6771. [Google Scholar] [CrossRef] [PubMed]
- Khandelia, R.; Bhandari, S.; Pan, U.N.; Ghosh, S.S.; Chattopadhyay, A. Gold nanocluster embedded albumin nanoparticles for two-photon imaging of cancer cells accompanying drug delivery. Small 2015, 11, 4075–4081. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Byeon, H.J.; Choi, J.S.; Thao, L.; Kim, I.; Lee, E.S.; Shin, B.S.; Lee, K.C.; Youn, Y.S. Inhalable self-assembled albumin nanoparticles for treating drug-resistant lung cancer. J. Control. Release 2015, 197, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.D.; Alam, N.; Saneja, A.; Khare, V.; Kumar, A.; Vaidh, S.; Mahajan, G.; Sharma, P.R.; Singh, S.K.; Mondhe, D.M. Development and evaluation of folate functionalized albumin nanoparticles for targeted delivery of gemcitabine. Int. J. Pharm. 2015, 492, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Sun, S.; Gong, X. Preparation, characterization, and antiproliferative activities of biotin-decorated docetaxel-loaded bovine serum albumin nanoparticles. Braz. J. Pharm. Sci. 2018, 54, 54. [Google Scholar] [CrossRef]
- Karmali, P.P.; Kotamraju, V.R.; Kastantin, M.; Black, M.; Missirlis, D.; Tirrell, M.; Ruoslahti, E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 73–82. [Google Scholar] [CrossRef]
- Anhorn, M.G.; Wagner, S.; Kreuter, J.; Langer, K.; Von Briesen, H. Specific targeting of HER2 overexpressing breast cancer cells with doxorubicin-loaded trastuzumab-modified human serum albumin nanoparticles. Bioconjugate Chem. 2008, 19, 2321–2331. [Google Scholar] [CrossRef]
- Abbasi, S.; Paul, A.; Shao, W.; Prakash, S. Cationic albumin nanoparticles for enhanced drug delivery to treat breast cancer: Preparation and in vitro assessment. J. Drug Deliv. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Hartung, G.; Stehle, G.; Sinn, H.; Wunder, A.; Schrenk, H.H.; Heeger, S.; Kränzle, M.; Edler, L.; Frei, E.; Fiebig, H.H. Phase I Trial of Methotrexate-Albumin in a Weekly Intravenous Bolus Regimen in Cancer Patients. Clin. Cancer Res. 1999, 5, 753–759. [Google Scholar]
- Vis, A.; Van Der Gaast, A.; Van Rhijn, B.; Catsburg, T.; Schmidt, C.; Mickisch, G. A phase II trial of methotrexate-human serum albumin (MTX-HSA) in patients with metastatic renal cell carcinoma who progressed under immunotherapy. Cancer Chemother. Pharmacol. 2002, 49, 342–345. [Google Scholar]
- Kudla, A.; Adiwijaya, B.; Paragas, V.; Richards, D.; Braiteh, F.; Garcia, A.; Denlinger, C.; Conkling, P.; Edenfield, W.; Anthony, S. Biomarker Analysis of a Phase 1 Study of Mm-111, A Bispecific Her2/Her3 Antibody Fusion Protein, in Combination with Multiple Treatment Regimens in Patients with Advanced Her2 Positive Solid Tumors. Ann. Oncol. 2014, 25, iv81. [Google Scholar] [CrossRef]
- Richards, D.; Braiteh, F.; Anthony, S.; Edenfield, W.; Hellerstedt, B.; Raju, R.; Conkling, P.; McDonagh, C.; Frye, S.; Moyo, V. A phase 1 study of MM-111; a bispecific HER2/HER3 antibody fusion protein, combined with multiple treatment regimens in patients with advanced HER2 positive solid tumors. Ann. Oncol. 2012, 23, ix170. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Baird, R.D.; Linossi, C.; Middleton, M.; Lord, S.; Harris, A.; Rodón, J.; Zitt, C.; Fiedler, U.; Dawson, K.M.; Leupin, N. First-in-human phase i study of MP0250, A first-in-class DARPin drug candidate targeting VEGF and HGF, in patients with advanced solid tumors. J. Clin. Oncol. 2021, 39, 145–154. [Google Scholar] [CrossRef]
- Azaro, A.; Rodon, J.; Middleton, M.R.; Baird, R.D.; Herrmann, R.; Fiedler, U.; Haunschild, J.; Häuptle, M.; Hermann, F.J.; Schreiner, S. First-in-class phase I study evaluating MP0250, a VEGF and HGF neutralizing DARPIN molecule, in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, 2520. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; De Diego Puente, T. A compressive review about taxol®: History and future challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Li, C.; Li, Y.; Gao, Y.; Wei, N.; Zhao, X.; Wang, C.; Li, Y.; Xiu, X.; Cui, J. Direct comparison of two albumin-based paclitaxel-loaded nanoparticle formulations: Is the crosslinked version more advantageous? Int. J. Pharm. 2014, 468, 15–25. [Google Scholar] [CrossRef]
- Desai, N.; Trieu, V.; Damascelli, B.; Soon-Shiong, P. SPARC expression correlates with tumor response to albumin-bound paclitaxel in head and neck cancer patients. Transl. Oncol. 2009, 2, 59–64. [Google Scholar] [CrossRef]
- FDA. ABRAXANE® for Injectable Suspension (Paclitaxel Protein-Bound Particles for Injectable Suspension)(Albumin-Bound). 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021660s040lbl.pdf (accessed on 19 January 2022).
- Ibrahim, N.K.; Desai, N.; Legha, S.; Soon-Shiong, P.; Theriault, R.L.; Rivera, E.; Esmaeli, B.; Ring, S.E.; Bedikian, A.; Hortobagyi, G.N. Phase I and pharmacokinetic study of ABI-007, A Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel. Clin. Cancer Res. 2002, 8, 1038–1044. [Google Scholar]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. Int. J. Nanomed. 2009, 4, 99. [Google Scholar]
- Hersh, E.; O’Day, S.; Gonzalez, R.; Ribas, A.; Samlowski, W.; Gordon, M. Phase II trial of ABI-007 (Abraxane) in previously treated and chemotherapy naive patients with metastatic melanoma. Melanoma Res. 2006, 16, S78. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil–based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef] [PubMed]
- Roy, V.; LaPlant, B.; Gross, G.; Bane, C.; Palmieri, F.; Group, N.C.C.T. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel)(Abraxane®) in combination with gemcitabine in patients with metastatic breast cancer (N0531). Ann. Oncol. 2009, 20, 449–453. [Google Scholar] [CrossRef]
- Socinski, M.A.; Bondarenko, I.; Karaseva, N.; Makhson, A.; Vynnichenko, I.; Okamoto, I.; Hon, J.; Hirsh, V.; Bhar, P.; Iglesias, J. Results of a randomized, phase III trial of nab-paclitaxel (nab-P) and carboplatin (C) compared with cremophor-based paclitaxel (P) and carboplatin as first-line therapy in advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2010, 28, LBA7511. [Google Scholar] [CrossRef]
- Lobo, C.; Lopes, G.; Silva, O.; Gluck, S. Paclitaxel albumin-bound particles (abraxane™) in combination with bevacizumab with or without gemcitabine: Early experience at the University of Miami/Braman Family Breast Cancer Institute. Biomed. Pharmacother. 2007, 61, 531–533. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, Z.-S.; Zeng, Z. Clinical efficacy and safety of combination of abraxane and trastuzumab in treatment of recurrent ovarian cancer. Pak. J. Pharm. Sci. 2018, 31, 2831–2834. [Google Scholar]
- Robidoux, A.; Buzdar, A.U.; Quinaux, E.; Jacobs, S.; Rastogi, P.; Fourchotte, V.; Younan, R.J.; Pajon, E.R.; Shalaby, I.A.; Desai, A.M. A phase II neoadjuvant trial of sequential nanoparticle albumin-bound paclitaxel followed by 5-fluorouracil/epirubicin/cyclophosphamide in locally advanced breast cancer. Clin. Breast Cancer 2010, 10, 81–86. [Google Scholar] [CrossRef]
- De, T.; Trieu, V.; Yim, Z.; Cordia, J.; Yang, A.; Beals, B.; Ci, S.; Louie, L.; Desai, N. Nanoparticle albumin-bound (nab) rapamycin as an anticancer agent. AACR 2007, 67, 4719. [Google Scholar]
- Desai, N.; Trieu, V.; Yang, A.; De, T.; Cordia, J.; Yim, Z.; Ci, S.; Louie, L.; Grim, B.B.; Azoulay, J. Enhanced efficacy and safety of nanoparticle albumin-bound nab-docetaxel versus taxotere. AACR 2006, 66, 1277–1278. [Google Scholar]
- Trieu, V.; De, T.; Labao, E.; Ci, S.; Louie, L.; Tao, C.; Wang, Q.; Yim, Z.; Hawkins, M.; Soon-Shiong, P. Anti-angiogenic and antitumor activity of nanoparticle albumin bound (nab) thiocolchicine dimer (IDN5404) with a novel dual mechanism of action on Tubulin and Topoisomerase-1. AACR 2006, 66, 899. [Google Scholar]
- Avisar, N.; Pukac, L.; Adar, L.; Barash, S.; Clark, S.; Liu, P.; Bock, J.; Shen, W.D. Recombinant Albumin-Partnering Technology: Development Of Balugrastim, a Novel Long-Acting Granulocyte Colony-Stimulating Factor. Blood 2013, 122, 4854. [Google Scholar] [CrossRef]
- Hao, L.; Zhou, Q.; Piao, Y.; Zhou, Z.; Tang, J.; Shen, Y. Albumin-binding prodrugs via reversible iminoboronate forming nanoparticles for cancer drug delivery. J. Control. Release 2021, 330, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Yousefpour, P.; Ahn, L.; Tewksbury, J.; Saha, S.; Costa, S.A.; Bellucci, J.J.; Li, X.; Chilkoti, A. Conjugate of Doxorubicin to Albumin-Binding Peptide Outperforms Aldoxorubicin. Small 2019, 15, 1804452. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Mita, M.M.; Natale, R.B.; Wolin, E.M.; Laabs, B.; Dinh, H.; Wieland, S.; Levitt, D.J.; Mita, A.C. Pharmacokinetic study of aldoxorubicin in patients with solid tumors. Investig. New Drugs 2015, 33, 341–348. [Google Scholar] [CrossRef]
- Gong, J.; Yan, J.; Forscher, C.; Hendifar, A. Aldoxorubicin: A tumor-targeted doxorubicin conjugate for relapsed or refractory soft tissue sarcomas. Drug Des. Devel. Ther. 2018, 12, 777–786. [Google Scholar] [CrossRef]
- Sun, I.-C.; Yoon, H.Y.; Lim, D.-K.; Kim, K. Recent Trends in In Situ Enzyme-Activatable Prodrugs for Targeted Cancer Therapy. Bioconjugate Chem. 2020, 31, 1012–1024. [Google Scholar] [CrossRef]
- Brown, J.M. Tumor Microenvironment and the Response to Anticancer Therapy. Cancer Biol. Ther. 2002, 1, 453–458. [Google Scholar] [CrossRef]
- Shim, M.K.; Moon, Y.; Yang, S.; Kim, J.; Cho, H.; Lim, S.; Yoon, H.Y.; Seong, J.-K.; Kim, K. Cancer-specific drug-drug nanoparticles of pro-apoptotic and cathepsin B-cleavable peptide-conjugated doxorubicin for drug-resistant cancer therapy. Biomaterials 2020, 261, 120347. [Google Scholar] [CrossRef]
- Lee, S.; Xie, J.; Chen, X.J.B. Peptide-based probes for targeted molecular imaging. Biochemistry 2010, 49, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Um, W.; Park, J.; Ko, H.; Lim, S.; Yoon, H.Y.; Shim, M.K.; Lee, S.; Ko, Y.J.; Kim, M.J.; Park, J.H.; et al. Visible light-induced apoptosis activatable nanoparticles of photosensitizer-DEVD-anticancer drug conjugate for targeted cancer therapy. Biomaterials 2019, 224, 119494. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Shim, M.K.; Kim, W.J.; Choi, J.; Nam, G.-H.; Kim, J.; Kim, J.; Moon, Y.; Kim, H.Y.; Park, J.; et al. Cancer-activated doxorubicin prodrug nanoparticles induce preferential immune response with minimal doxorubicin-related toxicity. Biomaterials 2021, 272, 120791. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Sloane, B.F. Cathepsin B: Multiple roles in cancer. PROTEOMICS Clin. Appl. 2014, 8, 427–437. [Google Scholar] [CrossRef]
- Moon, Y.; Shim, M.K.; Choi, J.; Yang, S.; Kim, J.; Yun, W.S.; Cho, H.; Park, J.Y.; Kim, Y.; Seong, J.-K.; et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics 2022, 12, 1999–2014. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Kim, H.Y.; Um, S.H.; Sung, Y.; Shim, M.K.; Yang, S.; Park, J.; Kim, E.S.; Kim, K.; Kwon, I.C.; Ryu, J.H. Epidermal growth factor (EGF)-based activatable probe for predicting therapeutic outcome of an EGF-based doxorubicin prodrug. J. Control. Release 2020, 328, 222–236. [Google Scholar] [CrossRef]
- Kim, J.; Shim, M.K.; Cho, Y.-J.; Jeon, S.; Moon, Y.; Choi, J.; Kim, J.; Lee, J.; Lee, J.-W.; Kim, K. The safe and effective intraperitoneal chemotherapy with cathepsin B-specific doxorubicin prodrug nanoparticles in ovarian cancer with peritoneal carcinomatosis. Biomaterials 2021, 279, 121189. [Google Scholar] [CrossRef]
- Du, J.; Lane, L.A.; Nie, S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J. Control. Release 2015, 219, 205–214. [Google Scholar] [CrossRef]
- Kim, J.; Shim, M.K.; Yang, S.; Moon, Y.; Song, S.; Choi, J.; Kim, J.; Kim, K. Combination of cancer-specific prodrug nanoparticle with Bcl-2 inhibitor to overcome acquired drug resistance. J. Control. Release 2021, 330, 920–932. [Google Scholar] [CrossRef]
- Cho, H.; Shim, M.K.; Yang, S.; Song, S.; Moon, Y.; Kim, J.; Byun, Y.; Ahn, C.-H.; Kim, K. Cathepsin B-Overexpressed Tumor Cell Activatable Albumin-Binding Doxorubicin Prodrug for Cancer-Targeted Therapy. Pharmaceutics 2022, 14, 83. [Google Scholar] [CrossRef] [PubMed]
- Abu Ajaj, K.; Graeser, R.; Fichtner, I.; Kratz, F. In vitro and in vivo study of an albumin-binding prodrug of doxorubicin that is cleaved by cathepsin B. Cancer Chemother. Pharmacol. 2009, 64, 413. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Timmer, J.C.; Salvesen, G.S. Caspase substrates. Cell Death Differ. 2007, 14, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Keane, M.M.; Ettenberg, S.A.; Nau, M.M.; Russell, E.K.; Lipkowitz, S. Chemotherapy Augments TRAIL-induced Apoptosis in Breast Cell Lines. Cancer Res. 1999, 59, 734. [Google Scholar]
- Chung, S.W.; Choi, J.u.; Lee, B.S.; Byun, J.; Jeon, O.-C.; Kim, S.W.; Kim, I.-S.; Kim, S.Y.; Byun, Y. Albumin-binding caspase-cleavable prodrug that is selectively activated in radiation exposed local tumor. Biomaterials 2016, 94, 1–8. [Google Scholar] [CrossRef]
- Zhao, N.; Wu, B.; Hu, X.; Xing, D. NIR-triggered high-efficient photodynamic and chemo-cascade therapy using caspase-3 responsive functionalized upconversion nanoparticles. Biomaterials 2017, 141, 40–49. [Google Scholar] [CrossRef]
- Chung, S.W.; Choi, J.U.; Cho, Y.S.; Kim, H.R.; Won, T.H.; Dimitrion, P.; Jeon, O.-C.; Kim, S.W.; Kim, I.-S.; Kim, S.Y.; et al. Self-Triggered Apoptosis Enzyme Prodrug Therapy (STAEPT): Enhancing Targeted Therapies via Recurrent Bystander Killing Effect by Exploiting Caspase-Cleavable Linker. Adv. Sci. 2018, 5, 1800368. [Google Scholar] [CrossRef]
- Wang, Y.; Du, W.; Zhang, T.; Zhu, Y.; Ni, Y.; Wang, C.; Sierra Raya, F.M.; Zou, L.; Wang, L.; Liang, G. A Self-Evaluating Photothermal Therapeutic Nanoparticle. ACS Nano 2020, 14, 9585–9593. [Google Scholar] [CrossRef]
- Chung, S.W.; Cho, Y.S.; Choi, J.U.; Kim, H.R.; Won, T.H.; Kim, S.Y.; Byun, Y. Highly potent monomethyl auristatin E prodrug activated by caspase-3 for the chemoradiotherapy of triple-negative breast cancer. Biomaterials 2019, 192, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Bloomston, M.; Zervos, E.E.; Rosemurgy, A.S. Matrix metalloproteinases and their role in pancreatic cancer: A review of preclinical studies and clinical trials. Ann. Surg. Oncol. 2002, 9, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Alaseem, A.; Alhazzani, K.; Dondapati, P.; Alobid, S.; Bishayee, A.; Rathinavelu, A. Matrix Metalloproteinases: A challenging paradigm of cancer management. Semin. Cancer Biol. 2019, 56, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Mansour, A.M.; Drevs, J.; Esser, N.; Hamada, F.M.; Badary, O.A.; Unger, C.; Fichtner, I.; Kratz, F. A New Approach for the Treatment of Malignant Melanoma: Enhanced Antitumor Efficacy of an Albumin-binding Doxorubicin Prodrug That Is Cleaved by Matrix Metalloproteinase 21. Cancer Res. 2003, 63, 4062–4066. [Google Scholar]
- Stamey, T.A.; Yang, N.; Hay, A.R.; McNeal, J.E.; Freiha, F.S.; Redwine, E. Prostate-Specific Antigen as a Serum Marker for Adenocarcinoma of the Prostate. New Engl. J. Med. 1987, 317, 909–916. [Google Scholar] [CrossRef]
- Kratz, F.; Mansour, A.; Soltau, J.; Warnecke, A.; Fichtner, I.; Unger, C.; Drevs, J. Development of Albumin-binding Doxorubicin Prodrugs that are Cleaved by Prostate-specific Antigen. Arch. Der Pharm. 2005, 338, 462–472. [Google Scholar] [CrossRef]
- Cook, J.A.; Gius, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 2004, 14, 259–266. [Google Scholar] [CrossRef]
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.-C.; Zhou, G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, K.; Na, K.; Li, D.; Li, Z.; Zhao, D.; Zhong, L.; Wang, M.; Kou, L.; Luo, C.; et al. Striking a Balance between Carbonate/Carbamate Linkage Bond- and Reduction-Sensitive Disulfide Bond-Bearing Linker for Tailored Controlled Release: In Situ Covalent-Albumin-Binding Gemcitabine Prodrugs Promote Bioavailability and Tumor Accumulation. J. Med. Chem. 2018, 61, 4904–4917. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.H.; Shim, M.K.; Kim, G.L.; Kim, S.; Kang, H.; Kim, J.-H. Hypoxia-responsive folic acid conjugated glycol chitosan nanoparticle for enhanced tumor targeting treatment. Int. J. Pharm. 2020, 580, 119237. [Google Scholar] [CrossRef]
- Jang, E.H.; Kim, G.L.; Park, M.G.; Shim, M.K.; Kim, J.-H. Hypoxia-responsive, organic-inorganic hybrid mesoporous silica nanoparticles for triggered drug release. J. Drug Deliv. Sci. Technol. 2020, 56, 101543. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef]
- Patel, A.; Sant, S. Hypoxic tumor microenvironment: Opportunities to develop targeted therapies. Biotechnol. Adv. 2016, 34, 803–812. [Google Scholar] [CrossRef]
- Cheng, Z.; Huang, Y.; Shao, P.; Wang, L.; Zhu, S.; Yu, J.; Lu, W. Hypoxia-Activated Albumin-Binding Exatecan Prodrug for Cancer Therapy. ACS Omega 2022, 7, 1082–1089. [Google Scholar] [CrossRef]
- Bolling, C.; Graefe, T.; Lübbing, C.; Jankevicius, F.; Uktveris, S.; Cesas, A.; Meyer-Moldenhauer, W.-H.; Starkmann, H.; Weigel, M.; Burk, K. Phase II study of MTX-HSA in combination with cisplatin as first line treatment in patients with advanced or metastatic transitional cell carcinoma. Investigational new drugs 2006, 24, 521–527. [Google Scholar] [CrossRef]
- Richards, D.A.; Braiteh, F.S.; Garcia, A.; Denlinger, C.S.; Conkling, P.R.; Edenfield, W.J.; Anthony, S.P.; Hellerstedt, B.A.; Raju, R.N.; Becerra, C. A phase 1 study of MM-111, a bispecific HER2/HER3 antibody fusion protein, combined with multiple treatment regimens in patients with advanced HER2-positive solid tumors. J. Clin. Oncol. 2014, 32, 651. [Google Scholar] [CrossRef]
- Denlinger, C.; Beeram, M.; Tolcher, A.; Goldstein, L.; Slichenmyer, W.; Murray, J.; McDonagh, C.; Andreas, K.; Moyo, V. A phase I/II and pharmacologic study of MM-111 in patients with advanced, refractory HER2-positive (HER2+) cancers. J. Clin. Oncol. 2010, 28, TPS169. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.; Jeon, S.I.; Ahn, C.-H.; Shim, M.K.; Kim, K. Emerging Albumin-Binding Anticancer Drugs for Tumor-Targeted Drug Delivery: Current Understandings and Clinical Translation. Pharmaceutics 2022, 14, 728. https://doi.org/10.3390/pharmaceutics14040728
Cho H, Jeon SI, Ahn C-H, Shim MK, Kim K. Emerging Albumin-Binding Anticancer Drugs for Tumor-Targeted Drug Delivery: Current Understandings and Clinical Translation. Pharmaceutics. 2022; 14(4):728. https://doi.org/10.3390/pharmaceutics14040728
Chicago/Turabian StyleCho, Hanhee, Seong Ik Jeon, Cheol-Hee Ahn, Man Kyu Shim, and Kwangmeyung Kim. 2022. "Emerging Albumin-Binding Anticancer Drugs for Tumor-Targeted Drug Delivery: Current Understandings and Clinical Translation" Pharmaceutics 14, no. 4: 728. https://doi.org/10.3390/pharmaceutics14040728
APA StyleCho, H., Jeon, S. I., Ahn, C.-H., Shim, M. K., & Kim, K. (2022). Emerging Albumin-Binding Anticancer Drugs for Tumor-Targeted Drug Delivery: Current Understandings and Clinical Translation. Pharmaceutics, 14(4), 728. https://doi.org/10.3390/pharmaceutics14040728