
Nanotechnology in Immunotherapy for Type 1 Diabetes: Promising Innovations and Future Advances

 , ,
, ,  and
and 
Abstract
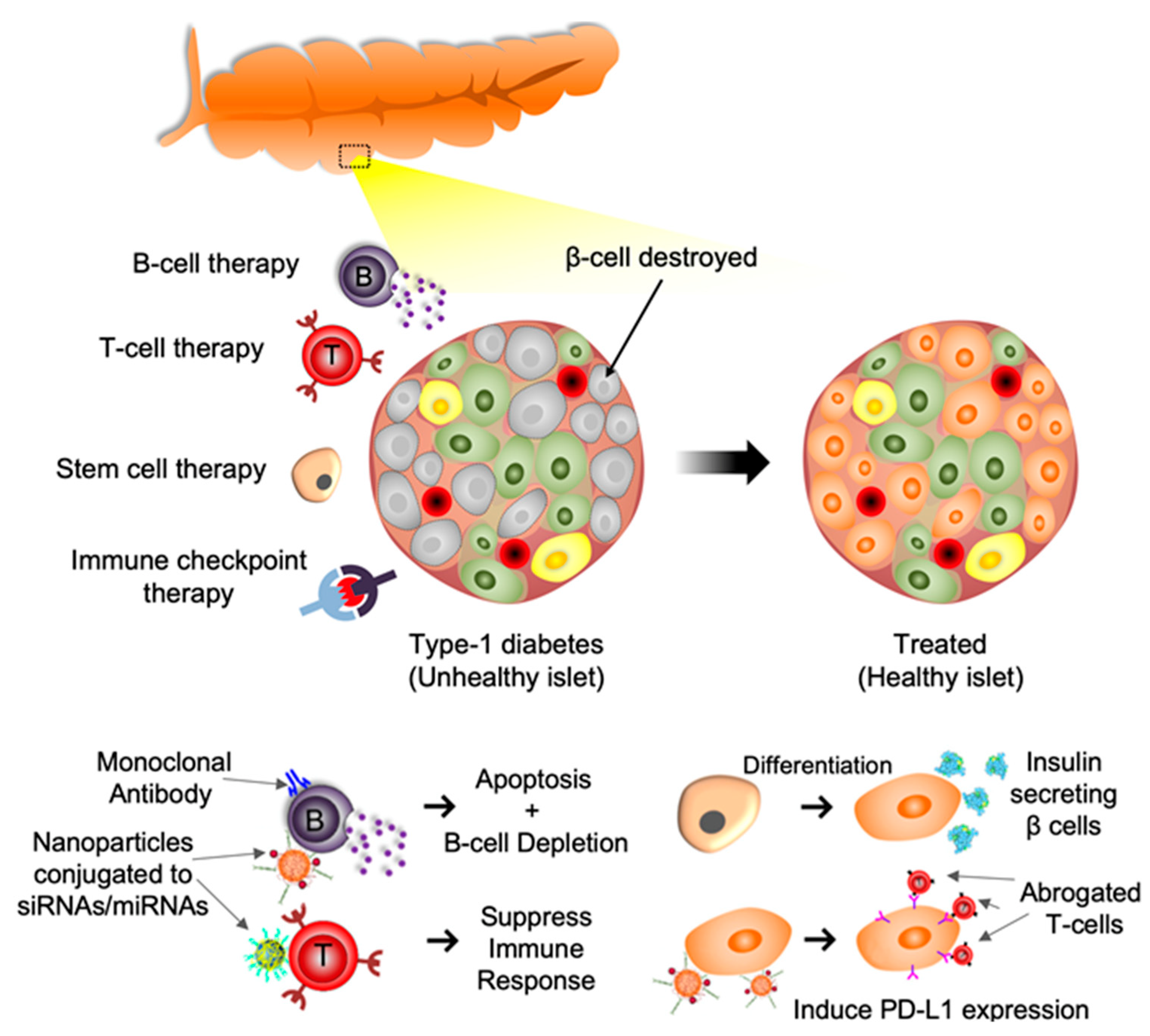
1. Introduction
1.1. Understanding T1D: Etiology and Current Clinical Scenario
1.2. T Cell Based Therapy
1.3. B Cell Based Therapy
1.4. Immune Checkpoint Molecules-Based Therapy
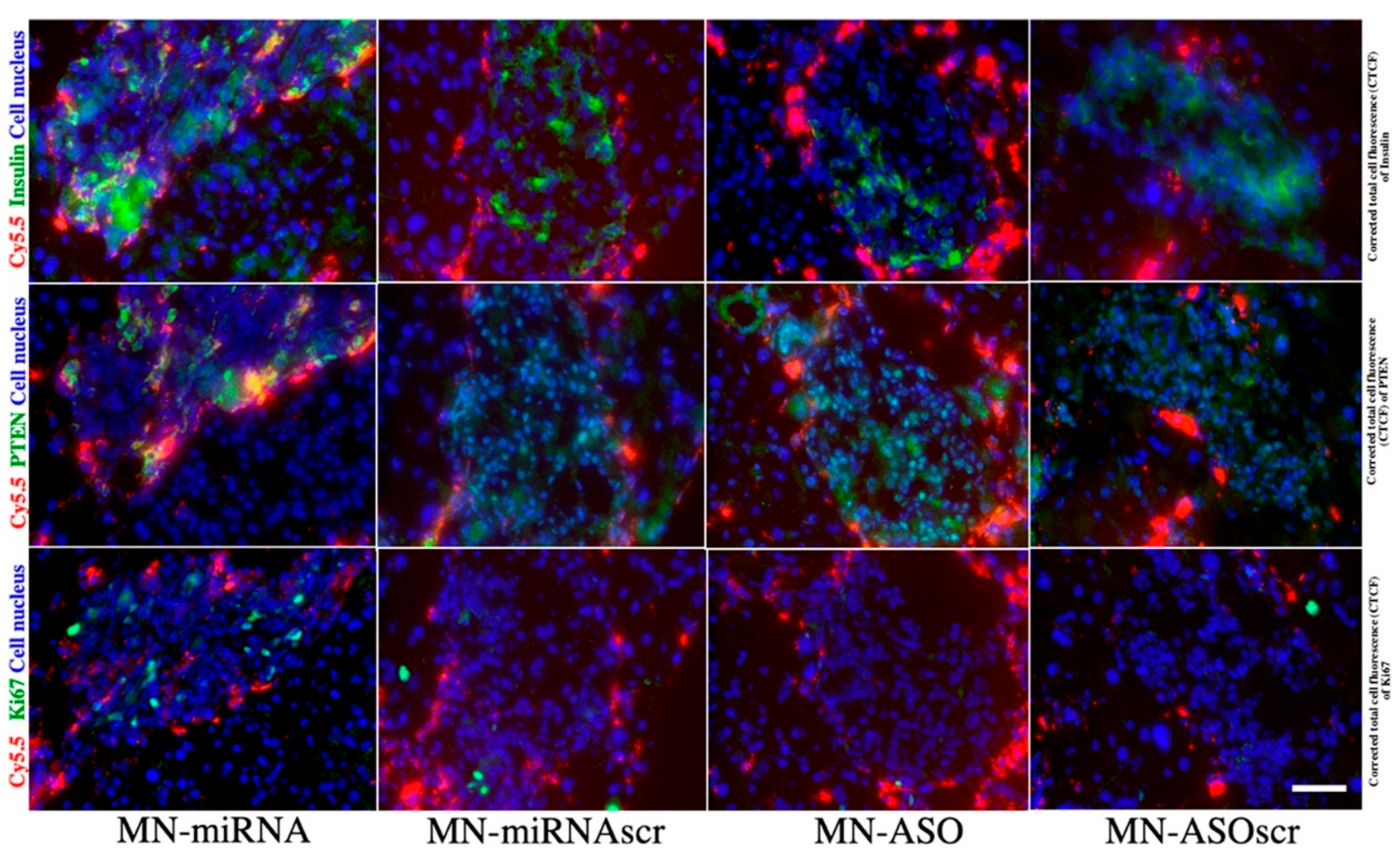
1.5. Extracellular Vesicles and miRNA-Based Therapy
1.6. Stem Cell Targeted Therapy
2. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Kappler, J.; Kotzin, B.L. Autoimmune disease: Why and where it occurs. Nat. Med. 2001, 7, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.-F. Insulin-Dependent Diabetes Mellitus as an Autoimmune Disease. Endocr. Rev. 1994, 15, 516–542. [Google Scholar] [CrossRef] [PubMed]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Riddell, M.C.; Gallen, I.W.; Smart, C.E.; Taplin, C.E.; Adolfsson, P.; Lumb, A.N.; Kowalski, A.; Rabasa-Lhoret, R.; McCrimmon, R.J.; Hume, C.; et al. Exercise management in type 1 diabetes: A consensus statement. Lancet Diabetes Endocrinol. 2017, 5, 377–390. [Google Scholar] [CrossRef]
- Lennerz, B.S.; Barton, A.; Bernstein, R.K.; Dikeman, R.D.; Diulus, C.; Hallberg, S.; Rhodes, E.T.; Ebbeling, C.B.; Westman, E.C.; Yancy, W.S.; et al. Management of Type 1 Diabetes With a Very Low–Carbohydrate Diet. Pediatrics 2018, 141, e20173349. [Google Scholar] [CrossRef]
- Pickup, J.C. Insulin-Pump Therapy for Type 1 Diabetes Mellitus. N. Engl. J. Med. 2012, 366, 1616–1624. [Google Scholar] [CrossRef]
- Zinman, B. Newer insulin analogs: Advances in basal insulin replacement. Diabetes Obes. Metab. 2013, 15, 6–10. [Google Scholar] [CrossRef]
- Vardi, M.; Jacobson, E.; Nini, A.; Bitterman, H. Intermediate acting versus long acting insulin for type 1 diabetes mellitus. Cochrane Database Syst. Rev. 2008, 2008, CD006297. [Google Scholar] [CrossRef]
- Brawerman, G.; Thompson, P.J. Beta Cell Therapies for Preventing Type 1 Diabetes: From Bench to Bedside. Biomolecules 2020, 10, 1681. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, S.; Sawatani, T.; Van Mulders, A.; De Leu, N.; Heremans, Y.; Heimberg, H.; Cnop, M.; Staels, W. Towards a Functional Cure for Diabetes Using Stem Cell-Derived Beta Cells: Are We There Yet? Cells 2021, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Barra, J.M.; Kozlovskaya, V.; Kharlampieva, E.; Tse, H.M. Localized Immunosuppression With Tannic Acid Encapsulation Delays Islet Allograft and Autoimmune-Mediated Rejection. Diabetes 2020, 69, 1948. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Immunotherapy: Past, present and future. Nat. Med. 2003, 9, 269–277. [Google Scholar] [CrossRef]
- Till, S.J.; Francis, J.N.; Nouri-Aria, K.; Durham, S.R. Mechanisms of immunotherapy. J. Allergy Clin. Immunol. 2004, 113, 1025–1034. [Google Scholar] [CrossRef]
- Kopan, C.; Tucker, T.; Alexander, M.; Mohammadi, M.R.; Pone, E.J.; Lakey, J.R.T. Approaches in Immunotherapy, Regenerative Medicine, and Bioengineering for Type 1 Diabetes. Front. Immunol. 2018, 9, 1354. [Google Scholar] [CrossRef]
- Garciafigueroa, Y.; Trucco, M.; Giannoukakis, N. A brief glimpse over the horizon for type 1 diabetes nanotherapeutics. Clin. Immunol. 2015, 160, 36–45. [Google Scholar] [CrossRef]
- Tang, Z.; Kong, N.; Ouyang, J.; Feng, C.; Kim, N.Y.; Ji, X.; Wang, C.; Farokhzad, O.C.; Zhang, H.; Tao, W. Phosphorus Science-Oriented Design and Synthesis of Multifunctional Nanomaterials for Biomedical Applications. Matter 2020, 2, 297–322. [Google Scholar] [CrossRef]
- Makvandi, P.; Wang, C.-y.; Zare, E.N.; Borzacchiello, A.; Niu, L.-n.; Tay, F.R. Metal-Based Nanomaterials in Biomedical Applications: Antimicrobial Activity and Cytotoxicity Aspects. Adv. Funct. Mater. 2020, 30, 1910021. [Google Scholar] [CrossRef]
- Duan, C.; Liang, L.; Li, L.; Zhang, R.; Xu, Z.P. Recent progress in upconversion luminescence nanomaterials for biomedical applications. J. Mater. Chem. B 2018, 6, 192–209. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, J. Antioxidative nanomaterials and biomedical applications. Nano Today 2019, 27, 146–177. [Google Scholar] [CrossRef]
- Gim, S.; Zhu, Y.; Seeberger, P.H.; Delbianco, M. Carbohydrate-based nanomaterials for biomedical applications. WIREs Nanomed. Nanobiotechnol. 2019, 11, e1558. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-L.; Zou, M.-Z.; Qin, S.-Y.; Cheng, Y.-J.; Ma, Y.-H.; Sun, Y.-X.; Zhang, X.-Z. Recent Advances of Cell Membrane-Coated Nanomaterials for Biomedical Applications. Adv. Funct. Mater. 2020, 30, 2003559. [Google Scholar] [CrossRef]
- Stabler, C.L.; Li, Y.; Stewart, J.M.; Keselowsky, B.G. Engineering immunomodulatory biomaterials for type 1 diabetes. Nat. Rev. Mater. 2019, 4, 429–450. [Google Scholar] [CrossRef]
- Thondawada, M.; Wadhwani, A.D.; Palanisamy, D.S.; Rathore, H.S.; Gupta, R.C.; Chintamaneni, P.K.; Samanta, M.K.; Dubala, A.; Varma, S.; Krishnamurthy, P.T.; et al. An effective treatment approach of DPP-IV inhibitor encapsulated polymeric nanoparticles conjugated with anti-CD-4 mAb for type 1 diabetes. Drug Dev. Ind. Pharm. 2018, 44, 1120–1129. [Google Scholar] [CrossRef]
- Li, X.; Zhen, M.; Zhou, C.; Deng, R.; Yu, T.; Wu, Y.; Shu, C.; Wang, C.; Bai, C. Gadofullerene Nanoparticles Reverse Dysfunctions of Pancreas and Improve Hepatic Insulin Resistance for Type 2 Diabetes Mellitus Treatment. ACS Nano 2019, 13, 8597–8608. [Google Scholar] [CrossRef]
- Volpatti, L.R.; Matranga, M.A.; Cortinas, A.B.; Delcassian, D.; Daniel, K.B.; Langer, R.; Anderson, D.G. Glucose-Responsive Nanoparticles for Rapid and Extended Self-Regulated Insulin Delivery. ACS Nano 2020, 14, 488–497. [Google Scholar] [CrossRef]
- Song, M.; Wang, H.; Chen, K.; Zhang, S.; Yu, L.; Elshazly, E.H.; Ke, L.; Gong, R. Oral insulin delivery by carboxymethyl-β-cyclodextrin-grafted chitosan nanoparticles for improving diabetic treatment. Artif. Cells Nanomed. Biotechnol. 2018, 46, S774–S782. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, Y.; Zhang, M.; Zhou, W.; Chai, Z.; Wang, H.; Sun, C.; Huang, F. Glycopolypeptide Nanocarriers Based on Dynamic Covalent Bonds for Glucose Dual-Responsiveness and Self-Regulated Release of Insulin in Diabetic Rats. Biomacromolecules 2020, 21, 1507–1515. [Google Scholar] [CrossRef]
- Neef, T.; Miller, S.D. Tolerogenic Nanoparticles to Treat Islet Autoimmunity. Curr. Diabetes Rep. 2017, 17, 84. [Google Scholar] [CrossRef]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Moorman, C.D.; Sohn, S.J.; Phee, H. Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T cell Therapy, and IL-2 Therapy. Front. Immunol. 2021, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Yeste, A.; Takenaka, M.C.; Mascanfroni, I.D.; Nadeau, M.; Kenison, J.E.; Patel, B.; Tukpah, A.-M.; Babon, J.A.B.; DeNicola, M.; Kent, S.C.; et al. Tolerogenic nanoparticles inhibit T cell–mediated autoimmunity through SOCS2. Sci. Signal. 2016, 9, ra61. [Google Scholar] [CrossRef] [PubMed]
- Rabinowe, S.L.; Eisenbarth, G.S. Type I Diabetes Mellitus: A Chronic Autoimmune Disease? Pediatr. Clin. N. Am. 1984, 31, 531–543. [Google Scholar] [CrossRef]
- Mein, C.; Esposito, L.; Dunn, M.G.; Johnson, G.C.L.; Timms, A.E.; Goy, J.V.; Smith, A.N.; Sebag-Montefiore, L.; Merriman, M.E.; Wilson, A.J.; et al. A search for type 1 diabetes susceptibility genes in families from the United Kingdom. Nat. Genet. 1998, 19, 297–300. [Google Scholar] [CrossRef]
- Aly, T.A.; Ide, A.; Jahromi, M.M.; Barker, J.M.; Fernando, M.S.; Babu, S.R.; Yu, L.; Miao, D.; Erlich, H.A.; Fain, P.R.; et al. Extreme genetic risk for type 1A diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 14074. [Google Scholar] [CrossRef]
- Kavvoura, F.K.; Ioannidis, J.P.A. CTLA-4 Gene Polymorphisms and Susceptibility to Type 1 Diabetes Mellitus: A HuGE Review and Meta-Analysis. Am. J. Epidemiol. 2005, 162, 3–16. [Google Scholar] [CrossRef]
- Redondo, M.J.; Geyer, S.; Steck, A.K.; Sharp, S.; Wentworth, J.M.; Weedon, M.N.; Antinozzi, P.; Sosenko, J.; Atkinson, M.; Pugliese, A.; et al. A Type 1 Diabetes Genetic Risk Score Predicts Progression of Islet Autoimmunity and Development of Type 1 Diabetes in Individuals at Risk. Diabetes Care 2018, 41, 1887. [Google Scholar] [CrossRef]
- Paschou, S.A.; Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. On type 1 diabetes mellitus pathogenesis. Endocr. Connect. 2018, 7, R38–R46. [Google Scholar] [CrossRef]
- Ablamuntis, V.; Elias, D.; Cohen, I.R. The pathogenicity of islet-infiltrating lymphocytes in the non-obese diabetic (NOD) mouse. Clin. Exp. Immunol. 1999, 115, 260–267. [Google Scholar] [CrossRef]
- Graham, K.L.; Krishnamurthy, B.; Fynch, S.; Ayala-Perez, R.; Slattery, R.M.; Santamaria, P.; Thomas, H.E.; Kay, T.W.H. Intra-islet proliferation of cytotoxic T lymphocytes contributes to insulitis progression. Eur. J. Immunol. 2012, 42, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Usher-Smith, J.A.; Thompson, M.J.; Sharp, S.J.; Walter, F.M. Factors associated with the presence of diabetic ketoacidosis at diagnosis of diabetes in children and young adults: A systematic review. BMJ 2011, 343, d4092. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.W.; Eisenbarth, G.S. Immunologic endocrine disorders. J. Allergy Clin. Immunol. 2010, 125, S226–S237. [Google Scholar] [CrossRef] [PubMed]
- Smigoc Schweiger, D.; Mendez, A.; Kunilo Jamnik, S.; Bratanic, N.; Bratina, N.; Battelino, T.; Brecelj, J.; Vidan-Jeras, B. High-risk genotypes HLA-DR3-DQ2/DR3-DQ2 and DR3-DQ2/DR4-DQ8 in co-occurrence of type 1 diabetes and celiac disease. Autoimmunity 2016, 49, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Devendra, D.; Eisenbarth, G.S. Immunologic endocrine disorders. J. Allergy Clin. Immunol. 2003, 111, S624–S636. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.S.; Kudva, Y.C.; Wilson, S.B.; Strominger, J.L.; David, C.S. Co-expression of HLA DR3 and DQ8 results in the development of spontaneous insulitis and loss of tolerance to GAD65 in transgenic mice. Diabetes 2000, 49, 548. [Google Scholar] [CrossRef][Green Version]
- Campbell-Thompson, M.L.; Atkinson, M.A.; Butler, A.E.; Chapman, N.M.; Frisk, G.; Gianani, R.; Giepmans, B.N.; von Herrath, M.G.; Hyöty, H.; Kay, T.W.; et al. The diagnosis of insulitis in human type 1 diabetes. Diabetologia 2013, 56, 2541–2543. [Google Scholar] [CrossRef]
- Culina, S.; Lalanne, A.I.; Afonso, G.; Cerosaletti, K.; Pinto, S.; Sebastiani, G.; Kuranda, K.; Nigi, L.; Eugster, A.; Østerbye, T.; et al. Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci. Immunol. 2018, 3, eaao4013. [Google Scholar] [CrossRef]
- Keenan, H.A.; Sun, J.K.; Levine, J.; Doria, A.; Aiello, L.P.; Eisenbarth, G.; Bonner-Weir, S.; King, G.L. Residual Insulin Production and Pancreatic β-Cell Turnover After 50 Years of Diabetes: Joslin Medalist Study. Diabetes 2010, 59, 2846–2853. [Google Scholar] [CrossRef]
- Mobasseri, M.; Shirmohammadi, M.; Amiri, T.; Vahed, N.; Hosseini Fard, H.; Ghojazadeh, M. Prevalence and incidence of type 1 diabetes in the world: A systematic review and meta-analysis. Health Promot. Perspect. 2020, 10, 98–115. [Google Scholar] [CrossRef]
- Svoren, B.M.; Volkening, L.K.; Wood, J.R.; Laffel, L.M.B. Significant Vitamin D Deficiency in Youth with Type 1 Diabetes Mellitus. J. Pediatr. 2009, 154, 132–134. [Google Scholar] [CrossRef][Green Version]
- Cooper, J.D.; Smyth, D.J.; Walker, N.M.; Stevens, H.; Burren, O.S.; Wallace, C.; Greissl, C.; Ramos-Lopez, E.; Hyppönen, E.; Dunger, D.B.; et al. Inherited Variation in Vitamin D Genes Is Associated With Predisposition to Autoimmune Disease Type 1 Diabetes. Diabetes 2011, 60, 1624. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Bhalla, A.K.; Amento, E.P.; Serog, B.; Glimcher, L.H. 1,25-Dihydroxyvitamin D3 inhibits antigen-induced T cell activation. J. Immunol. 1984, 133, 1748. [Google Scholar] [PubMed]
- Kamen, D.; Aranow, C. Vitamin D in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2008, 20, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, H.; Søvik, O. Dead in Bed Syndrome in Young Diabetic Patients in Norway. Diabet. Med. 1995, 12, 782–787. [Google Scholar] [CrossRef]
- Brazg, R.L.; Bailey, T.S.; Garg, S.; Buckingham, B.A.; Slover, R.H.; Klonoff, D.C.; Nguyen, X.; Shin, J.; Welsh, J.B.; Lee, S.W. The ASPIRE Study: Design and Methods of an In-Clinic Crossover Trial on the Efficacy of Automatic Insulin Pump Suspension in Exercise-Induced Hypoglycemia. J. Diabetes Sci. Technol. 2011, 5, 1466–1471. [Google Scholar] [CrossRef]
- Garg, S.K.; Voelmle, M.K.; Beatson, C.R.; Miller, H.A.; Crew, L.B.; Freson, B.J.; Hazenfield, R.M. Use of Continuous Glucose Monitoring in Subjects with Type 1 Diabetes on Multiple Daily Injections Versus Continuous Subcutaneous Insulin Infusion Therapy. Diabetes Care 2011, 34, 574. [Google Scholar] [CrossRef]
- Franek, E.; Haluzík, M.; Canecki Varžić, S.; Sargin, M.; Macura, S.; Zacho, J.; Christiansen, J.S. Twice-daily insulin degludec/insulin aspart provides superior fasting plasma glucose control and a reduced rate of hypoglycaemia compared with biphasic insulin aspart 30 in insulin-naïve adults with Type 2 diabetes. Diabet. Med. 2016, 33, 497–505. [Google Scholar] [CrossRef]
- Willner, S.; Whittemore, R.; Keene, D. “Life or death”: Experiences of insulin insecurity among adults with type 1 diabetes in the United States. SSM-Popul. Health 2020, 11, 100624. [Google Scholar] [CrossRef]
- Smith, M.J.; Simmons, K.M.; Cambier, J.C. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat. Rev. Nephrol. 2017, 13, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.S.; Wen, L.; Tang, M.; Ramanathan, M.; Visintin, I.; Daugherty, J.; Hannum, L.G.; Janeway, C.A.; Shlomchik, M.J. Investigation of the Role of B-Cells in Type 1 Diabetes in the NOD Mouse. Diabetes 2004, 53, 2581. [Google Scholar] [CrossRef] [PubMed]
- Hull, C.M.; Peakman, M.; Tree, T.I.M. Regulatory T cell dysfunction in type 1 diabetes: What’s broken and how can we fix it? Diabetologia 2017, 60, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, S.M.; DiLorenzo, T.P. A comprehensive guide to antibody and T-cell responses in type 1 diabetes. Tissue Antigens 2003, 62, 359–377. [Google Scholar] [CrossRef]
- Jacobsen, L.M.; Posgai, A.; Seay, H.R.; Haller, M.J.; Brusko, T.M. T Cell Receptor Profiling in Type 1 Diabetes. Curr. Diabetes Rep. 2017, 17, 118. [Google Scholar] [CrossRef]
- Gitelman, S.E.; Bluestone, J.A. Regulatory T cell therapy for type 1 diabetes: May the force be with you. J. Autoimmun. 2016, 71, 78–87. [Google Scholar] [CrossRef]
- Ahmed, S.; Cerosaletti, K.; James, E.; Long, S.A.; Mannering, S.; Speake, C.; Nakayama, M.; Tree, T.; Roep, B.O.; Herold, K.C.; et al. Standardizing T-Cell Biomarkers in Type 1 Diabetes: Challenges and Recent Advances. Diabetes 2019, 68, 1366–1379. [Google Scholar] [CrossRef]
- Pesenacker, A.M.; Chen, V.; Gillies, J.; Speake, C.; Marwaha, A.K.; Sun, A.; Chow, S.; Tan, R.; Elliott, T.; Dutz, J.P.; et al. Treg gene signatures predict and measure type 1 diabetes trajectory. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Couture, A.; Garnier, A.; Docagne, F.; Boyer, O.; Vivien, D.; Le-Mauff, B.; Latouche, J.-B.; Toutirais, O. HLA-Class II Artificial Antigen Presenting Cells in CD4+ T Cell-Based Immunotherapy. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Eichmann, M.; Baptista, R.; Ellis, R.J.; Heck, S.; Peakman, M.; Beam, C.A. Costimulation Blockade Disrupts CD4+ T Cell Memory Pathways and Uncouples Their Link to Decline in β-Cell Function in Type 1 Diabetes. J. Immunol. 2020, 204, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Thorpe, J.; Herold, K.C.; Ehlers, M.; Sanda, S.; Lim, N.; Linsley, P.S.; Nepom, G.T.; Harris, K.M. Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell. Immunol. 2017, 319, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.S.; Wen, L. A predictive CD8+ T cell phenotype for T1DM progression. Nat. Rev. Endocrinol. 2020, 16, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Wiedeman, A.E.; Muir, V.S.; Rosasco, M.G.; DeBerg, H.A.; Presnell, S.; Haas, B.; Dufort, M.J.; Speake, C.; Greenbaum, C.J.; Serti, E.; et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J. Clin. Investig. 2020, 130, 480–490. [Google Scholar] [CrossRef]
- Ohkura, N.; Sakaguchi, S. Transcriptional and epigenetic basis of Treg cell development and function: Its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020, 30, 465–474. [Google Scholar] [CrossRef]
- Yeh, W.-I.; Seay, H.R.; Newby, B.; Posgai, A.L.; Moniz, F.B.; Michels, A.; Mathews, C.E.; Bluestone, J.A.; Brusko, T.M. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front. Immunol. 2017, 8, 1313. [Google Scholar] [CrossRef]
- Pellegrino, M.; Crinò, A.; Rosado, M.M.; Fierabracci, A. Identification and functional characterization of CD8+ T regulatory cells in type 1 diabetes patients. PLoS ONE 2019, 14, e0210839. [Google Scholar] [CrossRef]
- Serr, I.; Fürst, R.W.; Achenbach, P.; Scherm, M.G.; Gökmen, F.; Haupt, F.; Sedlmeier, E.-M.; Knopff, A.; Shultz, L.; Willis, R.A.; et al. Type 1 diabetes vaccine candidates promote human Foxp3+Treg induction in humanized mice. Nat. Commun. 2016, 7, 10991. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur. J. Immunol. 2018, 48, 751–756. [Google Scholar] [CrossRef]
- Baekkeskov, S.; Hubbell, J.A.; Phelps, E.A. Bioengineering strategies for inducing tolerance in autoimmune diabetes. Adv. Drug Deliv. Rev. 2017, 114, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zheng, X.; Chen, G.; Su, L.; Luo, S.; Wang, W.; Ye, S.; Weng, J.; Min, Y. Nanotechnology’s application in Type 1 diabetes. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1645. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, Y.S.; Hobson, D.; Hydbring, P. Nanoparticles for immune system targeting. Drug Discov. Today 2017, 22, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Kroger, C.J.; Tisch, R.M.; Bachelder, E.M.; Ainslie, K.M. Prevention of Type 1 Diabetes with Acetalated Dextran Microparticles Containing Rapamycin and Pancreatic Peptide P31. Adv. Health Mater. 2018, 7, 1800341. [Google Scholar] [CrossRef]
- Bergot, A.-S.; Buckle, I.; Cikaluru, S.; Naranjo, J.L.; Wright, C.M.; Zheng, G.; Talekar, M.; Hamilton-Williams, E.E.; Thomas, R. Regulatory T Cells Induced by Single-Peptide Liposome Immunotherapy Suppress Islet-Specific T Cell Responses to Multiple Antigens and Protect from Autoimmune Diabetes. J. Immunol. 2020, 204, 1787–1797. [Google Scholar] [CrossRef]
- Jamison, B.L.; Neef, T.; Goodspeed, A.; Bradley, B.; Baker, R.L.; Miller, S.D.; Haskins, K. Nanoparticles Containing an Insulin–ChgA Hybrid Peptide Protect from Transfer of Autoimmune Diabetes by Shifting the Balance between Effector T Cells and Regulatory T Cells. J. Immunol. 2019, 203, 48–57. [Google Scholar] [CrossRef]
- Aboelnazar, S.; Ghoneim, H.; Shalaby, T.; Bahgat, E.; Moaaz, M. Effect of low dose IL-2 loaded chitosan nanoparticles on natural killer and regulatory T cell expression in experimentally induced autoimmune type 1 diabetes mellitus. Cent. Eur. J. Immunol. 2020, 45, 382–392. [Google Scholar] [CrossRef]
- Greenbaum, C.J.; Serti, E.; Lambert, K.; Weiner, L.J.; Kanaparthi, S.; Lord, S.; Gitelman, S.E.; Wilson, D.M.; Gaglia, J.L.; Griffin, K.J.; et al. IL-6 receptor blockade does not slow β cell loss in new-onset type 1 diabetes. JCI Insight 2021, 6, 6. [Google Scholar] [CrossRef]
- Xu, X.; Bian, L.; Shen, M.; Li, X.; Zhu, J.; Chen, S.; Xiao, L.; Zhang, Q.; Chen, H.; Xu, K.; et al. Multipeptide-coupled nanoparticles induce tolerance in ‘humanised’ HLA-transgenic mice and inhibit diabetogenic CD8+ T cell responses in type 1 diabetes. Diabetologia 2017, 60, 2418–2431. [Google Scholar] [CrossRef]
- Hamad, A.R.A.; Ahmed, R.; Donner, T.; Fousteri, G. B cell-targeted immunotherapy for type 1 diabetes: What can make it work? Discov. Med. 2016, 21, 213–219. [Google Scholar]
- Johnson, P.; Glennie, M. The mechanisms of action of rituximab in the elimination of tumor cells. Semin. Oncol. 2003, 30, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Huda, R. New Approaches to Targeting B Cells for Myasthenia Gravis Therapy. Front. Immunol. 2020, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K. B cells as a target of immune modulation. Ann. Indian Acad. Neurol. 2009, 12, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.S.; Peakman, M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64. [Google Scholar] [CrossRef]
- Sarikonda, G.; Sachithanantham, S.; Manenkova, Y.; Kupfer, T.; Posgai, A.; Wasserfall, C.; Bernstein, P.; Straub, L.; Pagni, P.P.; Schneider, D.; et al. Transient B-Cell Depletion with Anti-CD20 in Combination with Proinsulin DNA Vaccine or Oral Insulin: Immunologic Effects and Efficacy in NOD Mice. PLoS ONE 2013, 8, e54712. [Google Scholar] [CrossRef]
- Martucci, N.M.; Migliaccio, N.; Ruggiero, I.; Albano, F.; Calì, G.; Romano, S.; Terracciano, M.; Rea, I.; Arcari, P.; Lamberti, A. Nanoparticle-based strategy for personalized B-cell lymphoma therapy. Int. J. Nanomed. 2016, 11, 6089–6101. [Google Scholar] [CrossRef]
- Stensland, Z.C.; Cambier, J.C.; Smith, M.J. Therapeutic Targeting of Autoreactive B Cells: Why, How, and When? Biomedicines 2021, 9, 83. [Google Scholar] [CrossRef]
- Woldu, M.A.; Lenjisa, J.L. Nanoparticles and the new era in diabetes management. Int. J. Basic Clin. Pharmacol. 2017, 3, 8. [Google Scholar] [CrossRef]
- Temchura, V.V.; Kozlova, D.; Sokolova, V.; Überla, K.; Epple, M. Targeting and activation of antigen-specific B-cells by calcium phosphate nanoparticles loaded with protein antigen. Biomaterials 2014, 35, 6098–6105. [Google Scholar] [CrossRef]
- Hong, S.; Zhang, Z.; Liu, H.; Tian, M.; Zhu, X.; Zhang, Z.; Wang, W.; Zhou, X.; Zhang, F.; Ge, Q.; et al. B Cells Are the Dominant Antigen-Presenting Cells that Activate Naive CD4+ T Cells upon Immunization with a Virus-Derived Nanoparticle Antigen. Immunity 2018, 49, 695–708.e4. [Google Scholar] [CrossRef]
- Selim, M.E.; Abd-Elhakim, Y.M.; Al-Ayadhi, L.Y. Pancreatic Response to Gold Nanoparticles Includes Decrease of Oxidative Stress and Inflammation In Autistic Diabetic Model. Cell. Physiol. Biochem. 2015, 35, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, G.; Ippolito, E.; Ahmed, R.; Rahim, A.; Hamad, A. Beta-cell Specific Autoantibodies: Are they Just an Indicator of Type 1 Diabetes? Curr. Diabetes Rev. 2017, 13, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fan, Y.-N.; Chen, Z.-Y.; Luo, Y.-L.; Wang, Y.-C.; Lian, Z.-X.; Xu, C.-F.; Wang, J. Optimized nanoparticle-mediated delivery of CRISPR-Cas9 system for B cell intervention. Nano Res. 2018, 11, 6270–6282. [Google Scholar] [CrossRef]
- Chu, V.T.; Graf, R.; Wirtz, T.; Weber, T.; Favret, J.; Li, X.; Petsch, K.; Tran, N.T.; Sieweke, M.H.; Berek, C.; et al. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc. Natl. Acad. Sci. USA 2016, 113, 12514–12519. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Tai, X.; Van Laethem, F.; Pobezinsky, L.; Guinter, T.; Sharrow, S.O.; Adams, A.; Granger, L.; Kruhlak, M.; Lindsten, T.; Thompson, C.B.; et al. Basis of CTLA-4 function in regulatory and conventional CD4+ T cells. Blood 2012, 119, 5155–5163. [Google Scholar] [CrossRef]
- Taraban, V.Y.; Rowley, T.F.; O’Brien, L.; Chan, H.T.C.; Haswell, L.E.; Green, M.H.A.; Tutt, A.L.; Glennie, M.J.; Al-Shamkhani, A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur. J. Immunol. 2002, 32, 3617–3627. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, Y.; Wang, J.; Yan, J.; Chen, Q.; Cheng, H.; Huang, P.; Gu, Z. Engineered PD-L1-Expressing Platelets Reverse New-Onset Type 1 Diabetes. Adv. Mater. 2020, 32, 1907692. [Google Scholar] [CrossRef]
- Andersen, M.H. The Balance Players of the Adaptive Immune System. Cancer Res. 2018, 78, 1379. [Google Scholar] [CrossRef]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: A promising new anticancer strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Kapke, J.; Shaheen, Z.; Kilari, D.; Knudson, P.; Wong, S. Immune Checkpoint Inhibitor-Associated Type 1 Diabetes Mellitus: Case Series, Review of the Literature, and Optimal Management. Case Rep. Oncol. 2017, 10, 897–909. [Google Scholar] [CrossRef]
- Samoa, R.A.; Lee, H.S.; Kil, S.H.; Roep, B.O. Anti–PD-1 Therapy–Associated Type 1 Diabetes in a Pediatric Patient With Relapsed Classical Hodgkin Lymphoma. Diabetes Care 2020, 43, 2293. [Google Scholar] [CrossRef]
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; Coomans de Brachène, A.; Paula, F.M.M.; Op de Beeck, A.; Castela, A.; et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 2018, 36, 367–375. [Google Scholar] [CrossRef]
- Falcone, M.; Fousteri, G. Role of the PD-1/PD-L1 Dyad in the Maintenance of Pancreatic Immune Tolerance for Prevention of Type 1 Diabetes. Front. Endocrinol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Chou, F.-C.; Chu, C.-H.; Wu, J.-C.; Lin, S.-H.; Chang, D.-M.; Sytwu, H.-K. Protective Role of Programmed Death 1 Ligand 1 (PD-L1)in Nonobese Diabetic Mice. Diabetes 2008, 57, 1861. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Q.; Zhao, H.; Bishop, J.O.; Zhou, G.; Olson, L.K.; Moore, A. miR-216a-targeting theranostic nanoparticles promote proliferation of insulin-secreting cells in type 1 diabetes animal model. Sci. Rep. 2020, 10, 5302. [Google Scholar] [CrossRef]
- Qian, L.; Liu, F.; Chu, Y.; Zhai, Q.; Wei, X.; Shao, J.; Li, R.; Xu, Q.; Yu, L.; Liu, B.; et al. MicroRNA-200c Nanoparticles Sensitized Gastric Cancer Cells to Radiotherapy by Regulating PD-L1 Expression and EMT. Cancer Manag. Res. 2020, 12, 12215–12223. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Wei, J.; Nduom, E.; Kong, L.-Y.; Wang, F.; Xu, S.; Gabrusiewicz, K.; Alum, A.; Fuller, G.; Calin, G.; Heimberger, A.B. miR-138 exerts anti-glioma efficacy by targeting immune checkpoints. J. ImmunoTherapy Cancer 2013, 1, P177. [Google Scholar] [CrossRef]
- Lee, S.W.L.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef]
- Heninger, A.-K.; Eugster, A.; Kuehn, D.; Buettner, F.; Kuhn, M.; Lindner, A.; Dietz, S.; Jergens, S.; Wilhelm, C.; Beyerlein, A.; et al. A divergent population of autoantigen-responsive CD4+ T cells in infants prior to β cell autoimmunity. Sci. Transl. Med. 2017, 9, eaaf8848. [Google Scholar] [CrossRef]
- Chang, W.; Wang, J. Exosomes and Their Noncoding RNA Cargo Are Emerging as New Modulators for Diabetes Mellitus. Cells 2019, 8, 853. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xie, Z.; Lu, Q.; Chang, C.; Zhou, Z. Beyond Genetics: What Causes Type 1 Diabetes. Clin. Rev. Allergy Immunol. 2017, 52, 273–286. [Google Scholar] [CrossRef]
- Santos, A.S.; Cunha Neto, E.; Fukui, R.T.; Ferreira, L.R.P.; Silva, M.E.R. Increased Expression of Circulating microRNA 101-3p in Type 1 Diabetes Patients: New Insights Into miRNA-Regulated Pathophysiological Pathways for Type 1 Diabetes. Front. Immunol. 2019, 10, 1637. [Google Scholar] [CrossRef] [PubMed]
- Scherm, M.G.; Daniel, C. miRNA-Mediated Immune Regulation in Islet Autoimmunity and Type 1 Diabetes. Front. Endocrinol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Scherm, M.G.; Serr, I.; Zahm, A.M.; Schug, J.; Bellusci, S.; Manfredini, R.; Salb, V.K.; Gerlach, K.; Weigmann, B.; Ziegler, A.-G.; et al. miRNA142-3p targets Tet2 and impairs Treg differentiation and stability in models of type 1 diabetes. Nat. Commun. 2019, 10, 5697. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, P.; Syed, F.; Jiyun Kang, N.; Mirmira, R.G.; Evans-Molina, C. Profiling of RNAs from Human Islet-Derived Exosomes in a Model of Type 1 Diabetes. Int. J. Mol. Sci. 2019, 20, 5903. [Google Scholar] [CrossRef]
- Dini, S.; Zakeri, M.; Ebrahimpour, S.; Dehghanian, F.; Esmaeili, A. Quercetin-conjugated superparamagnetic iron oxide nanoparticles modulate glucose metabolism-related genes and miR-29 family in the hippocampus of diabetic rats. Sci. Rep. 2021, 11, 8618. [Google Scholar] [CrossRef]
- Serr, I.; Scherm, M.G.; Zahm, A.M.; Schug, J.; Flynn, V.K.; Hippich, M.; Kälin, S.; Becker, M.; Achenbach, P.; Nikolaev, A.; et al. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Sci. Transl. Med. 2018, 10, eaag1782. [Google Scholar] [CrossRef]
- Vasu, S.; Kumano, K.; Darden, C.M.; Rahman, I.; Lawrence, M.C.; Naziruddin, B. MicroRNA Signatures as Future Biomarkers for Diagnosis of Diabetes States. Cells 2019, 8, 1533. [Google Scholar] [CrossRef]
- Satake, E.; Pezzolesi, M.G.; Md Dom, Z.I.; Smiles, A.M.; Niewczas, M.A.; Krolewski, A.S. Circulating miRNA Profiles Associated With Hyperglycemia in Patients With Type 1 Diabetes. Diabetes 2018, 67, 1013. [Google Scholar] [CrossRef] [PubMed]
- Åkerman, L.; Casas, R.; Ludvigsson, J.; Tavira, B.; Skoglund, C. Serum miRNA levels are related to glucose homeostasis and islet autoantibodies in children with high risk for type 1 diabetes. PLoS ONE 2018, 13, e0191067. [Google Scholar] [CrossRef]
- Bertoccini, L.; Sentinelli, F.; Incani, M.; Bailetti, D.; Cimini, F.A.; Barchetta, I.; Lenzi, A.; Cavallo, M.G.; Cossu, E.; Baroni, M.G. Circulating miRNA-375 levels are increased in autoantibodies-positive first-degree relatives of type 1 diabetes patients. Acta Diabetol. 2019, 56, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Lakhter, A.J.; Pratt, R.E.; Moore, R.E.; Doucette, K.K.; Maier, B.F.; DiMeglio, L.A.; Sims, E.K. Beta cell extracellular vesicle miR-21-5p cargo is increased in response to inflammatory cytokines and serves as a biomarker of type 1 diabetes. Diabetologia 2018, 61, 1124–1134. [Google Scholar] [CrossRef]
- Chhabra, P.; Brayman, K.L. Stem Cell Therapy to Cure Type 1 Diabetes: From Hype to Hope. Stem Cells Transl. Med. 2013, 2, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Fiorina, P.; Jurewicz, M.; Augello, A.; Vergani, A.; Dada, S.; La Rosa, S.; Selig, M.; Godwin, J.; Law, K.; Placidi, C.; et al. Immunomodulatory Function of Bone Marrow-Derived Mesenchymal Stem Cells in Experimental Autoimmune Type 1 Diabetes. J. Immunol. 2009, 183, 993. [Google Scholar] [CrossRef]
- Carlsson, P.-O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved β-Cell Function in Type 1 Diabetes by Mesenchymal Stromal Cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Voltarelli, J.C.; Couri, C.E.B.; Stracieri, A.B.P.L.; Oliveira, M.C.; Moraes, D.A.; Pieroni, F.; Coutinho, M.; Malmegrim, K.C.R.; Foss-Freitas, M.C.; Simões, B.P.; et al. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation in Newly Diagnosed Type 1 Diabetes Mellitus. JAMA 2007, 297, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- D’Addio, F.; Valderrama Vasquez, A.; Ben Nasr, M.; Franek, E.; Zhu, D.; Li, L.; Ning, G.; Snarski, E.; Fiorina, P. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation in New-Onset Type 1 Diabetes: A Multicenter Analysis. Diabetes 2014, 63, 3041. [Google Scholar] [CrossRef]
- Cai, J.; Wu, Z.; Xu, X.; Liao, L.; Chen, J.; Huang, L.; Wu, W.; Luo, F.; Wu, C.; Pugliese, A.; et al. Umbilical Cord Mesenchymal Stromal Cell With Autologous Bone Marrow Cell Transplantation in Established Type 1 Diabetes: A Pilot Randomized Controlled Open-Label Clinical Study to Assess Safety and Impact on Insulin Secretion. Diabetes Care 2016, 39, 149. [Google Scholar] [CrossRef]
- Lennard, A.L. Science, medicine, and the future: Stem cell transplantation. BMJ 2000, 321, 1331. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.K.; Riegler, J.; Wu, J.C. Stem Cell Imaging: From Bench to Bedside. Cell Stem Cell 2014, 14, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tian, D.-C.; He, W.; Lv, W.; Fan, J.; Li, H.; Jin, W.-N.; Meng, X. Cellular and molecular imaging for stem cell tracking in neurological diseases. Stroke Vasc. Neurol. 2021, 6, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Gu, E.; Chen, W.-Y.; Gu, J.; Burridge, P.; Wu, J.C. Molecular Imaging of Stem Cells: Tracking Survival, Biodistribution, Tumorigenicity, and Immunogenicity. Theranostics 2012, 2, 335–345. [Google Scholar] [CrossRef]
- Tabbara, I.A.; Zimmerman, K.; Morgan, C.; Nahleh, Z. Allogeneic Hematopoietic Stem Cell Transplantation: Complications and Results. Arch. Intern. Med. 2002, 162, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Lijkwan, M.A.; Bos, E.J.; Wu, J.C.; Robbins, R.C. Role of Molecular Imaging in Stem Cell Therapy for Myocardial Restoration. Trends Cardiovasc. Med. 2010, 20, 183–188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ping, W.; Anna, M. Molecular Imaging of Stem Cell Transplantation for Neurodegenerative Diseases. Curr. Pharm. Des. 2012, 18, 4426–4440. [Google Scholar] [CrossRef]
- Abbas, F.; Wu, J.C.; Gambhir, S.S.; Rodriguez-Porcel, M. Molecular Imaging of Stem Cells. Stem J. 2019, 1, 27–46. [Google Scholar] [CrossRef]
- Pei, Z.; Zeng, J.; Song, Y.; Gao, Y.; Wu, R.; Chen, Y.; Li, F.; Li, W.; Zhou, H.; Yang, Y. In vivo imaging to monitor differentiation and therapeutic effects of transplanted mesenchymal stem cells in myocardial infarction. Sci. Rep. 2017, 7, 6296. [Google Scholar] [CrossRef]
- Zheng, B.; von See, M.P.; Yu, E.; Gunel, B.; Lu, K.; Vazin, T.; Schaffer, D.V.; Goodwill, P.W.; Conolly, S.M. Quantitative Magnetic Particle Imaging Monitors the Transplantation, Biodistribution, and Clearance of Stem Cells In Vivo. Theranostics 2016, 6, 291–301. [Google Scholar] [CrossRef]
- Ahrens, E.T.; Bulte, J.W.M. Tracking immune cells in vivo using magnetic resonance imaging. Nat. Rev. Immunol. 2013, 13, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-L.; Lu, P.-G.; Zhang, L.-J.; Li, F.; Chen, Z.; Wu, N.; Meng, H.; Lin, J.-K.; Feng, H. In vivo magnetic resonance imaging tracking of SPIO-labeled human umbilical cord mesenchymal stem cells. J. Cell. Biochem. 2012, 113, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, Y.; Yang, B.; Zhang, B.; Wu, Y. Islet Transplantation Imaging in vivo. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3301–3311. [Google Scholar] [CrossRef] [PubMed]
- Hayat, H.; Sun, A.; Hayat, H.; Liu, S.; Talebloo, N.; Pinger, C.; Bishop, J.O.; Gudi, M.; Dwan, B.F.; Ma, X.; et al. Artificial Intelligence Analysis of Magnetic Particle Imaging for Islet Transplantation in a Mouse Model. Mol. Imaging Biol. 2021, 23, 18–29. [Google Scholar] [CrossRef]
- Laurent, S.; Saei, A.A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: Opportunities and challenges. Expert Opin. Drug Deliv. 2014, 11, 1449–1470. [Google Scholar] [CrossRef]
- Blocki, A.; Beyer, S.; Dewavrin, J.-Y.; Goralczyk, A.; Wang, Y.; Peh, P.; Ng, M.; Moonshi, S.S.; Vuddagiri, S.; Raghunath, M.; et al. Microcapsules engineered to support mesenchymal stem cell (MSC) survival and proliferation enable long-term retention of MSCs in infarcted myocardium. Biomaterials 2015, 53, 12–24. [Google Scholar] [CrossRef]
- Guldris, N.; Argibay, B.; Gallo, J.; Iglesias-Rey, R.; Carbó-Argibay, E.; Kolen’ko, Y.V.; Campos, F.; Sobrino, T.; Salonen, L.M.; Bañobre-López, M.; et al. Magnetite Nanoparticles for Stem Cell Labeling with High Efficiency and Long-Term in Vivo Tracking. Bioconjugate Chem. 2017, 28, 362–370. [Google Scholar] [CrossRef]
- Wang, P.; Yoo, B.; Yang, J.; Zhang, X.; Ross, A.; Pantazopoulos, P.; Dai, G.; Moore, A. GLP-1R–Targeting Magnetic Nanoparticles for Pancreatic Islet Imaging. Diabetes 2014, 63, 1465–1474. [Google Scholar] [CrossRef]
- Yao, Y.; Li, Y.; Ma, G.; Liu, N.; Ju, S.; Jin, J.; Chen, Z.; Shen, C.; Teng, G. In Vivo Magnetic Resonance Imaging of Injected Endothelial Progenitor Cells after Myocardial Infarction in Rats. Mol. Imaging Biol. 2011, 13, 303–313. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| APPROACH | TARGET | REFERENCE/S | |
|---|---|---|---|
| 1. | Teplizumab | CD4+ and CD8+ cells | [72] |
| 2. | Population alteration | Autoreactive CD8+ T cells | [73] |
| 3. | Functional correction | Treg cells | [66,75,76,77] |
| 4. | Chimeric antigen receptors | Treg cells | [79] |
| 5. | Rapamycin | Selective effector T cells and CD4+ T cells | [84] |
| 6. | Liposomal formulation of Autoantigen + 1α,25-dihydroxyvitamin D3 | ChgA-specific Foxp3+ CD4+ T cells | [85] |
| 7. | poly(lactide-co-glycolide) nanoparticles loaded Insulin–ChgA hybrid peptide | Balance population of effector and regulatory T cells | [86] |
| 8. | interleukin-2 (IL-2) | Treg cells | [86,87] |
| 9. | tocilizumab | interleukin-2 (IL-6) | [88] |
| 10. | Carboxylated polystyrene beads with peptide HLA-A*02:01-restricted epitopes | Antigen-specific T cell immune tolerance | [89] |
| APPROACH | TARGET | REFERENCE/S | |
|---|---|---|---|
| 1. | Rituximab | Autoreactive B cells | [61,90] |
| 2. | Combination therapy (Antigens + Antibodies) | CD20+ B cells | [62,94,95] |
| 3. | Nanoparticles + siRNA gene silencing | Autoreactive B cells | [99,100] |
| 4. | Depletion | Autoreactive B cells | [101,102,104] |
| 5. | Nanoparticles + CRISPR-cas9 (Gene editing) | Autoreactive B cells | [103] |
| APPROACH | TARGET | REFERENCE/S | |
|---|---|---|---|
| 1. | CD8+ T cell activation | CD70 and CD137 or CD134 | [107] |
| 2. | T cell suppression | programmed cell death protein 1 (PD-1) + ligand (PD-L1, PD-L2) upregulation | [108,109,110,111] |
| 3. | T cell suppression | cytotoxic T lymphocytes-associated antigen 4 (CTLA-4) upregulation | [112] |
| 4. | PD-L1 upregulation | Interferons: IFNα and IFNγ | [120,121] |
| 5. | transplanted human islet-like organoids (HILOs) | PD-L1 upregulation | [123] |
| 6. | SPIONs + miRNA | overexpression of co-inhibitory molecules | [124] |
| 7. | Nanoparticles + miRNA (miR-200c, miR-138-5p, miR-513, miR-200a, and miR-34a) | PD-L1 and CTLA-4 regulation | [126,127,128] |
| Approach | Target | Reference/S | |
|---|---|---|---|
| 1. | SPIONs + miR-216a | Expression modulation | [124] |
| 2. | SPIONs + miR-29 family | miR-29a, miR-29b, and miR-29c levels’ modulation | [138] |
| 3. | Treg induction | Block miRNA181a | [139] |
| 4. | Diagnosis and prognosis of T1D | miRNA in systemic circulation | [140] |
| 5. | Biomarker of beta-cell death | Circulating miR-375 | [143] |
| 6. | Beta cell survival | miR-21-5p upregulation | [144] |
| 7. | Diagnosis of T1D progression | circulating miR-101-3p and miR-204-5p | [134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigam, S.; Bishop, J.O.; Hayat, H.; Quadri, T.; Hayat, H.; Wang, P. Nanotechnology in Immunotherapy for Type 1 Diabetes: Promising Innovations and Future Advances. Pharmaceutics 2022, 14, 644. https://doi.org/10.3390/pharmaceutics14030644
Nigam S, Bishop JO, Hayat H, Quadri T, Hayat H, Wang P. Nanotechnology in Immunotherapy for Type 1 Diabetes: Promising Innovations and Future Advances. Pharmaceutics. 2022; 14(3):644. https://doi.org/10.3390/pharmaceutics14030644
Chicago/Turabian StyleNigam, Saumya, Jack Owen Bishop, Hanaan Hayat, Tahnia Quadri, Hasaan Hayat, and Ping Wang. 2022. "Nanotechnology in Immunotherapy for Type 1 Diabetes: Promising Innovations and Future Advances" Pharmaceutics 14, no. 3: 644. https://doi.org/10.3390/pharmaceutics14030644
APA StyleNigam, S., Bishop, J. O., Hayat, H., Quadri, T., Hayat, H., & Wang, P. (2022). Nanotechnology in Immunotherapy for Type 1 Diabetes: Promising Innovations and Future Advances. Pharmaceutics, 14(3), 644. https://doi.org/10.3390/pharmaceutics14030644