
One-Step Synthesis of Nanoliposomal Copper Diethyldithiocarbamate and Its Assessment for Cancer Therapy

 ,
,  , , ,
, , , 
Abstract
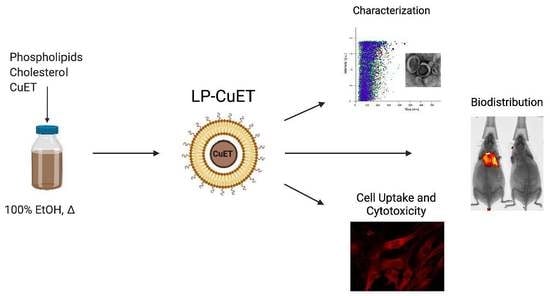
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CuET-encapsulated Nanoliposomes
2.3. Physicochemical Characterization
2.4. Encapsulation Efficiency
2.5. In Vitro Stability
2.6. Mouse Plasma and Hemolysis Study
2.7. Nanoliposome-Protein Complexes
2.8. Protein Corona Determination via SDS-PAGE
2.9. Evaluation of Cellular Toxicity
2.10. Cellular Uptake Assessment
2.11. Confocal Imaging
2.12. In Vivo Biodistribution
2.13. Statistical Analysis
3. Results
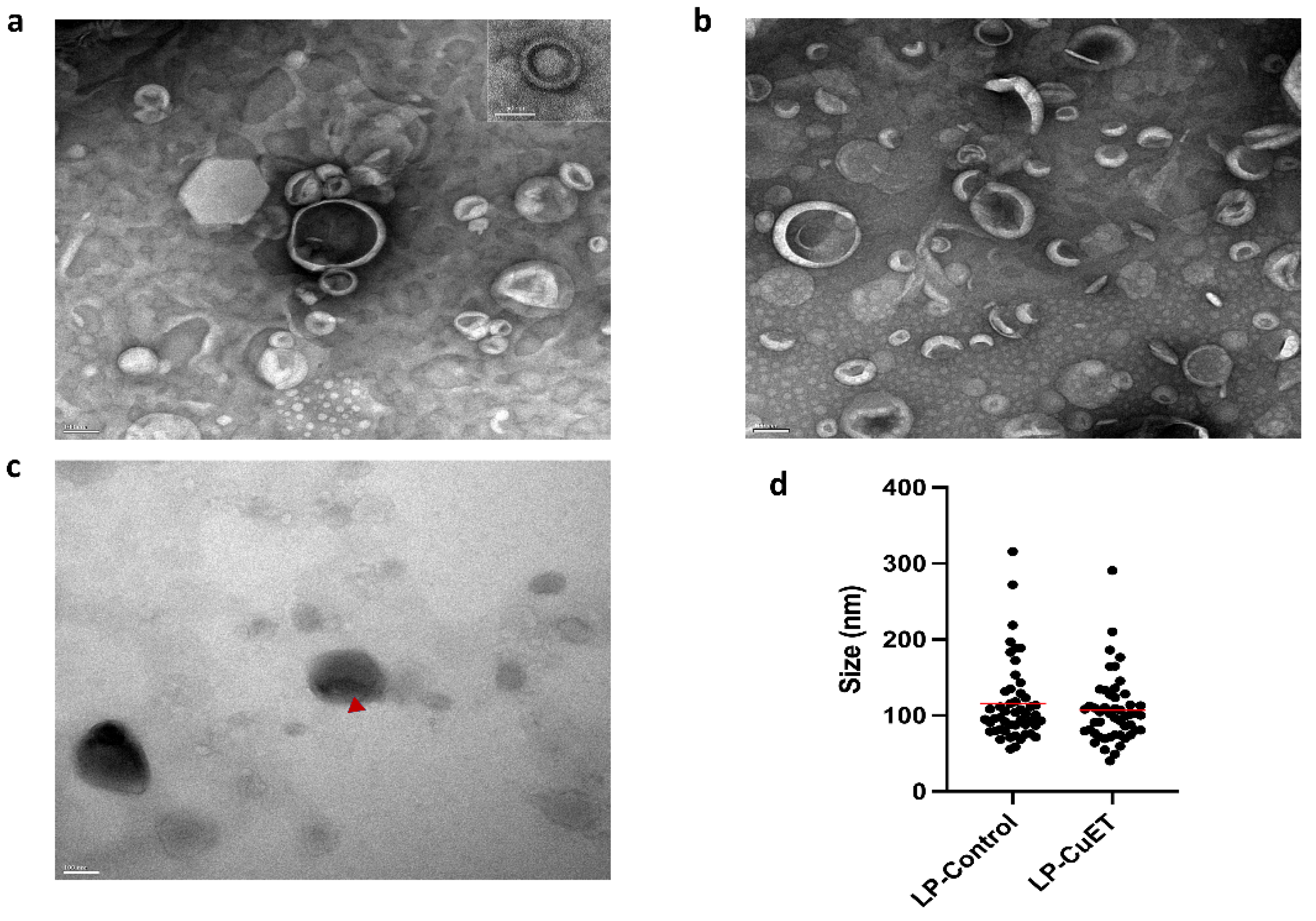
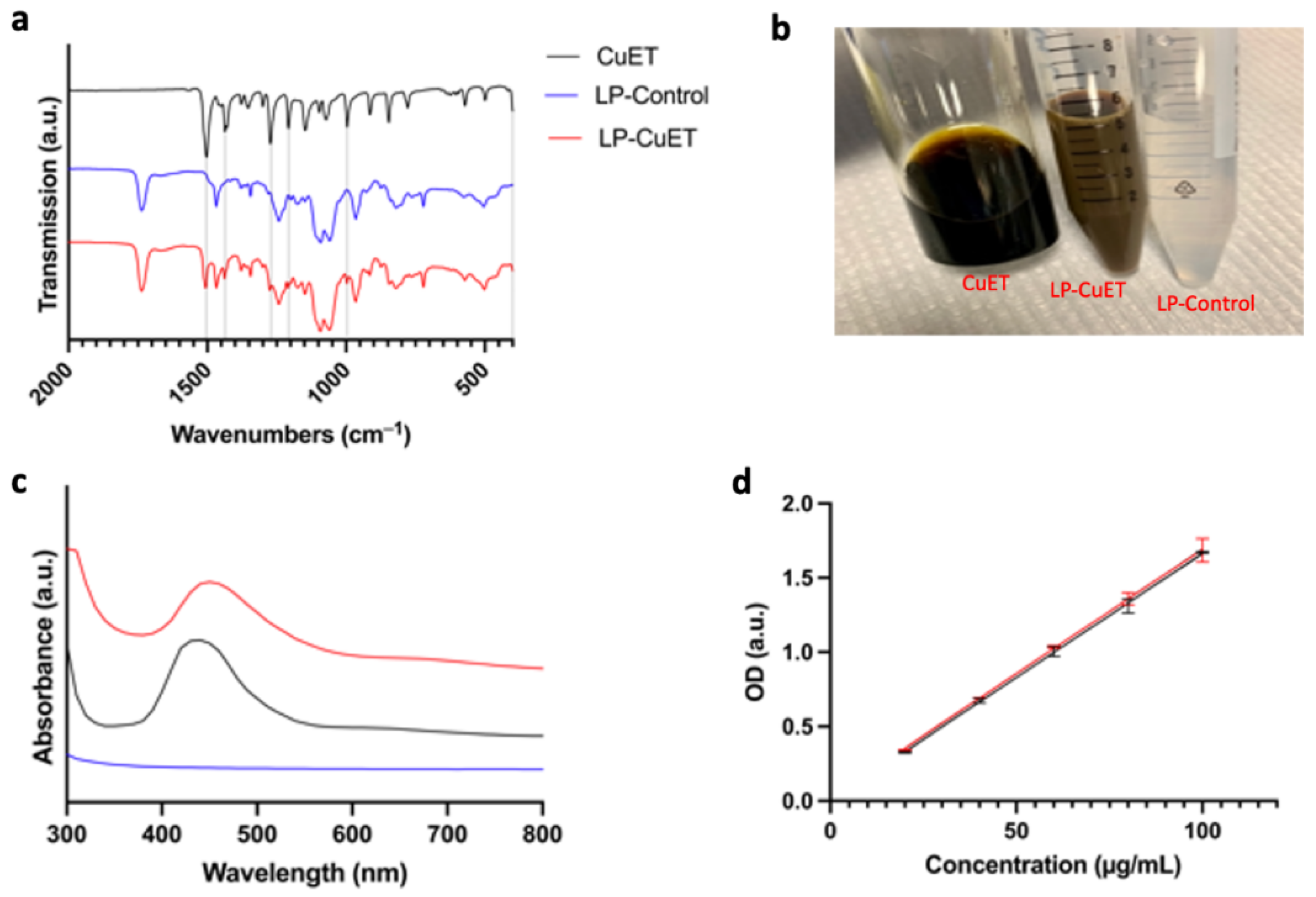
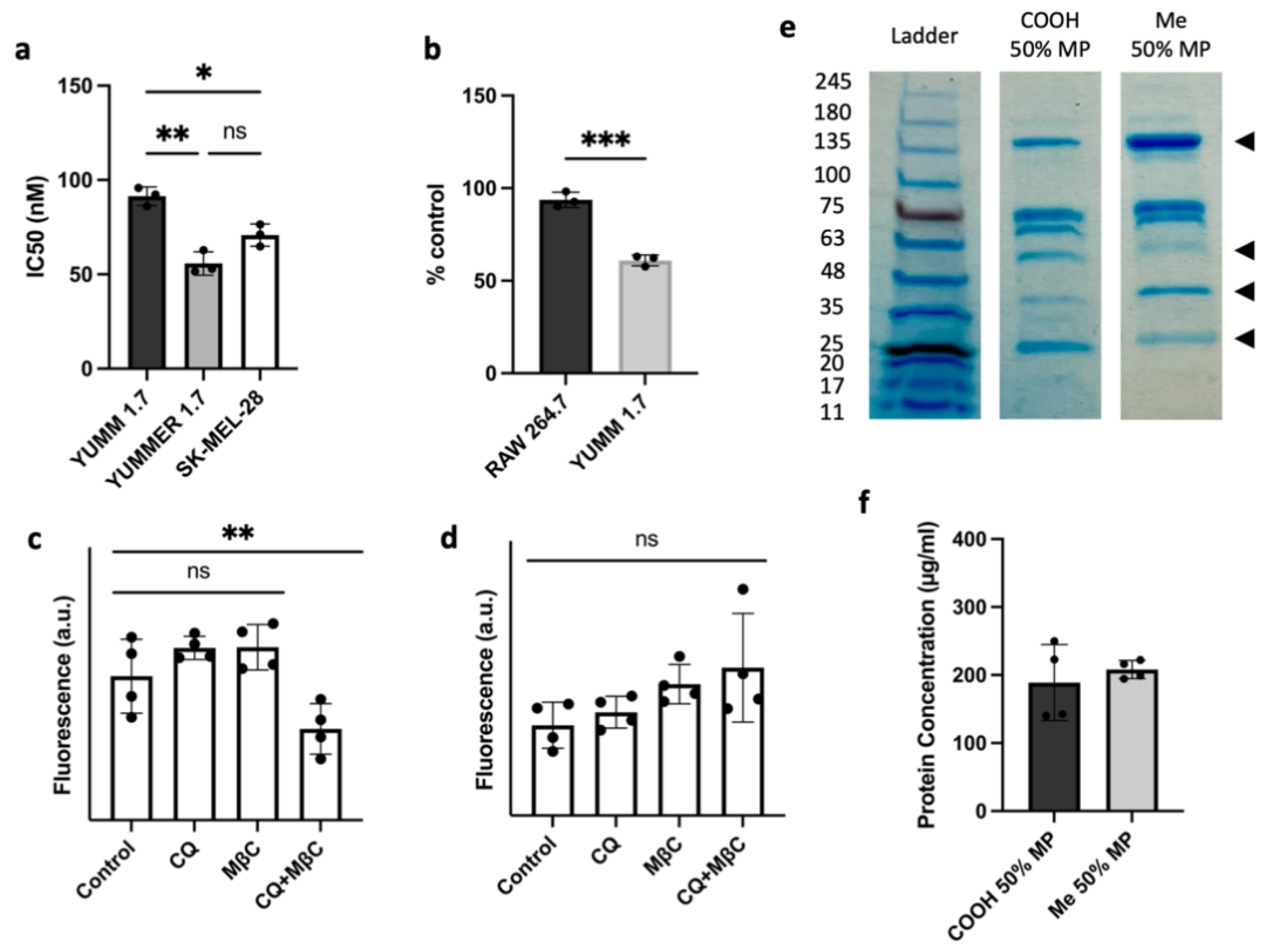
3.1. CuET Is Efficiently Encapsulated Inside Nanoliposomes
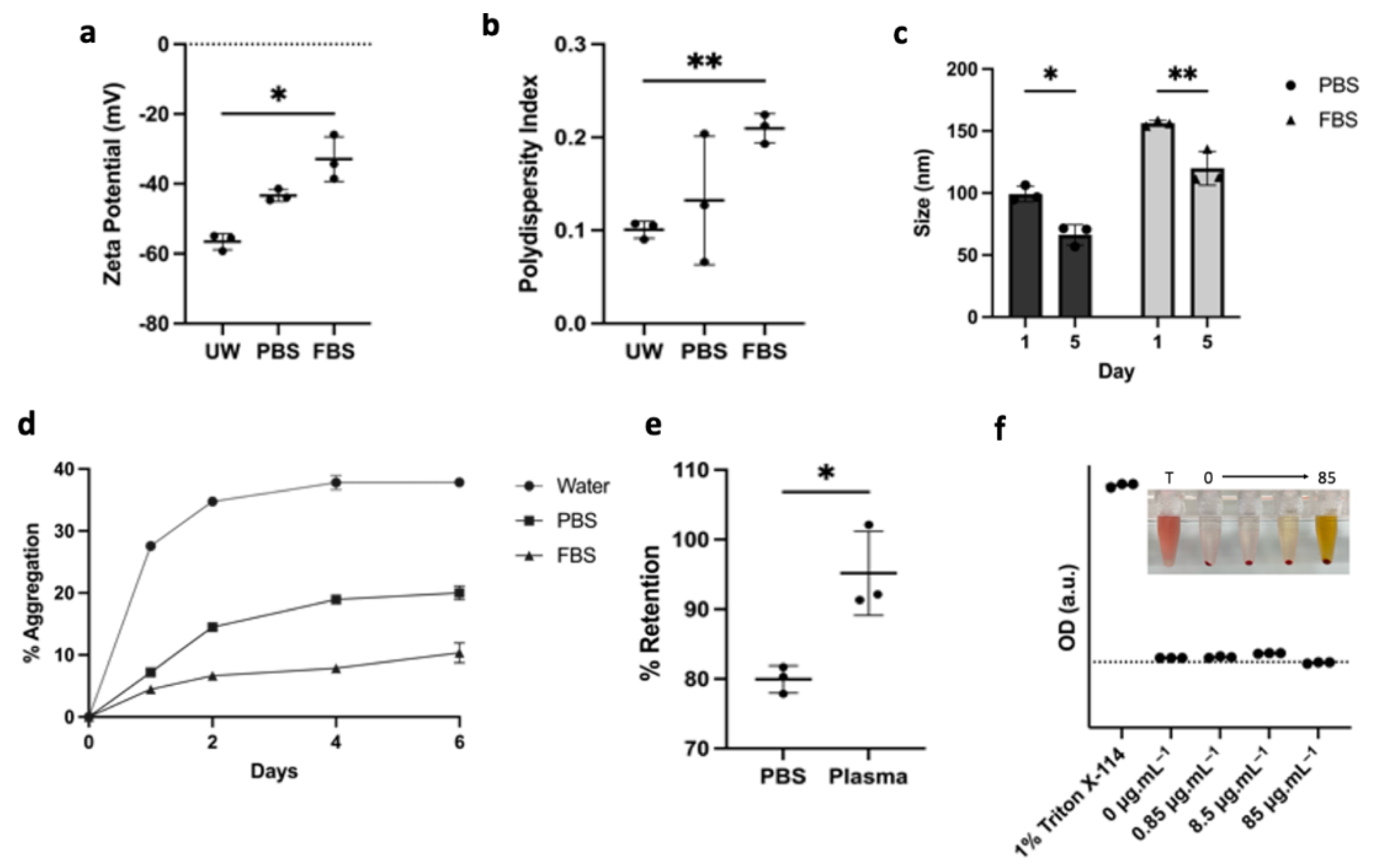
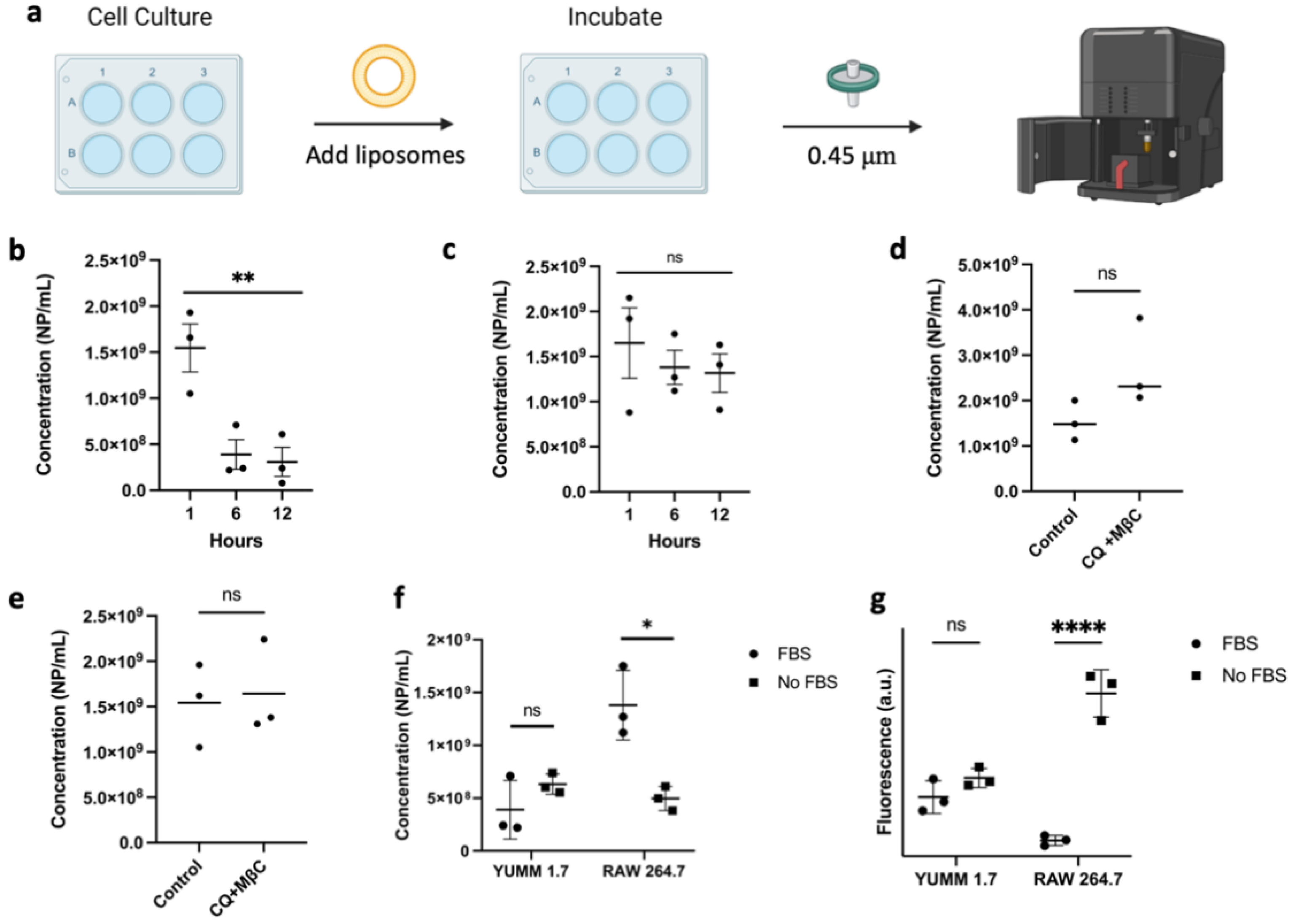
3.2. LP-CuET Nanoparticles Are Stable under Physiologically Relevant Conditions
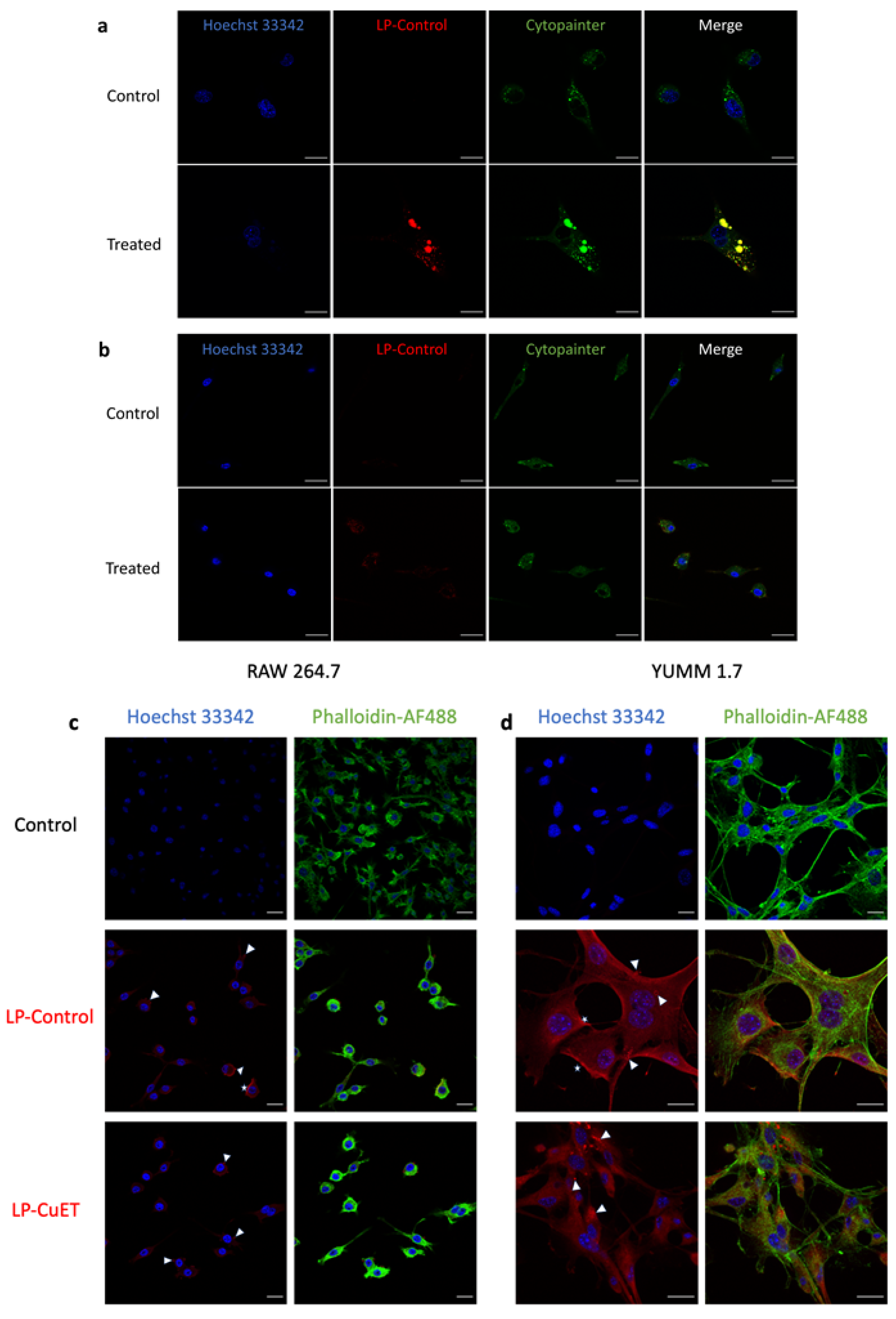
3.3. Cellular Uptake and Cytotoxicity in YUMM 1.7 and RAW 264.7 Cell Lines
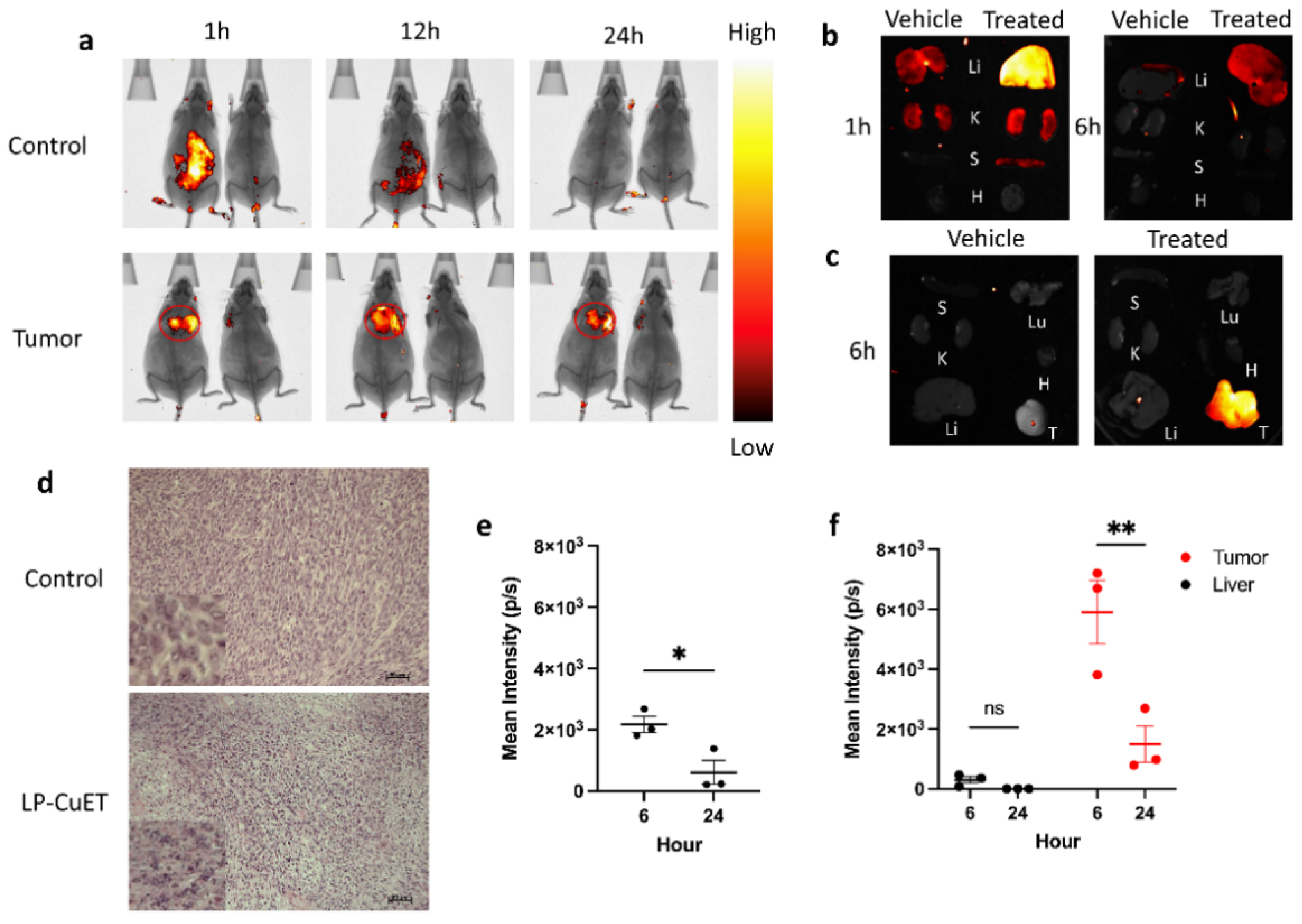
3.4. LP-CuET Nanoliposomes Preferentially Accumulate within YUMM 1.7 Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yang, W.; Chen, P.; Huang, Y.; Li, F. Disulfiram Copper Nanoparticles Prepared with a Stabilized Metal Ion Ligand Complex Method for Treating Drug-Resistant Prostate Cancers. ACS Appl. Mater. Interfaces 2018, 10, 41118–41128. [Google Scholar] [CrossRef]
- Ren, L.; Feng, W.; Shao, J.; Ma, J.; Xu, M.; Zhu, B.Z.; Zheng, N.; Liu, S. Diethyldithiocarbamate-copper nanocomplex reinforces disulfiram chemotherapeutic efficacy through light-triggered nuclear targeting. Theranostics 2020, 10, 6384–6398. [Google Scholar] [CrossRef]
- Said Suliman, A.; Khoder, M.; Tolaymat, I.; Webster, M.; Alany, R.G.; Wang, W.; Elhissi, A.; Najlah, M. Cyclodextrin Diethyldithiocarbamate Copper II Inclusion Complexes: A Promising Chemotherapeutic Delivery System against Chemoresistant Triple Negative Breast Cancer Cell Lines. Pharmaceutics 2021, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, M.; Anantha, M.; Shi, M.; Leung, A.W.; Dragowska, W.H.; Sanche, L.; Bally, M.B. Development and optimization of an injectable formulation of copper diethyldithiocarbamate, an active anticancer agent. Int. J. Nanomed. 2017, 12, 4129–4146. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Kong, Y.; Xu, J.; Li, X.; Gou, J.; Yin, T.; He, H.; Zhang, Y.; Tang, X. Doxorubicin intercalated copper diethyldithiocarbamate functionalized layered double hydroxide hybrid nanoparticles for targeted therapy of hepatocellular carcinoma. Biomater. Sci. 2020, 8, 897–911. [Google Scholar] [CrossRef]
- Li, X.; Du, K.; Sun, J.; Feng, F.D. Apoferritin as a Carrier of Cu(II) Diethyldithiocarbamate and Biomedical Application for Glutathione-Responsive Combination Chemotherapy. ACS Appl. Bio Mater. 2020, 3, 654–663. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal 2008, 10, 997–1030. [Google Scholar] [CrossRef] [Green Version]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.W.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- de Lazaro, I.; Mooney, D.J. A nanoparticle’s pathway into tumours. Nat. Mater. 2020, 19, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Kim, H.S.; Palanikumar, L.; Go, E.M.; Jana, B.; Park, S.A.; Kim, H.Y.; Kim, K.; Seo, J.K.; Kwak, S.K.; et al. Cloaking nanoparticles with protein corona shield for targeted drug delivery. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papi, M.; Caputo, D.; Palmieri, V.; Coppola, R.; Palchetti, S.; Bugli, F.; Martini, C.; Digiacomo, L.; Pozzi, D.; Caracciolo, G. Clinically approved PEGylated nanoparticles are covered by a protein corona that boosts the uptake by cancer cells. Nanoscale 2017, 9, 10327–10334. [Google Scholar] [CrossRef]
- Pattipeiluhu, R.; Crielaard, S.; Klein-Schiphorst, I.; Florea, B.I.; Kros, A.; Campbell, F. Unbiased Identification of the Liposome Protein Corona using Photoaffinity-based Chemoproteomics. ACS Cent. Sci. 2020, 6, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritz, S.; Schottler, S.; Kotman, N.; Baier, G.; Musyanovych, A.; Kuharev, J.; Landfester, K.; Schild, H.; Jahn, O.; Tenzer, S.; et al. Protein corona of nanoparticles: Distinct proteins regulate the cellular uptake. Biomacromolecules 2015, 16, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guan, J.; Jiang, Z.X.; Yang, Y.; Liu, J.C.; Hua, W.; Mao, Y.; Li, C.; Lu, W.Y.; Qian, J.; et al. Brain-targeted drug delivery by manipulating protein corona functions. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elechalawar, C.K.; Hossen, M.N.; McNally, L.; Bhattacharya, R.; Mukherjee, P. Analysing the nanoparticle-protein corona for potential molecular target identification. J. Control. Release 2020, 322, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Giulimondi, F.; Digiacomo, L.; Pozzi, D.; Palchetti, S.; Vulpis, E.; Capriotti, A.L.; Chiozzi, R.Z.; Lagana, A.; Amenitsch, H.; Masuelli, L.; et al. Interplay of protein corona and immune cells controls blood residency of liposomes. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehbe, M.; Anantha, M.; Backstrom, I.; Leung, A.; Chen, K.; Malhotra, A.; Edwards, K.; Bally, M.B. Nanoscale Reaction Vessels Designed for Synthesis of Copper-Drug Complexes Suitable for Preclinical Development. PLoS ONE 2016, 11, e0153416. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.; Koll-Weber, M.; Suss, R. Preclinical In Vitro Studies with 3D Spheroids to Evaluate Cu(DDC)2 Containing Liposomes for the Treatment of Neuroblastoma. Pharmaceutics 2021, 13, 894. [Google Scholar] [CrossRef]
- Gouda, A.; Sakr, O.S.; Nasr, M.; Sammour, O. Ethanol injection technique for liposomes formulation: An insight into development, influencing factors, challenges and applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102174. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res. 2010, 20, 228–243. [Google Scholar] [CrossRef]
- Meeth, K.; Wang, J.X.; Micevic, G.; Damsky, W.; Bosenberg, M.W. The YUMM lines: A series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment. Cell Melanoma Res. 2016, 29, 590–597. [Google Scholar] [CrossRef]
- Wang, J.; Perry, C.J.; Meeth, K.; Thakral, D.; Damsky, W.; Micevic, G.; Kaech, S.; Blenman, K.; Bosenberg, M. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment. Cell Melanoma Res. 2017, 30, 428–435. [Google Scholar] [CrossRef]
- Hayashi, Y.; Takamiya, M.; Jensen, P.B.; Ojea-Jimenez, I.; Claude, H.; Antony, C.; Kjaer-Sorensen, K.; Grabher, C.; Boesen, T.; Gilliland, D.; et al. Differential Nanoparticle Sequestration by Macrophages and Scavenger Endothelial Cells Visualized in Vivo in Real-Time and at Ultrastructural Resolution. ACS Nano 2020, 14, 1665–1681. [Google Scholar] [CrossRef]
- Tripuramallu, S.; Suthakaran, R. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening for Some Pyrazole-Quinazoline Derivatives. Indo Am. J. Pharm. Sci. 2018, 5, 16606–16611. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Marengo, A.; Forciniti, S.; Dando, I.; Dalla Pozza, E.; Stella, B.; Tsapis, N.; Yagoubi, N.; Fanelli, G.; Fattal, E.; Heeschen, C.; et al. Pancreatic cancer stem cell proliferation is strongly inhibited by diethyldithiocarbamate-copper complex loaded into hyaluronic acid decorated liposomes. Biochim. Biophys. Acta-Gen. Subj. 2019, 1863, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.E.; Mathiasen, S.; Bright, N.A.; Pierre, F.; Kelly, B.T.; Kladt, N.; Schauss, A.; Merrifield, C.J.; Stamou, D.; Honing, S.; et al. CALM Regulates Clathrin-Coated Vesicle Size and Maturation by Directly Sensing and Driving Membrane Curvature. Dev. Cell 2015, 33, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Pelt, J.; Busatto, S.; Ferrari, M.; Thompson, E.A.; Mody, K.; Wolfram, J. Chloroquine and nanoparticle drug delivery: A promising combination. Pharmacol. Ther. 2018, 191, 43–49. [Google Scholar] [CrossRef]
- Rodal, S.K.; Skretting, G.; Garred, O.; Vilhardt, F.; van Deurs, B.; Sandvig, K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell 1999, 10, 961–974. [Google Scholar] [CrossRef]
- Vercauteren, D.; Vandenbroucke, R.E.; Jones, A.T.; Rejman, J.; Demeester, J.; De Smedt, S.C.; Sanders, N.N.; Braeckmans, K. The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls. Mol. Ther. 2010, 18, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Pochapski, D.J.; Carvalho Dos Santos, C.; Leite, G.W.; Pulcinelli, S.H.; Santilli, C.V. Zeta Potential and Colloidal Stability Predictions for Inorganic Nanoparticle Dispersions: Effects of Experimental Conditions and Electrokinetic Models on the Interpretation of Results. Langmuir 2021, 37, 13379–13389. [Google Scholar] [CrossRef]
- Strauss, G.; Hauser, H. Stabilization of Lipid Bilayer Vesicles by Sucrose during Freezing. Proc. Natl. Acad. Sci. USA 1986, 83, 2422–2426. [Google Scholar] [CrossRef] [Green Version]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Sou, K.; Tsuchida, E. Electrostatic interactions and complement activation on the surface of phospholipid vesicle containing acidic lipids: Effect of the structure of acidic groups. Biochim. Et Biophys. Acta-Biomembr. 2008, 1778, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Wang, J.L.; Mu, Q.X.; Su, G.X. In vivo Protein Corona Formation: Characterizations, Effects on Engineered Nanoparticles’ Biobehaviors, and Applications. Front. Bioeng. Biotechnol. 2021, 9, 263. [Google Scholar] [CrossRef]
- Settanni, G.; Zhou, J.J.; Suo, T.C.; Schottler, S.; Landfester, K.; Schmid, F.; Mailander, V. Protein corona composition of poly(ethylene glycol)- and poly(phosphoester)-coated nanoparticles correlates strongly with the amino acid composition of the protein surface. Nanoscale 2017, 9, 2138–2144. [Google Scholar] [CrossRef] [Green Version]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Simon, J.; Kuhn, G.; Fichter, M.; Gehring, S.; Landfester, K.; Mailander, V. Unraveling the In Vivo Protein Corona. Cells 2021, 10, 132. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef]
- Naufer, A.; Hipolito, V.E.B.; Ganesan, S.; Prashar, A.; Zaremberg, V.; Botelho, R.J.; Terebiznik, M.R. pH of endophagosomes controls association of their membranes with Vps34 and Ptdlns(3)P levels. J. Cell Biol. 2018, 214, 329–346. [Google Scholar] [CrossRef] [Green Version]
- Le Poole, I.C.; van den Wijngaard, R.M.; Westerhof, W.; Verkruisen, R.P.; Dutrieux, R.P.; Dingemans, K.P.; Das, P.K. Phagocytosis by normal human melanocytes in vitro. Exp. Cell Res. 1993, 205, 388–395. [Google Scholar] [CrossRef]
- Pan, M.; Zheng, Q.; Yu, Y.; Ai, H.; Xie, Y.; Zeng, X.; Wang, C.; Liu, L.; Zhao, M. Seesaw conformations of Npl4 in the human p97 complex and the inhibitory mechanism of a disulfiram derivative. Nat. Commun. 2021, 12, 121. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Watson, I.R.; Wu, C.J.; Gershenwald, J.E.; Chin, L.; Grp, M.A.W.; Network, C.G.A.R. Comprehensive genomic characterization of cutaneous melanoma. Cancer Res. 2014, 74, e63597. [Google Scholar] [CrossRef]
- Wong, S.Q.; Li, J.; Salemi, R.; Sheppard, K.E.; Do, H.D.; Tothill, R.W.; McArthur, G.A.; Dobrovic, A. Targeted-capture massively-parallel sequencing enables robust detection of clinically informative mutations from formalin-fixed tumours. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennie, L.A.; McCarthy, H.O.; Coulter, J.A. Enhanced nanoparticle delivery exploiting tumour-responsive formulations. Cancer Nanotechnol. 2018, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Van der Meel, R.; Chen, X.Y.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Kingston, B.R.; Lin, Z.P.; Ouyang, B.; MacMillan, P.; Ngai, J.; Syed, A.M.; Sindhwani, S.; Chan, W.C.W. Specific Endothelial Cells Gouvern Nanoparticle Entry into Solid Tumors. ACS Nano 2021, 15, 14080–14094. [Google Scholar] [CrossRef]
- Bitto-Dotan, R.; Bohrisch, J.; Schmidt, C.; Tsuriel, M.; Tulichala, R.N.P.; Breuer, E.; Reich, R.; Hoffman, A.; Strosberg, J. The Effect of Chemical Modifications of Chitosan on Intestinal Permeability and Oral Bioavailability of Carbamoylphosphonate JS403. J. Bioequiv. Availab. 2020, 12, 392. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating Pharmacokinetics, Tumor Uptake and Biodistribution by Engineered Nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, L.; Alvarez-Puebla, R.A.; Pazos-Peres, N. Surface Modifications of Nanoparticles for Stability in Biological Fluids. Materials 2018, 11, 1154. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Size (nm) | Zeta (mV) | PDI | EE (%) |
|---|---|---|---|---|
| LP-Control | 123.5 ± 1.9 | −57.36 ± 0.019 | 0.240 ± 0.019 | N/A |
| LP-CuET | 111.9 ± 2.1 | −56.60 ± 0.009 | 0.132 ± 0.069 | 81.01 ± 1.16 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paun, R.A.; Dumut, D.C.; Centorame, A.; Thuraisingam, T.; Hajduch, M.; Mistrik, M.; Dzubak, P.; De Sanctis, J.B.; Radzioch, D.; Tabrizian, M. One-Step Synthesis of Nanoliposomal Copper Diethyldithiocarbamate and Its Assessment for Cancer Therapy. Pharmaceutics 2022, 14, 640. https://doi.org/10.3390/pharmaceutics14030640
Paun RA, Dumut DC, Centorame A, Thuraisingam T, Hajduch M, Mistrik M, Dzubak P, De Sanctis JB, Radzioch D, Tabrizian M. One-Step Synthesis of Nanoliposomal Copper Diethyldithiocarbamate and Its Assessment for Cancer Therapy. Pharmaceutics. 2022; 14(3):640. https://doi.org/10.3390/pharmaceutics14030640
Chicago/Turabian StylePaun, Radu A., Daciana C. Dumut, Amanda Centorame, Thusanth Thuraisingam, Marian Hajduch, Martin Mistrik, Petr Dzubak, Juan B. De Sanctis, Danuta Radzioch, and Maryam Tabrizian. 2022. "One-Step Synthesis of Nanoliposomal Copper Diethyldithiocarbamate and Its Assessment for Cancer Therapy" Pharmaceutics 14, no. 3: 640. https://doi.org/10.3390/pharmaceutics14030640
APA StylePaun, R. A., Dumut, D. C., Centorame, A., Thuraisingam, T., Hajduch, M., Mistrik, M., Dzubak, P., De Sanctis, J. B., Radzioch, D., & Tabrizian, M. (2022). One-Step Synthesis of Nanoliposomal Copper Diethyldithiocarbamate and Its Assessment for Cancer Therapy. Pharmaceutics, 14(3), 640. https://doi.org/10.3390/pharmaceutics14030640