
Development and Validation of the New Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Unbound Tacrolimus in the Plasma Ultrafiltrate of Transplant Recipients

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Patient Samples
2.2. Sample Preparation
2.3. LC–MS/MS Instrument Parameters
- (1)
- A total of 90% of solution A and 10% of solution B directly after injection—minute 1.
- (2)
- A total of 5% of solution A and 95% of solution B—minutes 1 to 3.
- (3)
- A total of 90% of solution A and 10% of solution B—minutes 3 to 5.
2.4. Method Validation
2.5. Statistics
3. Results
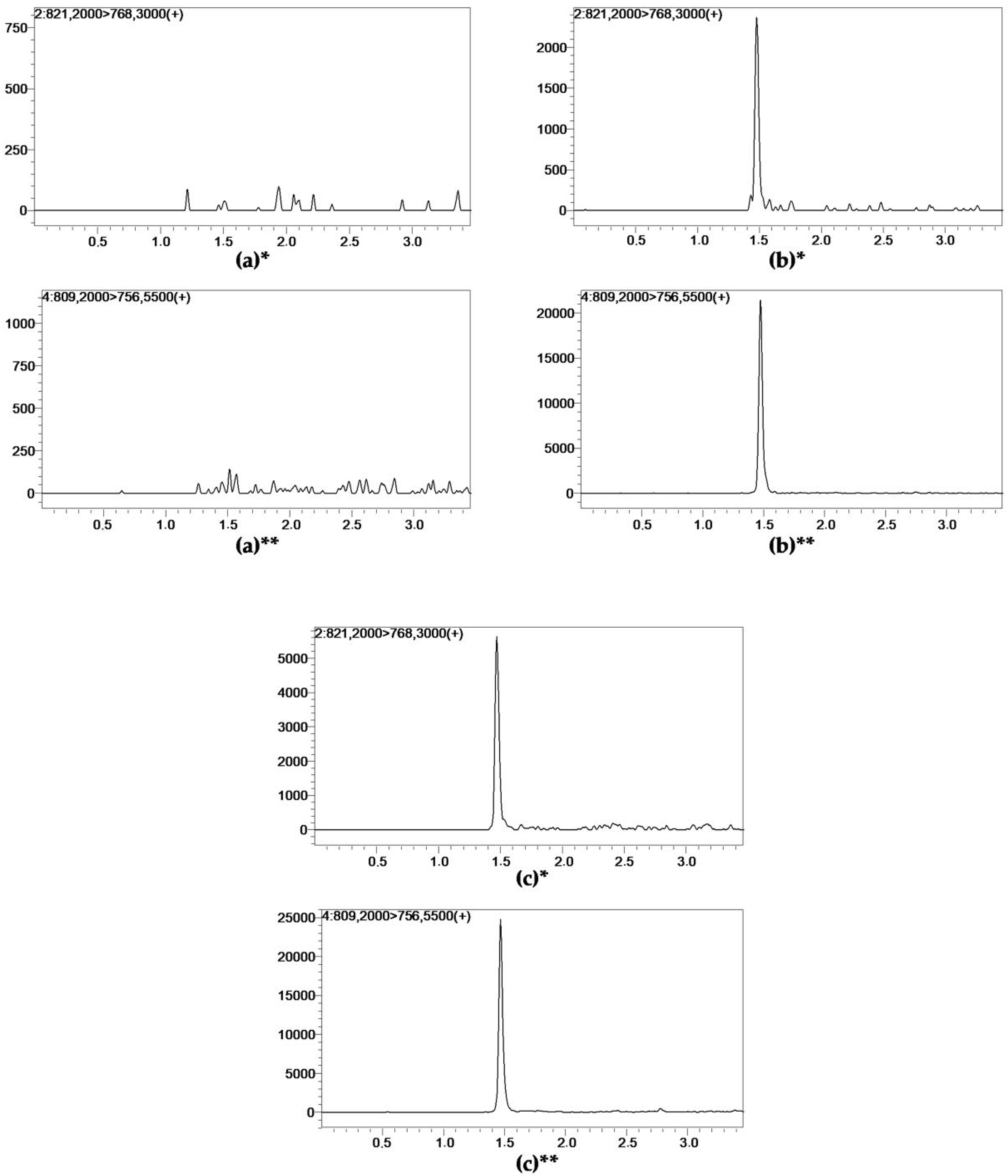
3.1. Method Development and Conditions
3.2. Method Validation
3.3. Patient Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2009, 9 (Suppl. 3), S1–S155. [Google Scholar] [CrossRef]
- Burra, P.; Burroughs, A.; Graziadei, I.; Pirenne, J.; Valdecasas, J.C.; Muiesan, P.; Samuel, D.; Forns, X. EASL Clinical Practice Guidelines: Liver transplantation. J. Hepatol. 2016, 64, 433–485. [Google Scholar] [CrossRef]
- Cecka, J.M.; Terasaki, P.I. Early rejection episodes. Clin. Transpl. 1989, 425–434. Available online: https://pubmed.ncbi.nlm.nih.gov/2487611/ (accessed on 8 February 2022).
- Kim, W.R.; Stock, P.G.; Smith, J.M.; Heimbach, J.K.; Skeans, M.A.; Edwards, E.B.; Harper, A.M.; Snyder, J.J.; Israni, A.K.; Kasiske, B.L. OPTN/SRTR 2011 Annual Data Report: Liver. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2013, 13 (Suppl. 1), 73–102. [Google Scholar] [CrossRef]
- Matas, A.J.; Smith, J.M.; Skeans, M.A.; Lamb, K.E.; Gustafson, S.K.; Samana, C.J.; Stewart, D.E.; Snyder, J.J.; Israni, A.K.; Kasiske, B.L. OPTN/SRTR 2011 Annual Data Report: Kidney. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2013, 13 (Suppl. 1), 11–46. [Google Scholar] [CrossRef] [Green Version]
- Brunet, M.; van Gelder, T.; Asberg, A.; Haufroid, V.; Hesselink, D.A.; Langman, L.; Lemaitre, F.; Marquet, P.; Seger, C.; Shipkova, M.; et al. Therapeutic Drug Monitoring of Tacrolimus-Personalized Therapy: Second Consensus Report. Ther. Drug Monit. 2019, 41, 261–307. [Google Scholar] [CrossRef]
- Sikma, M.A.; van Maarseveen, E.M.; van de Graaf, E.A.; Kirkels, J.H.; Verhaar, M.C.; Donker, D.W.; Kesecioglu, J.; Meulenbelt, J. Pharmacokinetics and Toxicity of Tacrolimus Early After Heart and Lung Transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2015, 15, 2301–2313. [Google Scholar] [CrossRef]
- Bouamar, R.; Shuker, N.; Hesselink, D.A.; Weimar, W.; Ekberg, H.; Kaplan, B.; Bernasconi, C.; van Gelder, T. Tacrolimus predose concentrations do not predict the risk of acute rejection after renal transplantation: A pooled analysis from three randomized-controlled clinical trials. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2013, 13, 1253–1261. [Google Scholar] [CrossRef]
- Kershner, R.P.; Fitzsimmons, W.E. Relationship of FK506 whole blood concentrations and efficacy and toxicity after liver and kidney transplantation. Transplantation 1996, 62, 920–926. [Google Scholar] [CrossRef]
- Rodríguez-Perálvarez, M.; Germani, G.; Darius, T.; Lerut, J.; Tsochatzis, E.; Burroughs, A.K. Tacrolimus trough levels, rejection and renal impairment in liver transplantation: A systematic review and meta-analysis. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2012, 12, 2797–2814. [Google Scholar] [CrossRef] [Green Version]
- Zahir, H.; McCaughan, G.; Gleeson, M.; Nand, R.A.; McLachlan, A.J. Changes in tacrolimus distribution in blood and plasma protein binding following liver transplantation. Ther. Drug Monit. 2004, 26, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Shuker, N.; Shuker, L.; van Rosmalen, J.; Roodnat, J.I.; Borra, L.C.; Weimar, W.; Hesselink, D.A.; van Gelder, T. A high intrapatient variability in tacrolimus exposure is associated with poor long-term outcome of kidney transplantation. Transpl. Int. Off. J. Eur. Soc. Organ Transplant. 2016, 29, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Sikma, M.A.; Van Maarseveen, E.M.; Hunault, C.C.; Moreno, J.M.; Van de Graaf, E.A.; Kirkels, J.H.; Verhaar, M.C.; Grutters, J.C.; Kesecioglu, J.; De Lange, D.W.; et al. Unbound Plasma, Total Plasma, and Whole-Blood Tacrolimus Pharmacokinetics Early After Thoracic Organ Transplantation. Clin. Pharmacokinet. 2020, 59, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahir, H.; Nand, R.A.; Brown, K.F.; Tattam, B.N.; McLachlan, A.J. Validation of methods to study the distribution and protein binding of tacrolimus in human blood. J. Pharmacol. Toxicol. Methods 2001, 46, 27–35. [Google Scholar] [CrossRef]
- Zheng, S.; Easterling, T.R.; Umans, J.G.; Miodovnik, M.; Calamia, J.C.; Thummel, K.E.; Shen, D.D.; Davis, C.L.; Hebert, M.F. Pharmacokinetics of tacrolimus during pregnancy. Ther. Drug Monit. 2012, 34, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capron, A.; Haufroid, V.; Wallemacq, P. Intra-cellular immunosuppressive drugs monitoring: A step forward towards better therapeutic efficacy after organ transplantation? Pharmacol. Res. 2016, 111, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Gerson, B. Free drug monitoring. Clin. Lab. Med. 1987, 7, 279–287. [Google Scholar] [CrossRef]
- Bittersohl, H.; Schniedewind, B.; Christians, U.; Luppa, P.B. A simple and highly sensitive on-line column extraction liquid chromatography-tandem mass spectrometry method for the determination of protein-unbound tacrolimus in human plasma samples. J. Chromatogr. A 2018, 1547, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, N.A.; Sikma, M.A.; van Dapperen, A.L.; de Lange, D.W.; van Maarseveen, E.M. Development of a Simple and Rapid Method to Measure the Free Fraction of Tacrolimus in Plasma Using Ultrafiltration and LC-MS/MS. Ther. Drug Monit. 2016, 38, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Bodnar-Broniarczyk, M.; Warzyszyńska, K. The New LC-MS/MS Method for Determination of Unbound Tacrolimus in Plasma (FreeTAC)—Method description. 2022. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Bioanalytical Method Validation. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 9 February 2022).
- Food and Drug Administration Guidance for Industry: Bioanalytical Method Validation. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 9 February 2022).
- Napoli, K.L.; Hammett-Stabler, C.; Taylor, P.J.; Lowe, W.; Franklin, M.E.; Morris, M.R.; Cooper, D.P. Multi-center evaluation of a commercial Kit for tacrolimus determination by LC/MS/MS. Clin. Biochem. 2010, 43, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Franklin, M.E.; Tai, C.H.; Pillans, P.I. Therapeutic drug monitoring of tacrolimus by liquid chromatography-tandem mass spectrometry: Is it truly a routine test? J. Chromatogr. B 2012, 883–884, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: A method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 1998, 70, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Seger, C.; Shipkova, M.; Christians, U.; Billaud, E.M.; Wang, P.; Holt, D.W.; Brunet, M.; Kunicki, P.K.; Pawiński, T.; Langman, L.J.; et al. Assuring the Proper Analytical Performance of Measurement Procedures for Immunosuppressive Drug Concentrations in Clinical Practice: Recommendations of the International Association of Therapeutic Drug Monitoring and Clinical Toxicology Immunosuppressive Drug Scientific Committee. Ther. Drug Monit. 2016, 38, 170–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar-Broniarczyk, M.; Warzyszyńska, K. The New LC-MS/MS Method for Determination of Unbound Tacrolimus in Plasma (FreeTAC)—FreeTAC dataset 1. 2022. [Google Scholar] [CrossRef]
- Howard, M.L.; Hill, J.J.; Galluppi, G.R.; McLean, M.A. Plasma protein binding in drug discovery and development. Comb. Chem. High Throughput Screen 2010, 13, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.W.; Lee, T.; Jones, K.; Johnston, A. Validation of an assay for routine monitoring of sirolimus using HPLC with mass spectrometric detection. Clin. Chem. 2000, 46, 1179–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keevil, B.G.; McCann, S.J.; Cooper, D.P.; Morris, M.R. Evaluation of a rapid micro-scale assay for tacrolimus by liquid chromatography-tandem mass spectrometry. Ann. Clin. Biochem. 2002, 39, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallemacq, P.E.; Vanbinst, R.; Asta, S.; Cooper, D.P. High-throughput liquid chromatography-tandem mass spectrometric analysis of sirolimus in whole blood. Clin. Chem. Lab. Med. 2003, 41, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Adaway, J.E.; Keevil, B.G. Therapeutic drug monitoring and LC-MS/MS. J. Chromatogr. B 2012, 883–884, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Christians, U.; Vinks, A.A.; Langman, L.J.; Clarke, W.; Wallemacq, P.; van Gelder, T.; Renjen, V.; Marquet, P.; Meyer, E.J. Impact of Laboratory Practices on Interlaboratory Variability in Therapeutic Drug Monitoring of Immunosuppressive Drugs. Ther. Drug Monit. 2015, 37, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Bodnar-Broniarczyk, M.; Pawiński, T.; Kunicki, P.K. Isotope-labeled versus analog internal standard in LC-MS/MS method for tacrolimus determination in human whole blood samples—A compensation of matrix effects. J. Chromatogr. B 2019, 1104, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical method validation: New FDA guidance vs. EMA guideline. Better or worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sample | Concentration Declared [pg/mL] | Within-Run (n = 6) | Between-Run (n = 6) | ||||
|---|---|---|---|---|---|---|---|
| Concentration [pg/mL] | Accuracy [%] | Imprecision [%] | Concentration [pg/mL] | Accuracy [%] | Imprecision [%] | ||
| LLOQ | 0.1 | 0.12 ± 0.02 | 109.72 | 7.48 | 0.11 ± 0.03 | 98.30 | 13.74 |
| LQC | 0.5 | 0.49 ± 0.04 | 97.75 | 8.67 | 0.54 ± 0.06 | 107.10 | 10.67 |
| MQC | 1.5 | 1.54 ± 0.16 | 102.20 | 10.56 | 1.49 ± 0.31 | 104.28 | 9.07 |
| HQC1 | 10 | 10.42 ± 0.27 | 100.48 | 2.67 | 10.07 ± 0.43 | 100.72 | 4.26 |
| HQC2 | 15 | 14.60 ± 0.92 | 97.33 | 6.29 | 15.11 ± 0.55 | 100.75 | 3.64 |
| Time | Low QC (0.5 pg/mL) | High QC1 (10 pg/mL) | ||
|---|---|---|---|---|
| Concentration Measured [pg/mL] | Stability [%] | Concentration Measured [pg/mL] | Stability [%] | |
| Autosampler stability at 4 °C (n = 3) | ||||
| 0 h | 0.51 ± 0.03 | 100.00 | 10.14 ± 0.32 | 100.00 |
| 4 h | 0.53 ± 0.03 | 104.56 | 9.80 ± 0.41 | 96.67 |
| 8 h | 0.53 ± 0.08 | 104.38 | 10.04 ± 0.22 | 99.05 |
| 12 h | 0.50 ± 0.06 | 97.96 | 10.26 ± 0.32 | 103.16 |
| Short-term stability at room temperature (n = 3) | ||||
| 0 h (standard procedure) | 0.52 ± 0.01 | 100.00 | 10.03 ± 0.54 | 100.00 |
| −4 h (before preparation) | 0.50 ± 0.02 | 98.43 | 9.47 ± 0.36 | 94.42 |
| +2 h (after preparation) | 0.49 ± 0.02 | 95.09 | 10.08 ± 0.58 | 100.52 |
| Long-term stability at −20 °C (n = 4) | ||||
| 1 week | 0.49 ± 0.03 | 100.00 | 10.16 ± 0.70 | 100.00 |
| 2 weeks | 0.48 ± 0.10 | 99.32 | 10.23 ± 0.25 | 100.71 |
| 3 weeks | 0.48 ± 0.07 | 98.54 | 10.26 ± 0.89 | 100.96 |
| 4 weeks | 0.47 ± 0.05 | 96.67 | 9.90 ± 0.50 | 97.42 |
| Parameter | Low QC (0.5 pg/mL) | High QC1 (10 pg/mL) | ||||
|---|---|---|---|---|---|---|
| TAC | IS ASC | F | TAC | IS ASC | F | |
| ME [%] (n = 6) | −46.26 ± 10.26 | −48.20 ± 9.98 | −1.03 ± 0.06 | −33.77 ± 21.63 | −42.78 ± 19.36 | 16.42 ± 15.19 |
| PE [%] (n = 6) | 47.26 ± 5.13 | 49.86 ± 5.67 | 95.10 ± 7.97 | 43.83 ± 3.76 | 47.14 ± 4.56 | 95.42 ± 8.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodnar-Broniarczyk, M.; Warzyszyńska, K.; Czerwińska, K.; Marszałek, D.; Dziewa, N.; Kosieradzki, M.; Pawiński, T. Development and Validation of the New Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Unbound Tacrolimus in the Plasma Ultrafiltrate of Transplant Recipients. Pharmaceutics 2022, 14, 632. https://doi.org/10.3390/pharmaceutics14030632
Bodnar-Broniarczyk M, Warzyszyńska K, Czerwińska K, Marszałek D, Dziewa N, Kosieradzki M, Pawiński T. Development and Validation of the New Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Unbound Tacrolimus in the Plasma Ultrafiltrate of Transplant Recipients. Pharmaceutics. 2022; 14(3):632. https://doi.org/10.3390/pharmaceutics14030632
Chicago/Turabian StyleBodnar-Broniarczyk, Magdalena, Karola Warzyszyńska, Katarzyna Czerwińska, Dorota Marszałek, Natalia Dziewa, Maciej Kosieradzki, and Tomasz Pawiński. 2022. "Development and Validation of the New Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Unbound Tacrolimus in the Plasma Ultrafiltrate of Transplant Recipients" Pharmaceutics 14, no. 3: 632. https://doi.org/10.3390/pharmaceutics14030632
APA StyleBodnar-Broniarczyk, M., Warzyszyńska, K., Czerwińska, K., Marszałek, D., Dziewa, N., Kosieradzki, M., & Pawiński, T. (2022). Development and Validation of the New Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Unbound Tacrolimus in the Plasma Ultrafiltrate of Transplant Recipients. Pharmaceutics, 14(3), 632. https://doi.org/10.3390/pharmaceutics14030632