
A Novel Deep Neural Network Technique for Drug–Target Interaction




Abstract
:1. Introduction
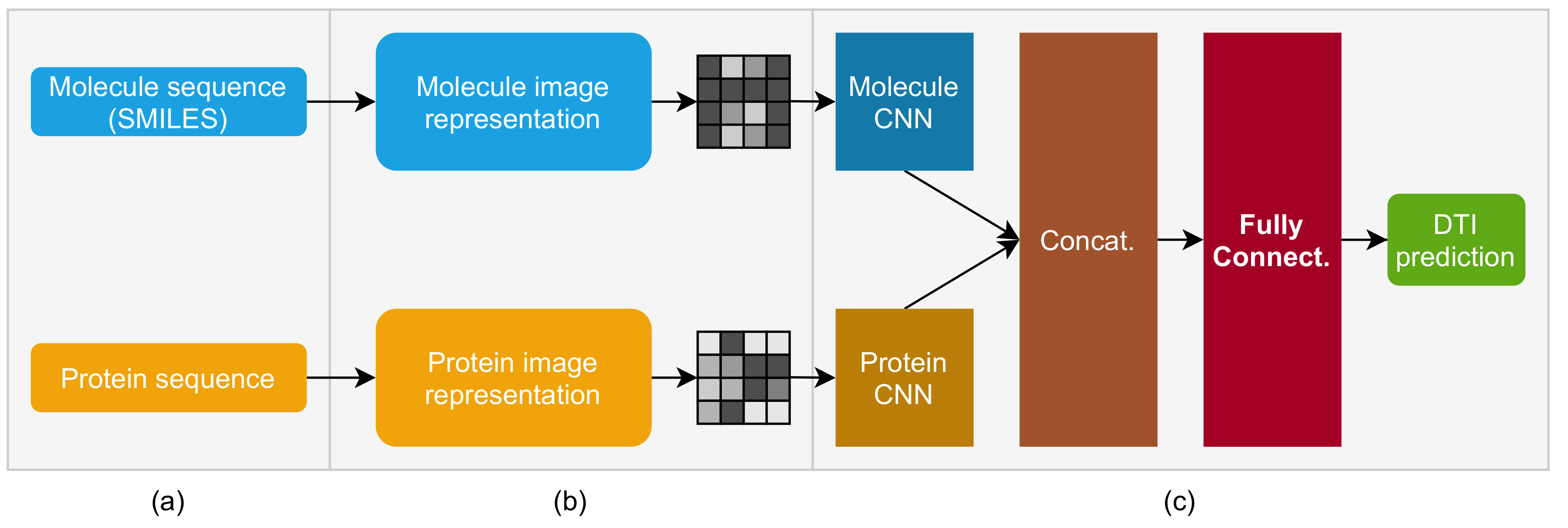
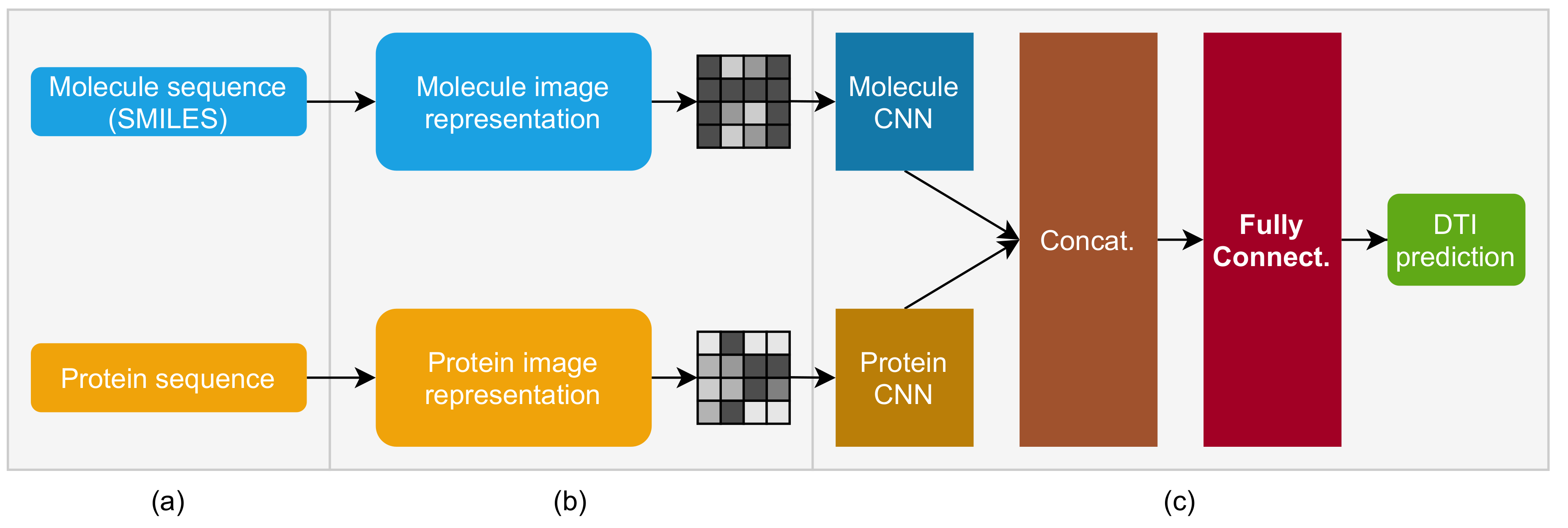
2. Molecule and Protein Sequence to Image Transformer DTI (MPS2IT-DTI)
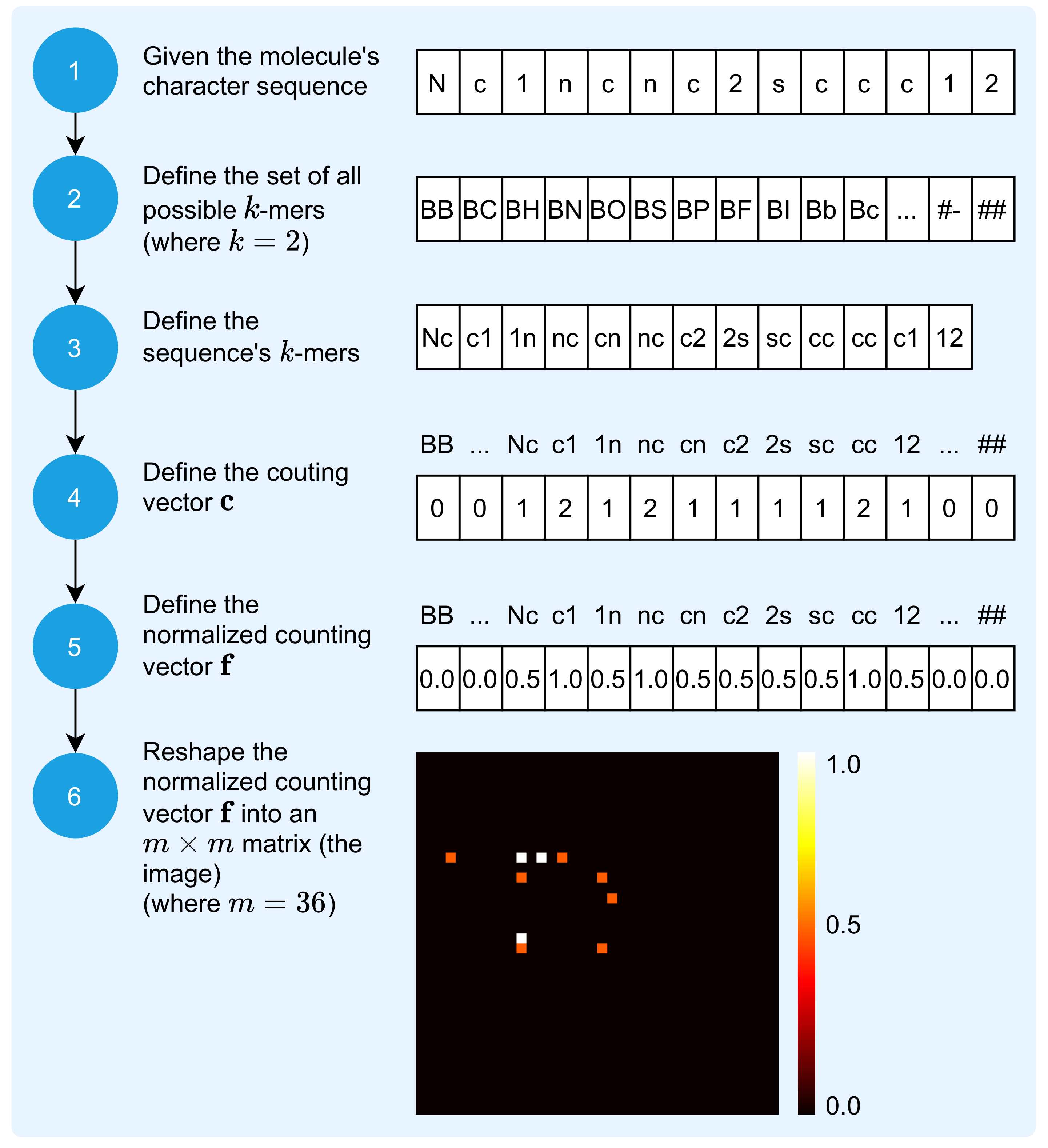
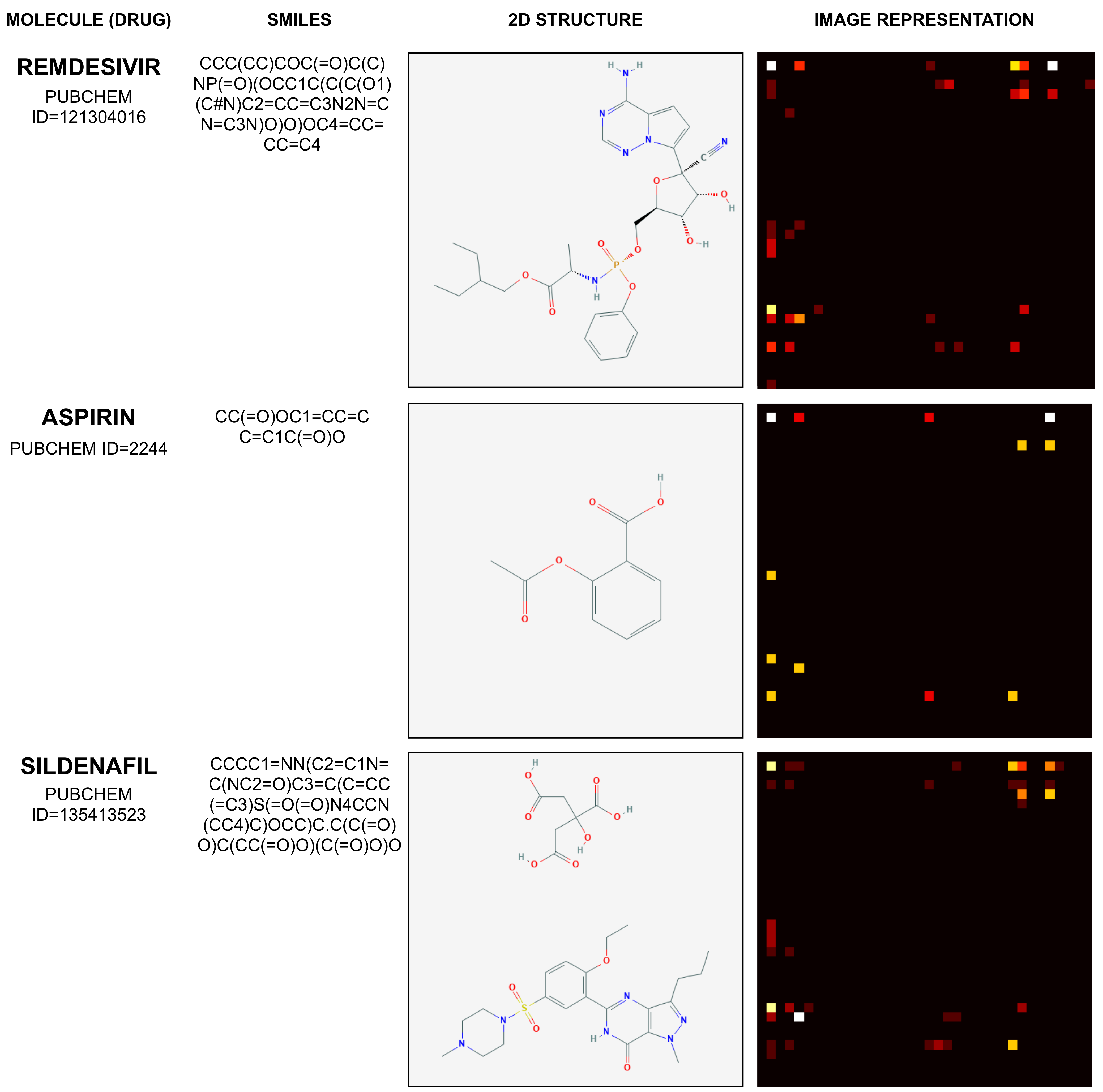
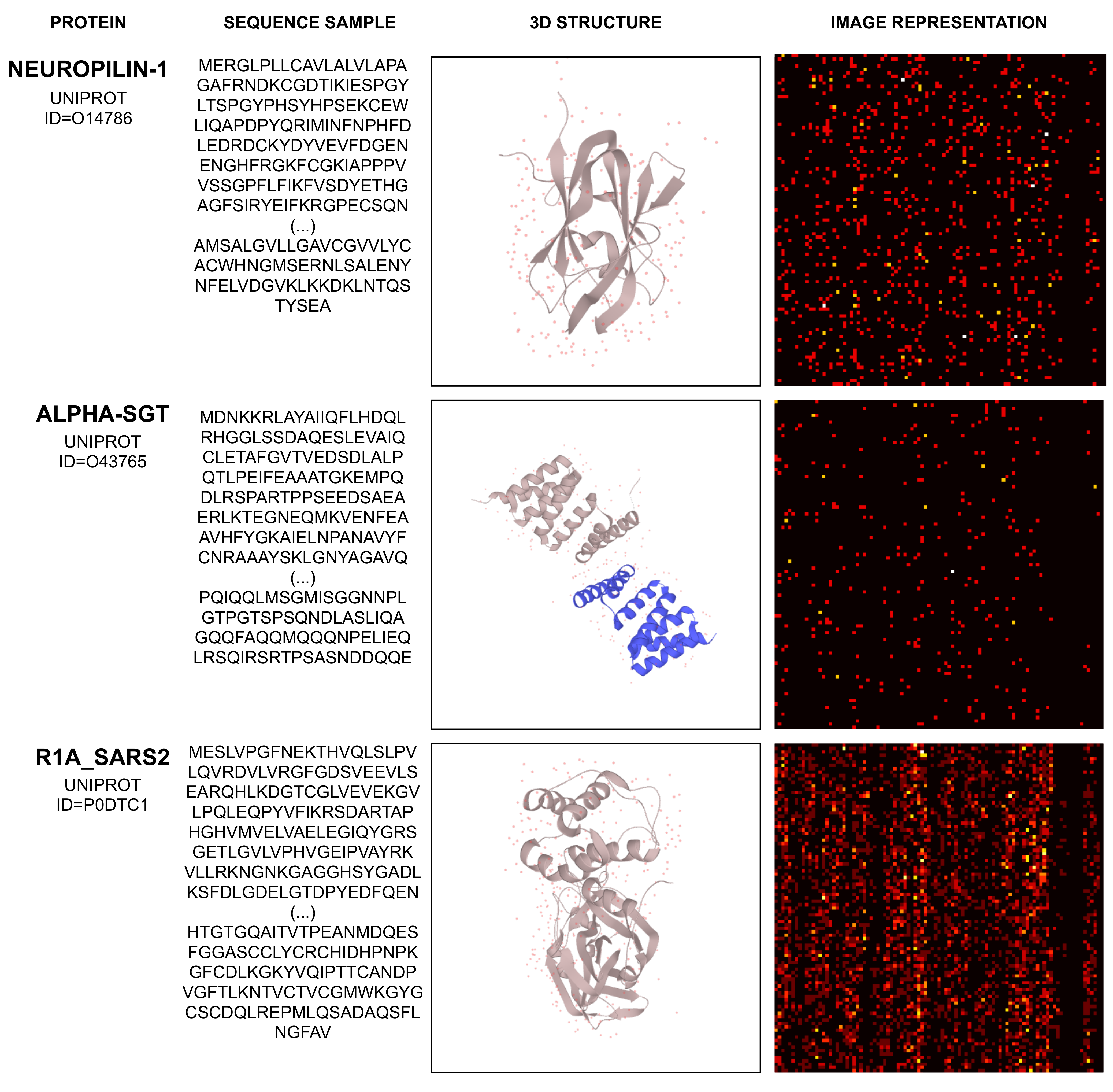
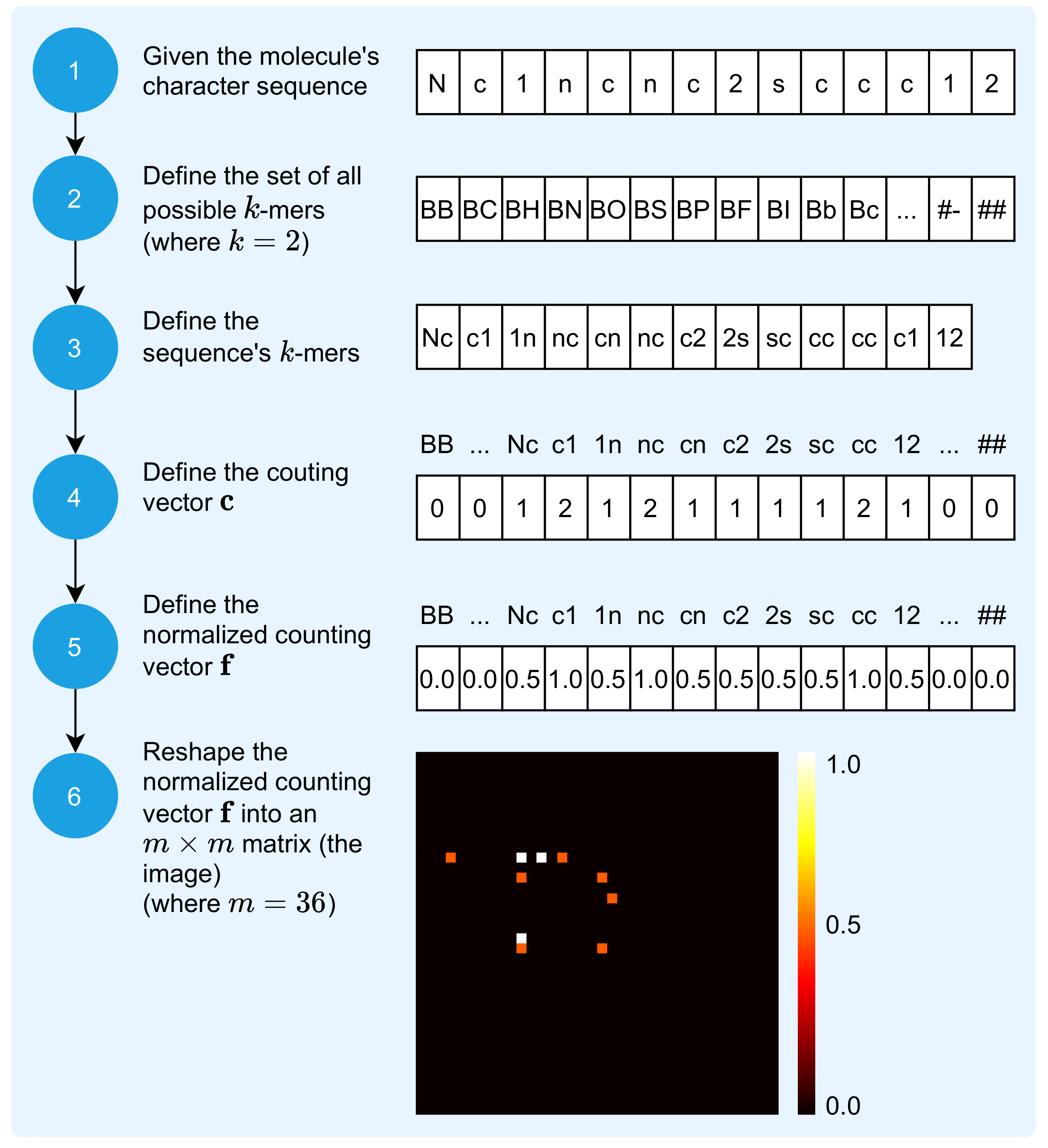
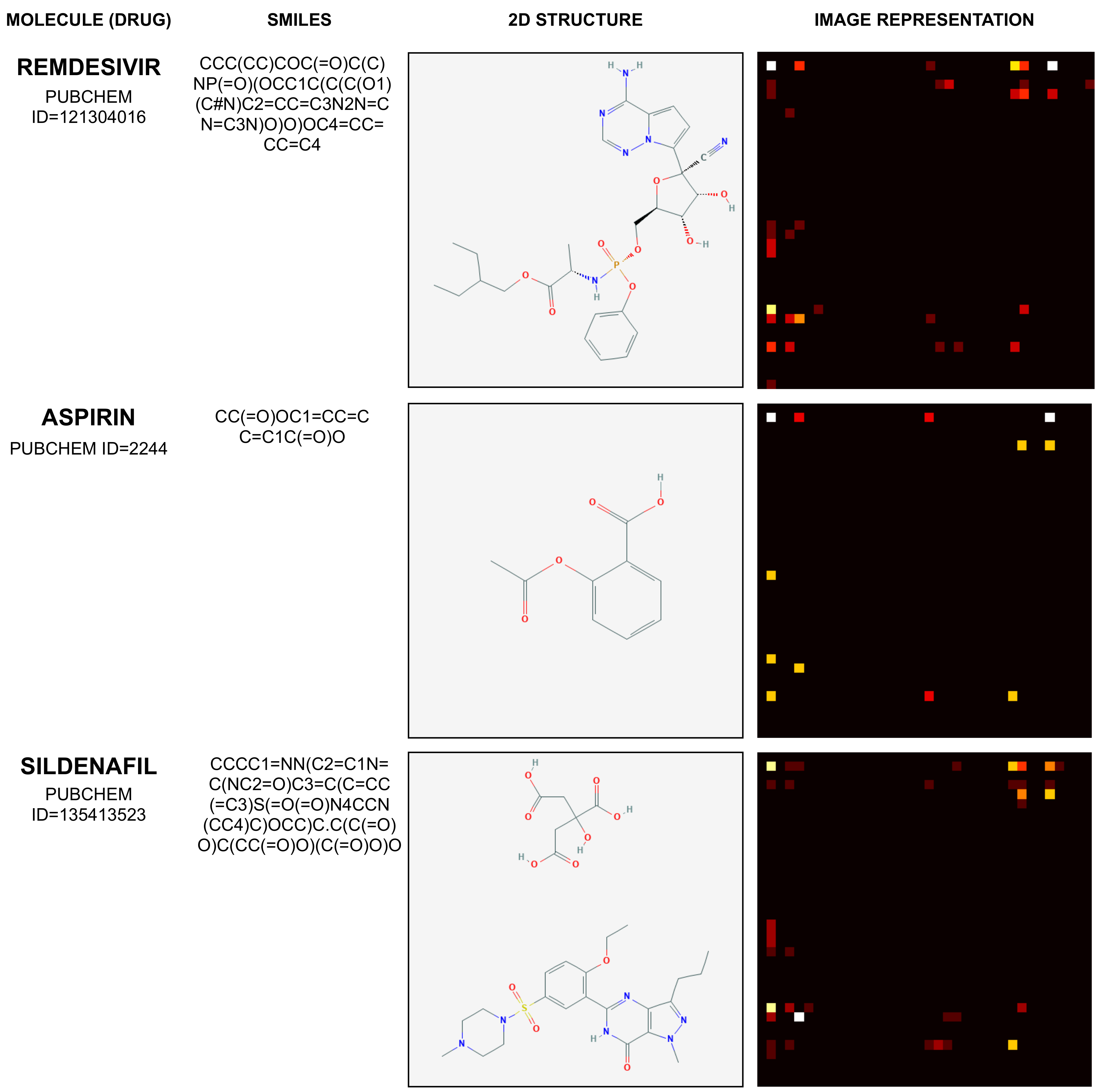
2.1. Mapping Molecule Sequence to an Image
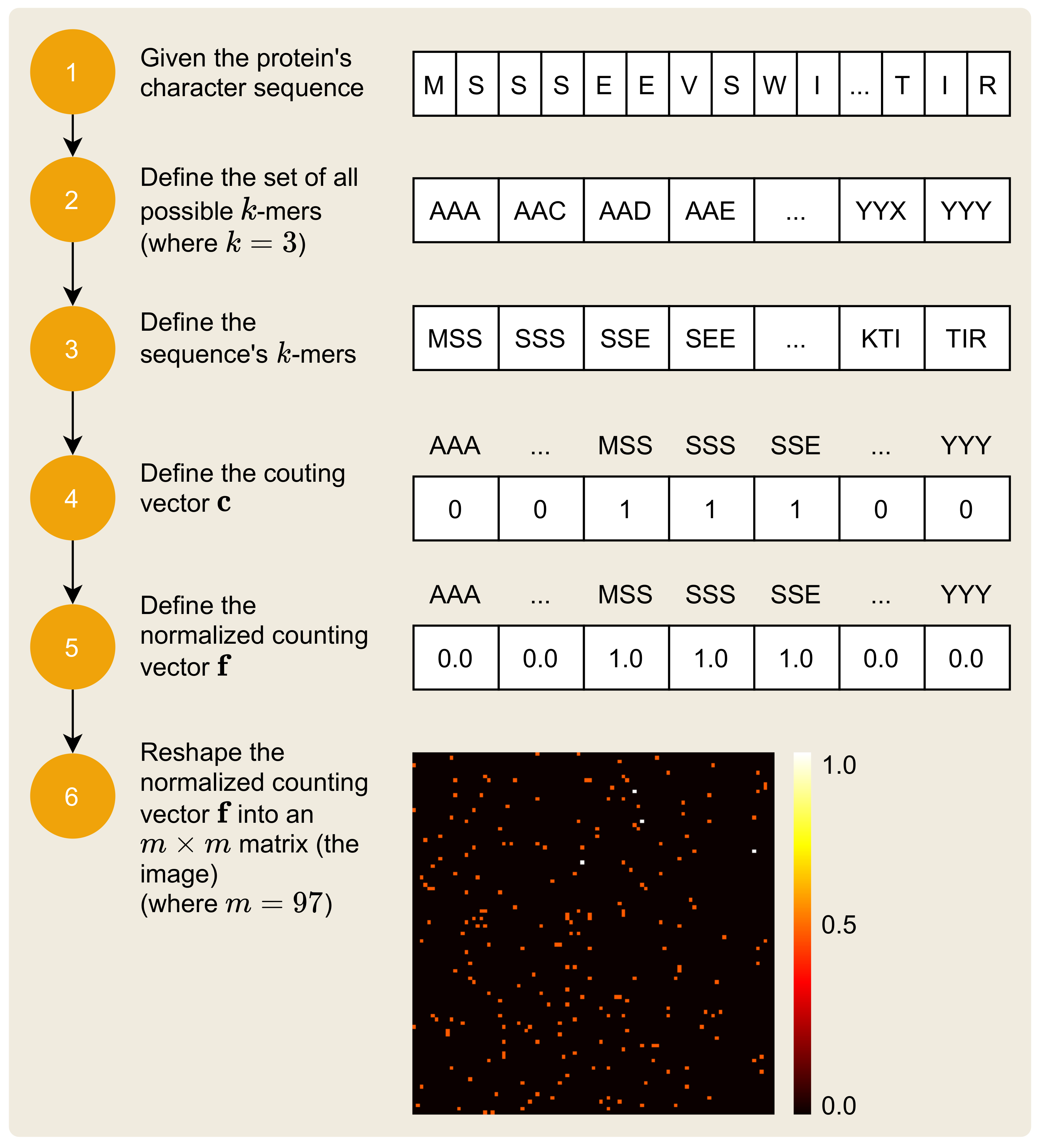
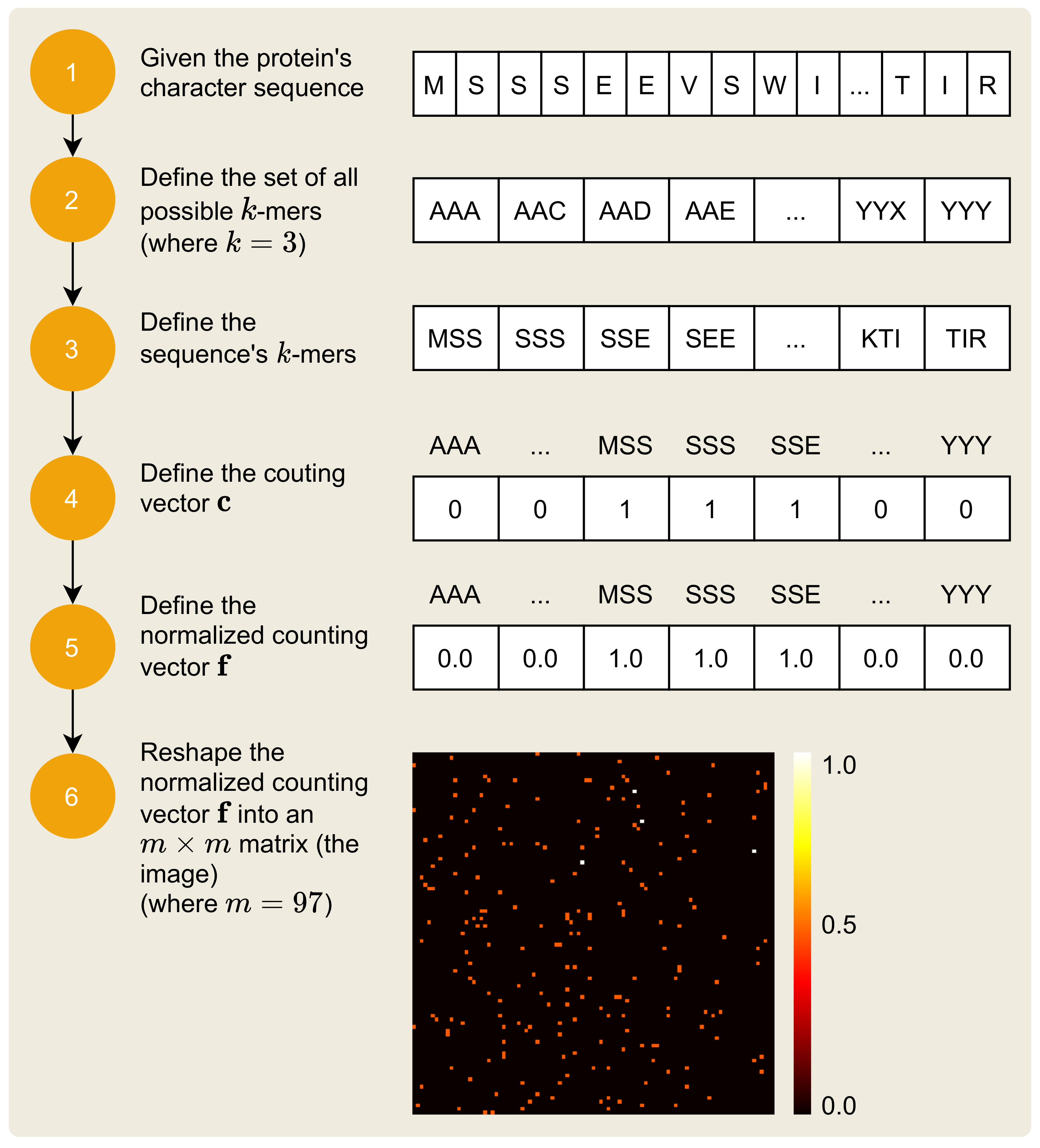
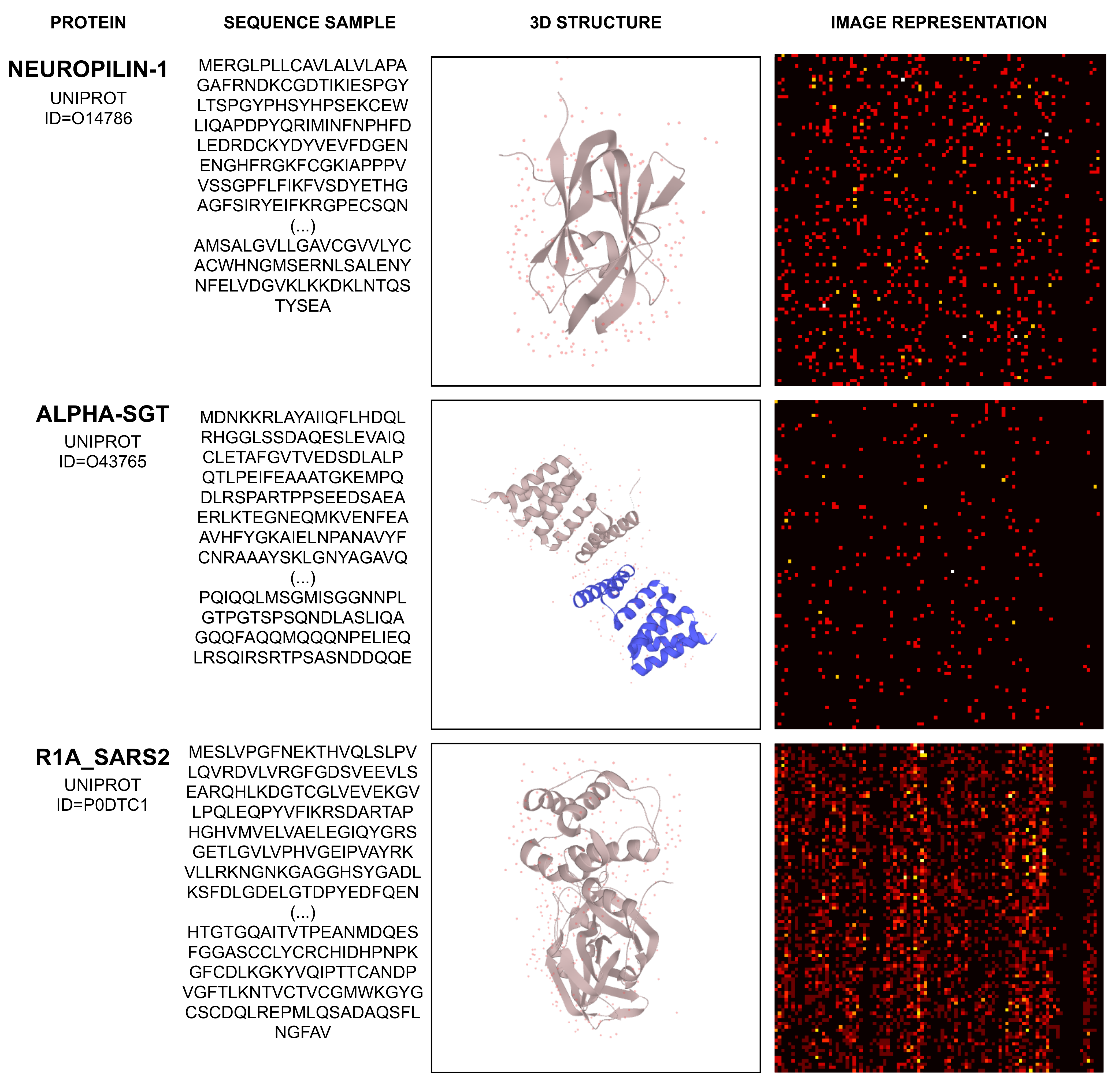
2.2. Mapping Protein Sequence to an Image
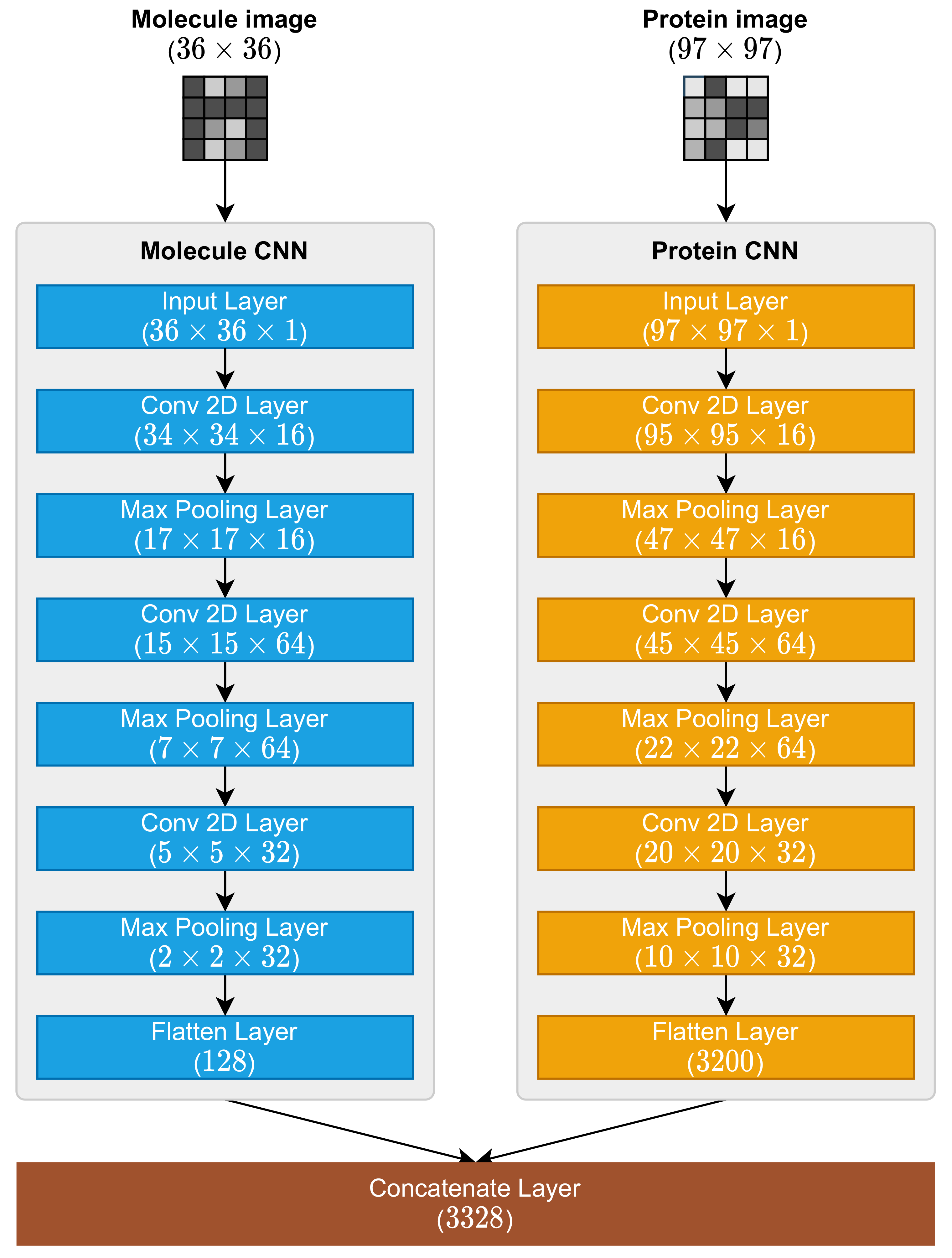
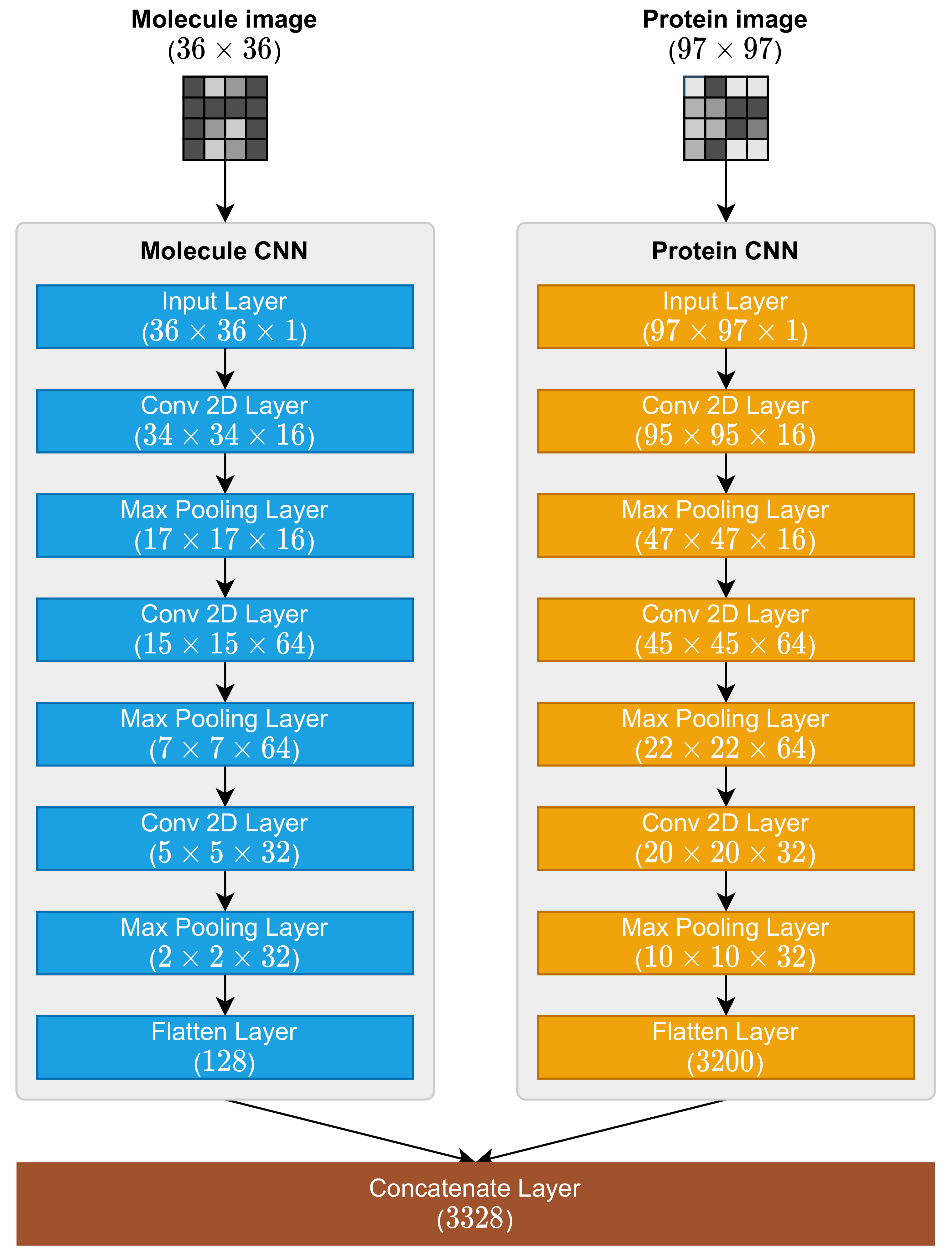
2.3. Molecule and Protein CNNs
- 1.
- Input Layer;
- 2.
- Conv 2D Layer (with ReLu activation function);
- 3.
- Max Pooling 2D Layer;
- 4.
- Conv 2D Layer (with ReLu activation function);
- 5.
- Max Pooling 2D Layer;
- 6.
- Conv 2D Layer (with ReLu activation function);
- 7.
- Max Pooling 2D Layer;
- 8.
- Flatten Layer.
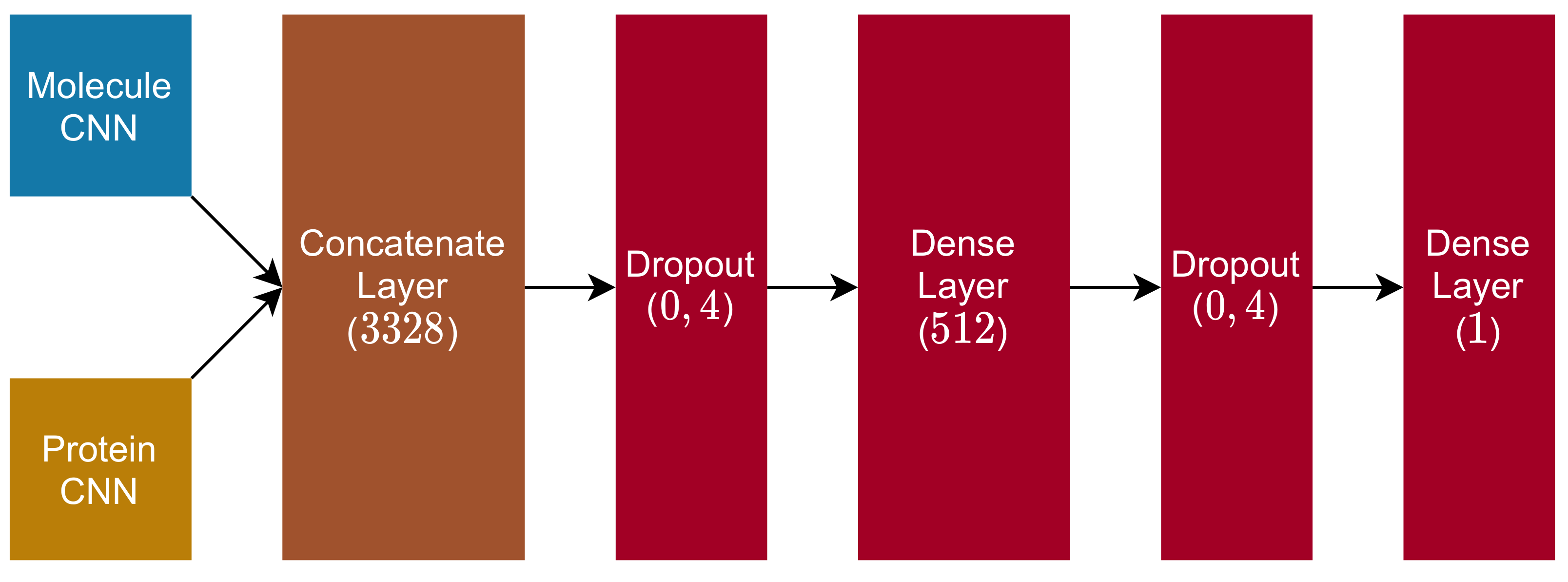
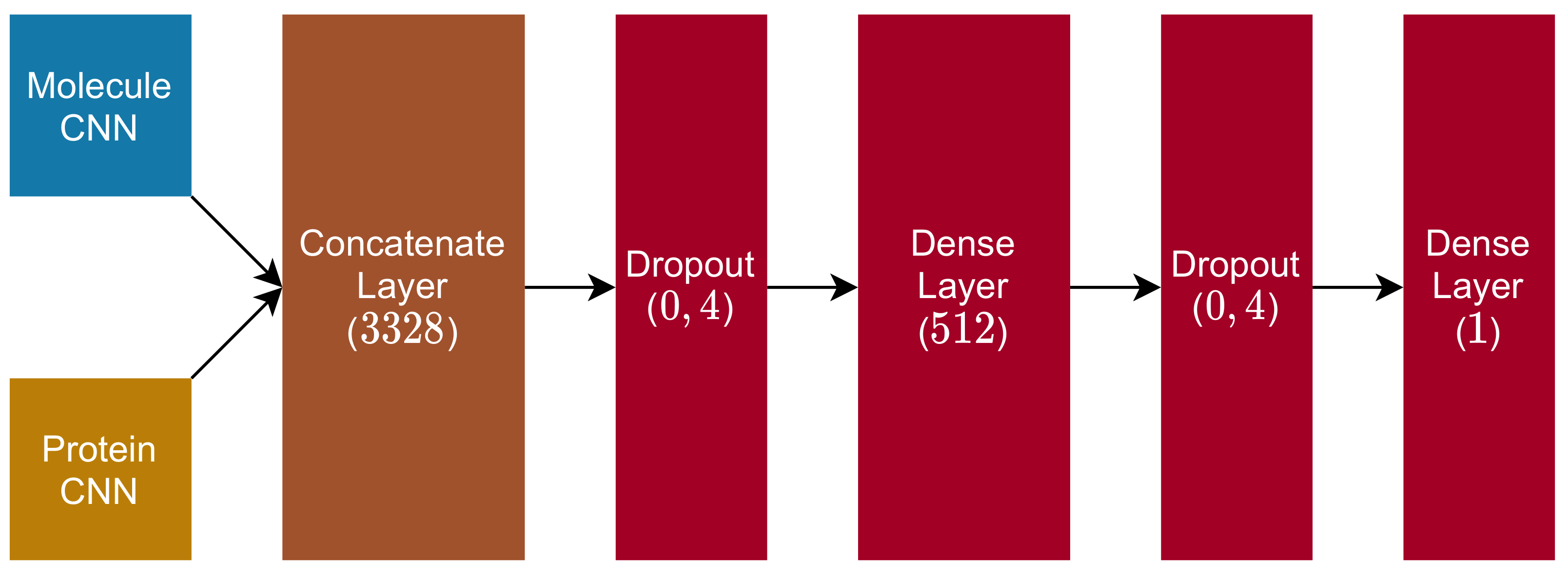
2.4. Fully Connected and Output Blocks
- 1.
- Dropout Layer ( rate);
- 2.
- Dense Layer (dimension , with ReLu activation function);
- 3.
- Dropout Layer ( rate);
- 4.
- Dense Layer (dimension , with linear activation function).
3. Materials and Methods
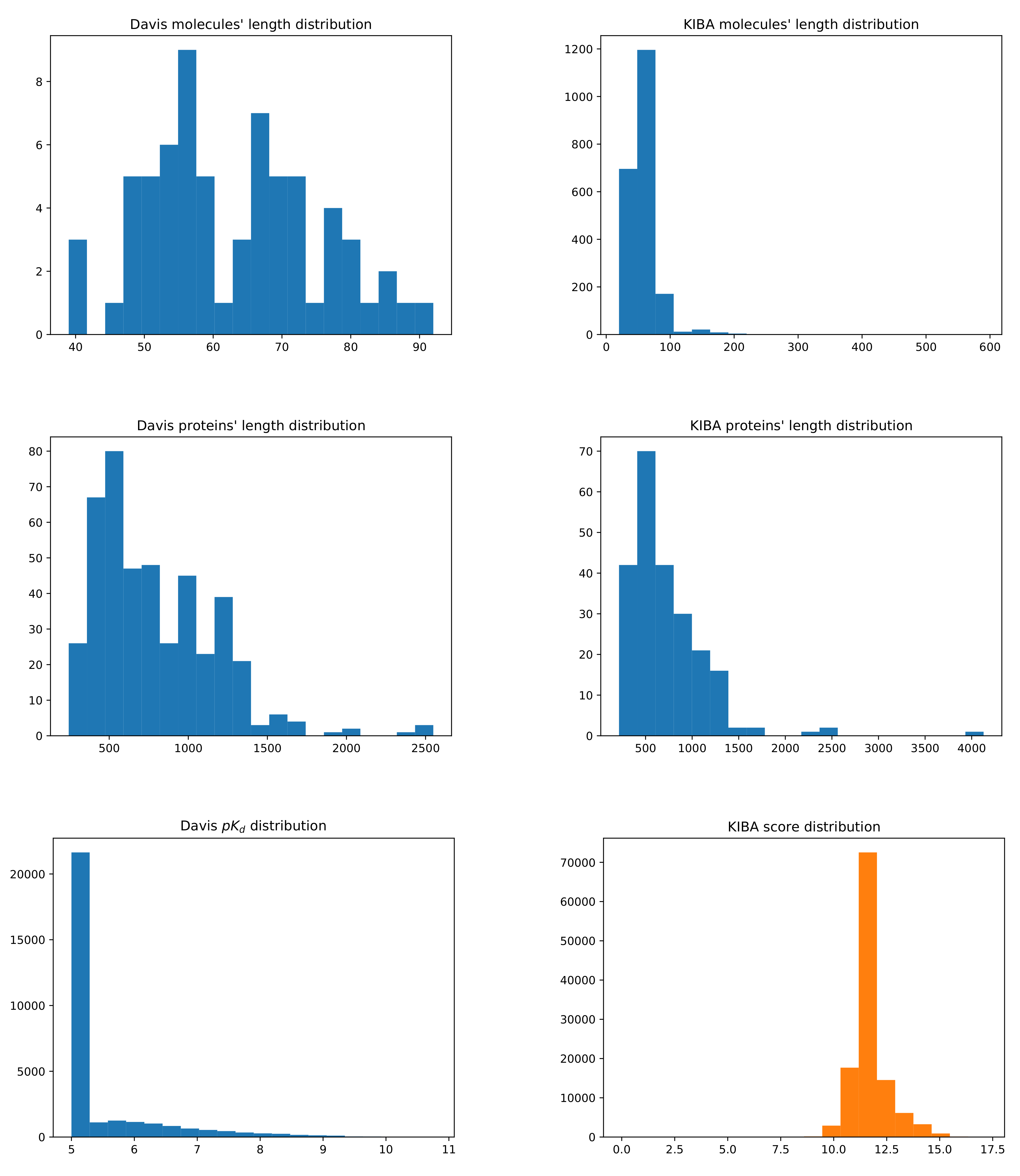
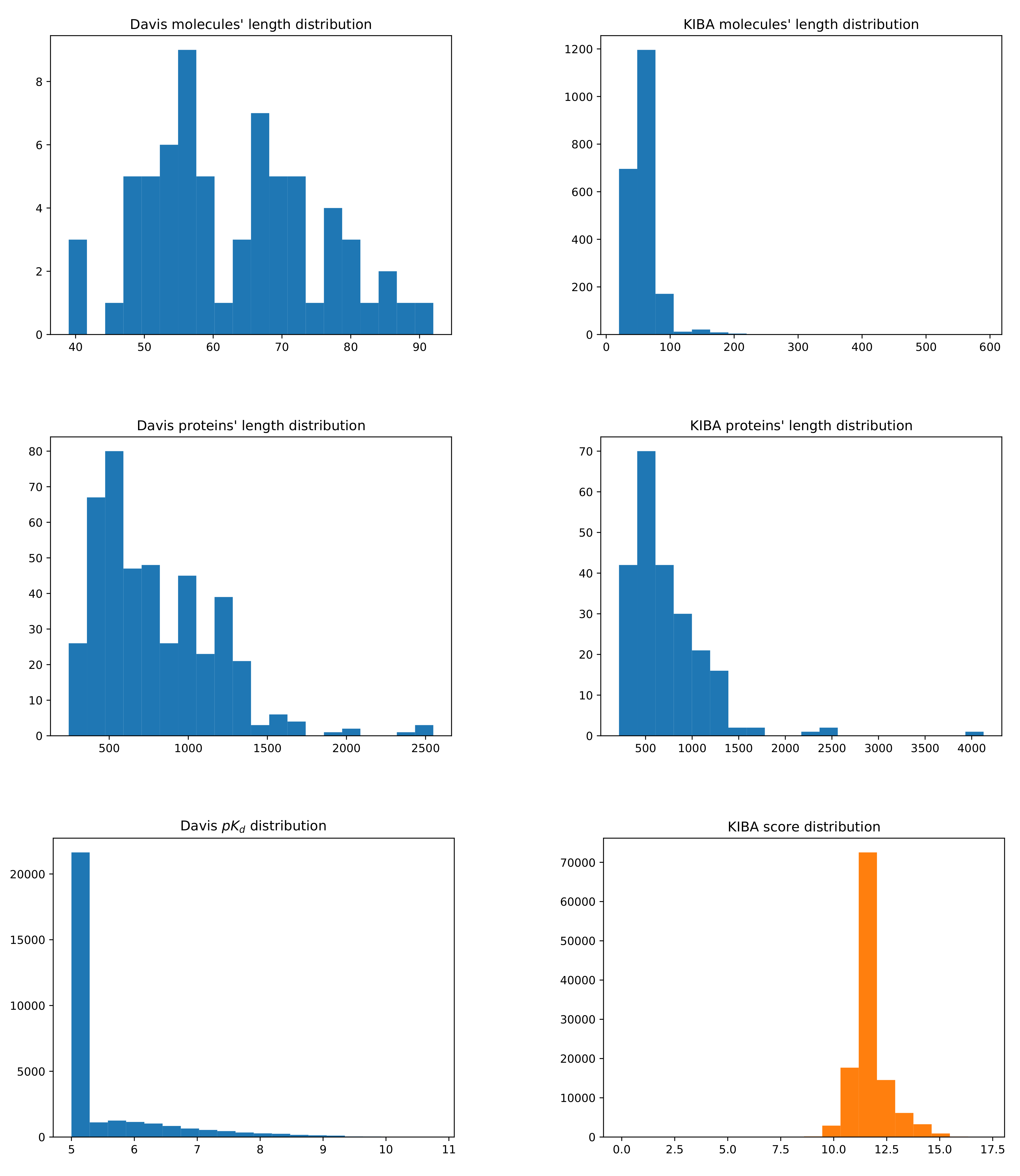
3.1. Datasets
3.2. Evaluation Metrics
3.3. Baselines
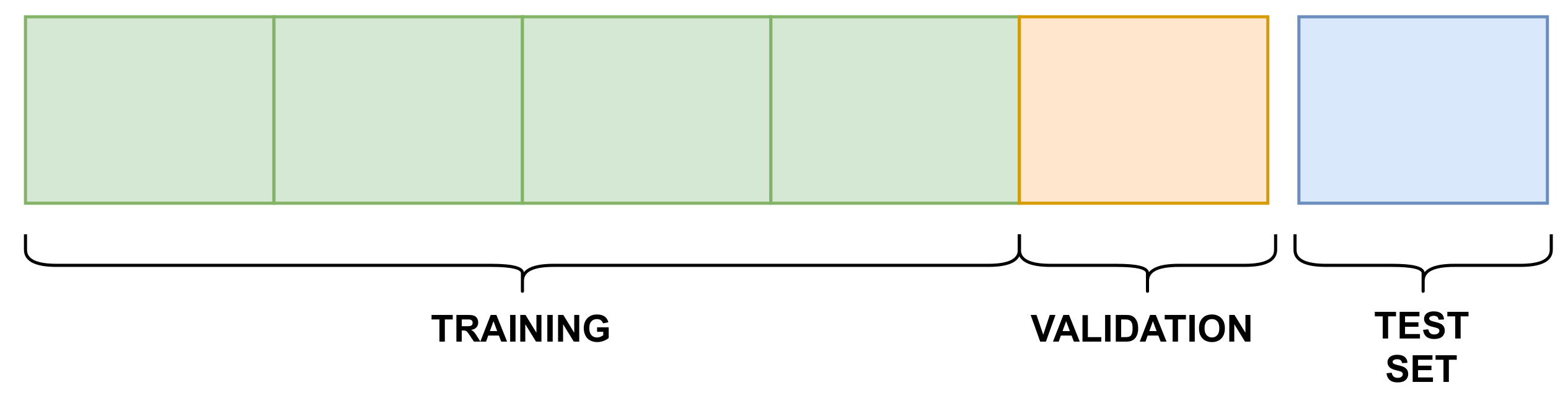
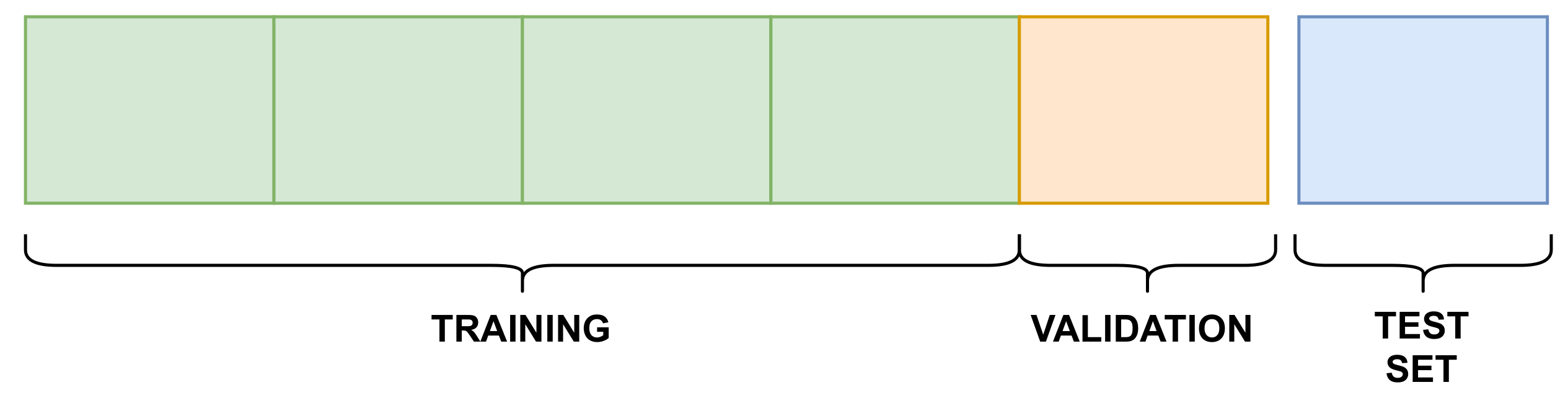
3.4. Training Details
- Training–validation phase: this phase used the training and validation splits, which resulted in a trained model, stored to be used in the next phase;
- Testing phase: the trained model was tested against the testing split, generating the results for each evaluation metric.
4. Results and Discussions
- MPS2IT-DTI employs an approach that represents molecule and protein sequences as two images that are fed into two CNNs;
- MT-DTI employs a different approach for molecule and protein sequences: the molecule representation is based on the BERT model, with a multi-layer transformation; the protein representation follows the same approach as did DeepDTA and WideDTA [15].
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anusuya, S.; Kesherwani, M.; Priya, K.V.; Vimala, A.; Shanmugam, G.; Velmurugan, D.; Gromiha, M.M. Drug-Target Interactions: Prediction Methods and Applications. Curr. Protein Pept. Sci. 2018, 19, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Translational research: 4 ways to fix the clinical trial. Nature 2011, 477, 526–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wu, Z. A Machine Learning-Based Biological Drug-Target Interaction Prediction Method for a Tripartite Heterogeneous Network. ACS Omega 2021, 6, 3037–3045. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Hao, M.; Takeda, T.; Bryant, S.H.; Wang, Y. Large-Scale Prediction of Drug-Target Interaction: A Data-Centric Review. AAPS J. 2017, 19, 1264–1275. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Strittmatter, S.M. Overcoming Drug Development Bottlenecks With Repurposing: Old drugs learn new tricks. Nat. Med. 2014, 20, 590–591. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Swamidass, S.J. Mining small-molecule screens to repurpose drugs. Brief. Bioinform. 2011, 12, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Moriaud, F.; Richard, S.B.; Adcock, S.A.; Chanas-Martin, L.; Surgand, J.S.; Ben Jelloul, M.; Delfaud, F. Identify drug repurposing candidates by mining the Protein Data Bank. Brief. Bioinform. 2011, 12, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Elkouzi, A.; Vedam-Mai, V.; Eisinger, R.S.; Okun, M.S. Emerging therapies in Parkinson disease—Repurposed drugs and new approaches. Nat. Rev. Neurol. 2019, 15, 204–223. [Google Scholar] [CrossRef]
- Gelosa, P.; Castiglioni, L.; Camera, M.; Sironi, L. Drug repurposing in cardiovascular diseases: Opportunity or hopeless dream? Biochem. Pharmacol. 2020, 177, 113894. [Google Scholar] [CrossRef] [PubMed]
- Nabirotchkin, S.; Peluffo, A.E.; Rinaudo, P.; Yu, J.; Hajj, R.; Cohen, D. Next-generation drug repurposing using human genetics and network biology. Curr. Opin. Pharmacol. 2020, 51, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, K.; Gupta, M.K. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019, 93, 103159. [Google Scholar] [CrossRef] [PubMed]
- Pliakos, K.; Vens, C. Drug-target interaction prediction with tree-ensemble learning and output space reconstruction. BMC Bioinform. 2020, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Park, S.; Kang, K.; Ho, J.C. Self-Attention Based Molecule Representation for Predicting Drug-Target Interaction. In Proceedings of the Machine Learning for Healthcare Conference, MLHC 2019. Ann Arbor, MI, USA, 9–10 August 2019; PMLR 2019. Volume 106, pp. 230–248. [Google Scholar]
- Wang, L.; You, Z.H.; Chen, X.; Xia, S.X.; Liu, F.; Yan, X.; Zhou, Y.; Song, K.J. A Computational-Based Method for Predicting Drug–Target Interactions by Using Stacked Autoencoder Deep Neural Network. J. Comput. Biol. 2018, 25, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790. [Google Scholar] [CrossRef]
- Nguyen, T.; Le, H.; Quinn, T.P.; Nguyen, T.; Le, T.D.; Venkatesh, S. GraphDTA: Predicting drug–target binding affinity with graph neural networks. Bioinformatics 2020, 37, 1140–1147. [Google Scholar] [CrossRef]
- Wu, Z.; Li, W.; Liu, G.; Tang, Y. Network-Based Methods for Prediction of Drug-Target Interactions. Front. Pharmacol. 2018, 9, 1134. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Mattes, W.; Mendrick, D.L.; Hong, H. Molecular Docking for Identification of Potential Targets for Drug Repurposing. Curr. Top. Med. Chem. 2016, 16, 3636–3645. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Takigawa, I.; Mamitsuka, H.; Zhu, S. Similarity-based machine learning methods for predicting drug–target interactions: A brief review. Brief. Bioinform. 2013, 15, 734–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahikkala, T.; Airola, A.; Pietilä, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward more realistic drug–target interaction predictions. Brief. Bioinform. 2014, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug-target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Dueva, E.; Cherkasov, A.; Ester, M. PADME: A Deep Learning-based Framework for Drug-Target Interaction Prediction. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug–target interaction: A survey paper. Brief. Bioinform. 2020, 22, 247–269. [Google Scholar] [CrossRef] [Green Version]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), Minneapolis, MN, USA, 2–7 June 2019; Association for Computational Linguistics: Minneapolis, MN, USA, 2019; pp. 4171–4186. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. SMILES-BERT: Large Scale Unsupervised Pre-Training for Molecular Property Prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; Association for Computing Machinery: New York, NY, USA, 2019. BCB ’19. pp. 429–436. [Google Scholar] [CrossRef]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2vec: Distributed representation of genes based on co-expression. BMC Genom. 2019, 20, 82. [Google Scholar] [CrossRef] [Green Version]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 8, 1735–1780. [Google Scholar] [CrossRef]
- Guo, Z.; Yu, W.; Zhang, C.; Jiang, M.; Chawla, N.V. GraSeq: Graph and Sequence Fusion Learning for Molecular Property Prediction. In Proceedings of the 29th ACM International Conference on Information & Knowledge Management, Virtual Event, Ireland, 19–23 October 2020; Association for Computing Machinery: New York, NY, USA, 2020. CIKM ’20. pp. 435–443. [Google Scholar] [CrossRef]
- Ozturk, H.; Ozkirimli, E.; Ozgur, A. WideDTA: Prediction of drug-target binding affinity. arXiv 2019, arXiv:1902.04166. [Google Scholar]
- Kwon, S.; Yoon, S. DeepCCI: End-to-End Deep Learning for Chemical-Chemical Interaction Prediction. In Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20–23 August 2017; Association for Computing Machinery: New York, NY, USA, 2017. ACM-BCB ’17. pp. 203–212. [Google Scholar] [CrossRef]
- Li, J.; Pu, Y.; Tang, J.; Zou, Q.; Guo, F. DeepAVP: A Dual-Channel Deep Neural Network for Identifying Variable-Length Antiviral Peptides. IEEE J. Biomed. Health Inform. 2020, 24, 3012–3019. [Google Scholar] [CrossRef] [PubMed]
- Bung, N.; Krishnan, S.R.; Bulusu, G.; Roy, A. De Novo Design of New Chemical Entities (NCEs) for SARS-CoV-2 Using Artificial Intelligence. Future Med. Chem. 2020, 13. [Google Scholar] [CrossRef]
- Coutinho, M.G.F.; Câmara, G.B.M.; de Melo Barbosa, R.; Fernandes, M.A.C. Deep learning based on stacked sparse autoencoder applied to viral genome classification of SARS-CoV-2 virus. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhou, Y. A Review of Text Classification Based on Deep Learning. In Proceedings of the 2020 3rd International Conference on Geoinformatics and Data Analysis, Marseille, France, 15–17 April 2020; Association for Computing Machinery: New York, NY, USA, 2020. ICGDA 2020. pp. 132–136. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Melsted, P.; Pritchard, J.K. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinform. 2011, 12, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizk, G.; Lavenier, D.; Chikhi, R. DSK: K-mer counting with very low memory usage. Bioinformatics 2013, 29, 652–653. [Google Scholar] [CrossRef]
- Sims, G.E.; Jun, S.R.; Wu, G.A.; Kim, S.H. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc. Natl. Acad. Sci. USA 2009, 106, 2677–2682. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making Sense of Large-Scale Kinase Inhibitor Bioactivity Data Sets: A Comparative and Integrative Analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Gönen, M.; Heller, G. Concordance Probability and Discriminatory Power in Proportional Hazards Regression. Biometrika 2005, 92, 965–970. [Google Scholar] [CrossRef]
- Davis, J.; Goadrich, M. The Relationship between Precision-Recall and ROC Curves. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; Association for Computing Machinery: New York, NY, USA, 2006. ICML ’06. pp. 233–240. [Google Scholar] [CrossRef] [Green Version]
- Boyd, K.; Eng, K.H.; Page, C.D. Area under the Precision-Recall Curve: Point Estimates and Confidence Intervals. In Machine Learning and Knowledge Discovery in Databases; Blockeel, H., Kersting, K., Nijssen, S., Železný, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 451–466. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2017, arXiv:1412.6980. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. Tensorflow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://github.com/fchollet/keras (accessed on 7 February 2022).
- Schwartz, R.; Dodge, J.; Smith, N.A.; Etzioni, O. Green AI. Commun. ACM 2020, 63, 54–63. [Google Scholar] [CrossRef]
- Sharir, O.; Peleg, B.; Shoham, Y. The Cost of Training NLP Models: A Concise Overview. arXiv 2020, arXiv:2004.08900. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | No. Drugs | No. Targets | No. Interactions |
|---|---|---|---|
| Davis | 68 | 442 | 30,056 |
| Kiba | 2111 | 229 | 118,254 |
| Dataset | Training | Validation | Testing |
|---|---|---|---|
| Davis | 20,037 | 5009 | 5010 |
| KIBA | 78,836 | 19,709 | 19,709 |
| Dataset | CI (std) | MSE | (std) | AUC-PR (std) | ACC (std) |
|---|---|---|---|---|---|
| Davis | 0.876 (0.002) | 0.276 | 0.637 (0.011) | 0.450 (0.029) | 0.944 (0.002) |
| KIBA | 0.836 (0.003) | 0.226 | 0.614 (0.011) | 0.601 (0.006) | 0.881 (0.002) |
| Dataset | Method | CI (std) | MSE | (std) | AUC-PR (std) |
|---|---|---|---|---|---|
| Davis | KronRLS | 0.871 (0.001) | 0.379 | 0.407 (0.005) | 0.661 (0.010) |
| SimBoost | 0.872 (0.002) | 0.282 | 0.644 (0.006) | 0.709 (0.008) | |
| MPS2IT-DTI | 0.876 (0.002) | 0.276 | 0.637 (0.011) | 0.450 (0.018) | |
| DeepDTA | 0.878 (0.004) | 0.261 | 0.630 (0.017) | 0.714 (0.010) | |
| WideDTA | 0.886 (0.003) | 0.262 | – | – | |
| MT-DTI | 0.887 (0.003) | 0.245 | 0.665 (0.014) | 0.730 (0.014) | |
| Kiba | KronRLS | 0.782 (0.001) | 0.411 | 0.342 (0.001) | 0.635 (0.004) |
| MPS2IT-DTI | 0.836 (0.003) | 0.226 | 0.614 (0.011) | 0.601 (0.006) | |
| SimBoost | 0.836 (0.001) | 0.222 | 0.629 (0.007) | 0.760 (0.003) | |
| DeepDTA | 0.863 (0.002) | 0.194 | 0.673 (0.009) | 0.788 (0.004) | |
| WideDTA | 0.875 (0.001) | 0.179 | – | – | |
| MT-DTI | 0.882 (0.001) | 0.152 | 0.738 (0.006) | 0.837 (0.003) |
| Inputs | MPS2IT-DTI | DeepDTA | MT-DTI | WideDTA |
|---|---|---|---|---|
| Num. of inputs | 2 | 2 | 2 | 4 |
| Molecule SMILES | • | • | • | • |
| Ligand Max. Common Structure | ◦ | ◦ | ◦ | • |
| Protein Sequence | • | • | • | • |
| Protein Motifs and Domains | ◦ | ◦ | ◦ | • |
| Approach | MPS2IT-DTI | DeepDTA | MT-DTI | WideDTA | ||||
|---|---|---|---|---|---|---|---|---|
| M | P | M | P | M | P | M | P | |
| Mers-based frequency | • | • | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ |
| Frequency to image | • | • | ◦ | ◦ | ◦ | ◦ | ◦ | ◦ |
| One-hot encoding | ◦ | ◦ | • | • | ◦ | • | • | • |
| Embedding layer | ◦ | ◦ | • | • | • | • | • | • |
| Self-attention layer | ◦ | ◦ | ◦ | ◦ | • | ◦ | ◦ | ◦ |
| Feed-forward layer | ◦ | ◦ | ◦ | ◦ | • | ◦ | ◦ | ◦ |
| Pre-training | ◦ | ◦ | ◦ | ◦ | • | ◦ | ◦ | ◦ |
| Fine tunning | ◦ | ◦ | ◦ | ◦ | • | ◦ | ◦ | ◦ |
| CNN | • | • | • | • | ◦ | • | • | • |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, J.G.; Fernandes, M.A.C.; de Melo Barbosa, R. A Novel Deep Neural Network Technique for Drug–Target Interaction. Pharmaceutics 2022, 14, 625. https://doi.org/10.3390/pharmaceutics14030625
de Souza JG, Fernandes MAC, de Melo Barbosa R. A Novel Deep Neural Network Technique for Drug–Target Interaction. Pharmaceutics. 2022; 14(3):625. https://doi.org/10.3390/pharmaceutics14030625
Chicago/Turabian Stylede Souza, Jackson G., Marcelo A. C. Fernandes, and Raquel de Melo Barbosa. 2022. "A Novel Deep Neural Network Technique for Drug–Target Interaction" Pharmaceutics 14, no. 3: 625. https://doi.org/10.3390/pharmaceutics14030625
APA Stylede Souza, J. G., Fernandes, M. A. C., & de Melo Barbosa, R. (2022). A Novel Deep Neural Network Technique for Drug–Target Interaction. Pharmaceutics, 14(3), 625. https://doi.org/10.3390/pharmaceutics14030625