
An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors




Abstract
1. Introduction
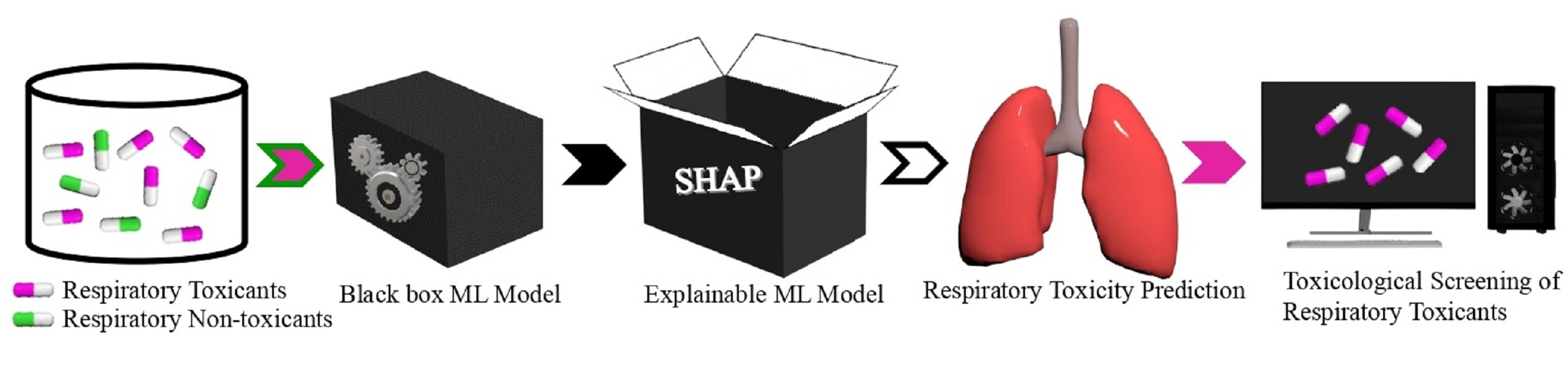
2. Materials and Methods
2.1. Dataset Preparation
2.2. Computation of Molecular Descriptors
2.3. Data Preprocessing and Feature Selection Methods
2.4. Model Development and Optimization
2.5. Model Validation and Evaluation Measures
2.6. Model Explainability
3. Results and Discussion
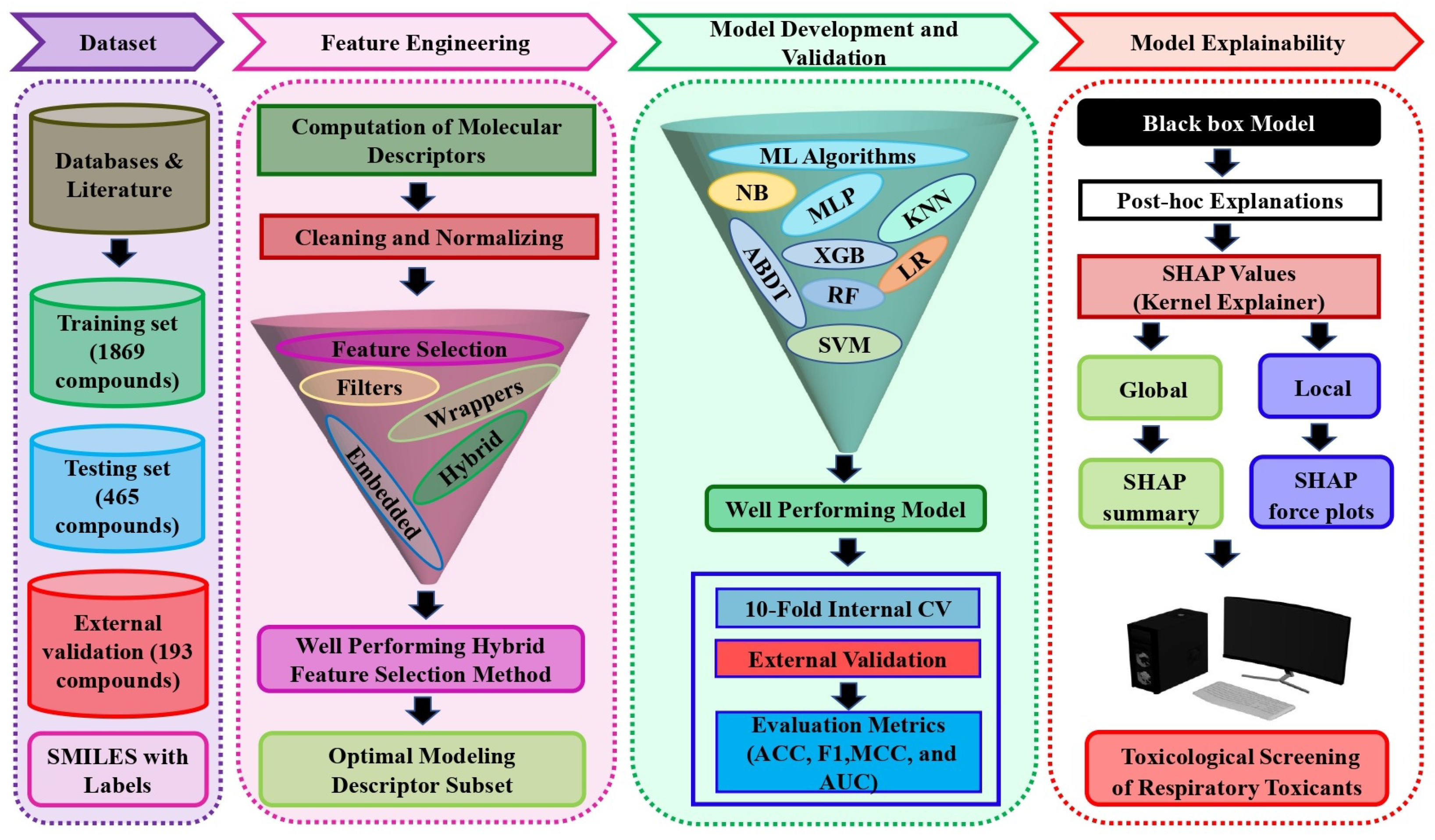
3.1. Data Distribution and Chemical Structural Diversity
3.2. Selection of Important Chemical Descriptors
3.2.1. Single Feature Selection Methods
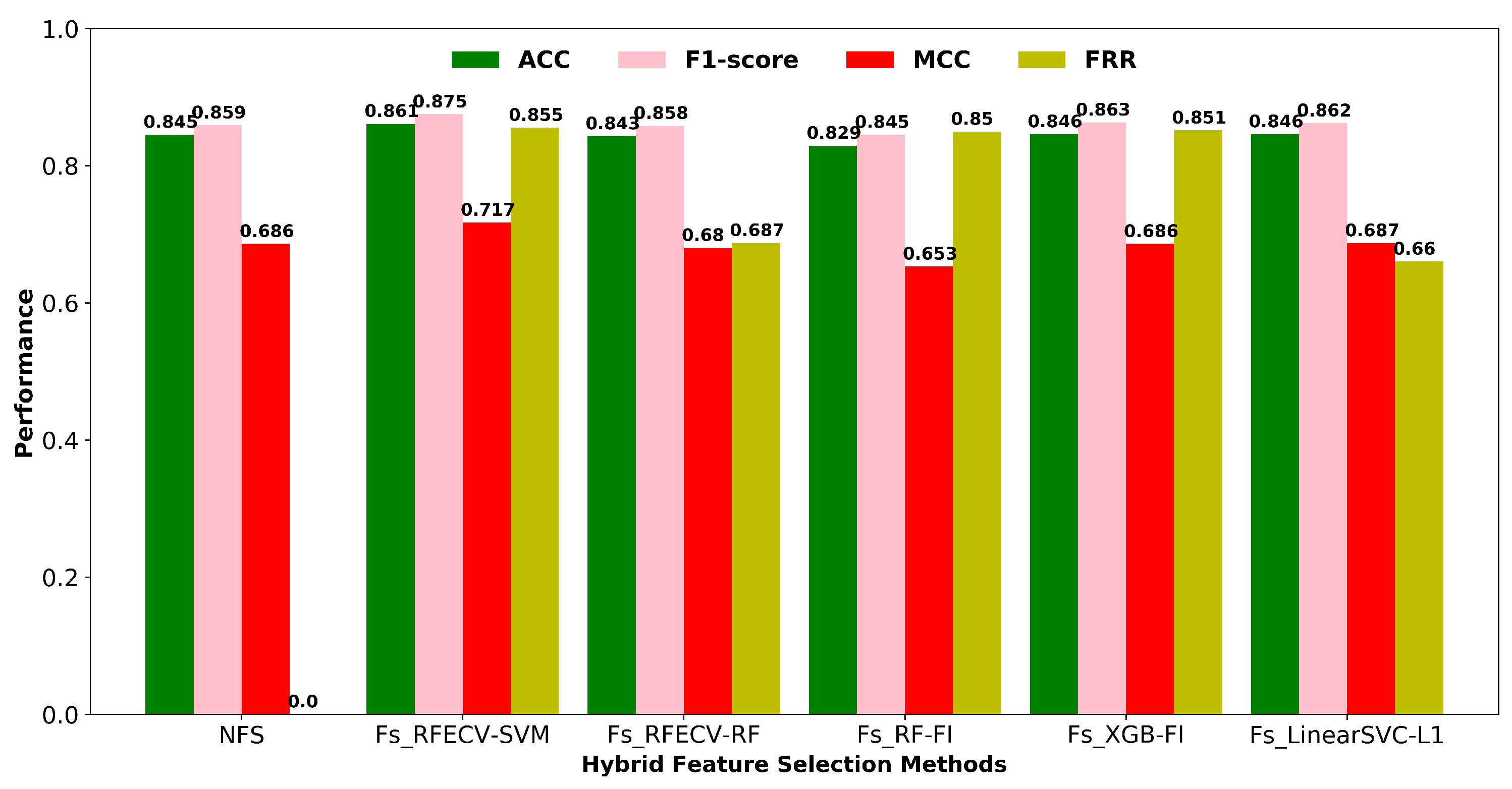
3.2.2. Hybrid Feature Selection Methods
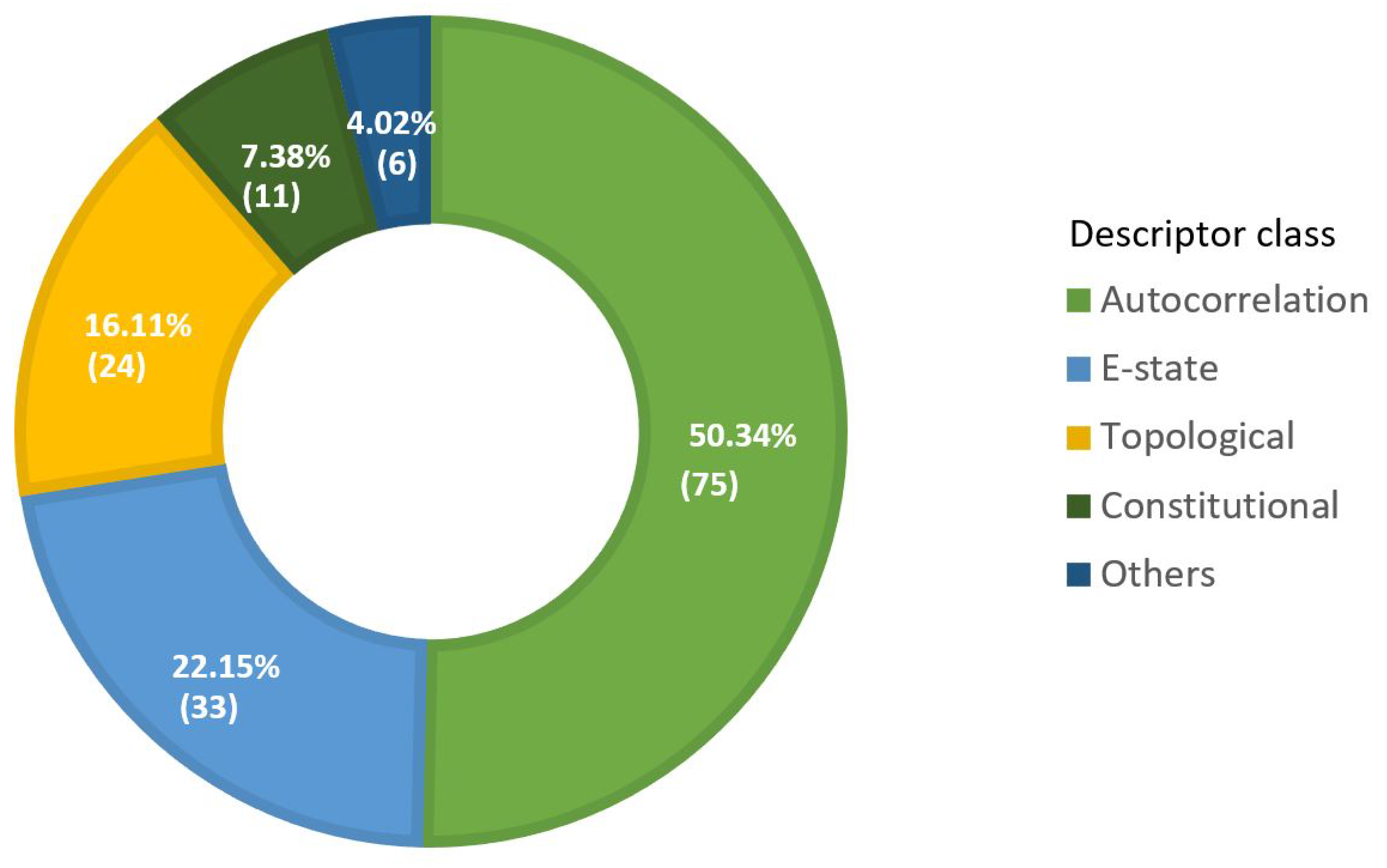
3.2.3. Selected Descriptors Analysis
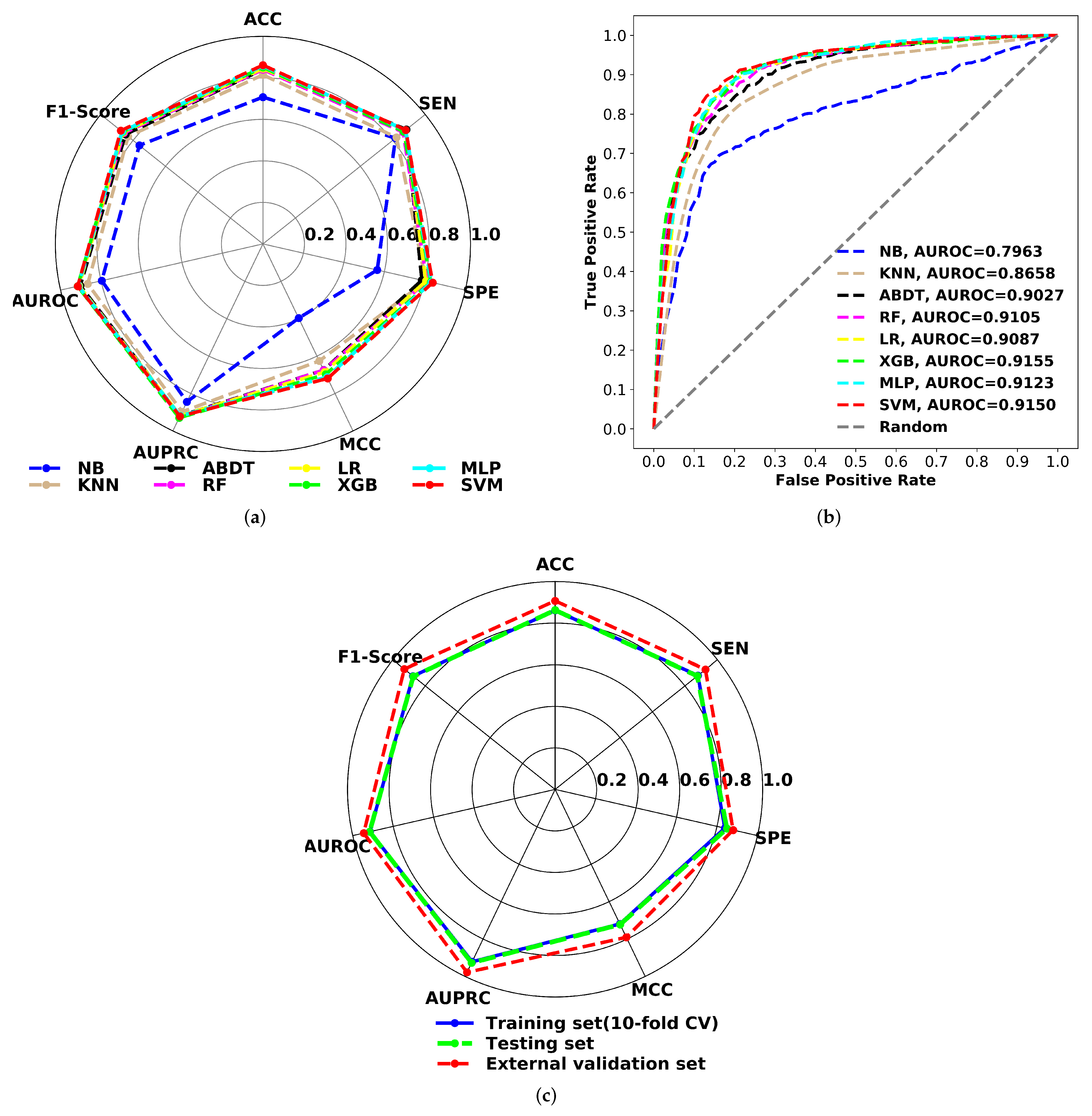
3.3. Prediction Performance of the Classification Models
3.4. Model Explainability
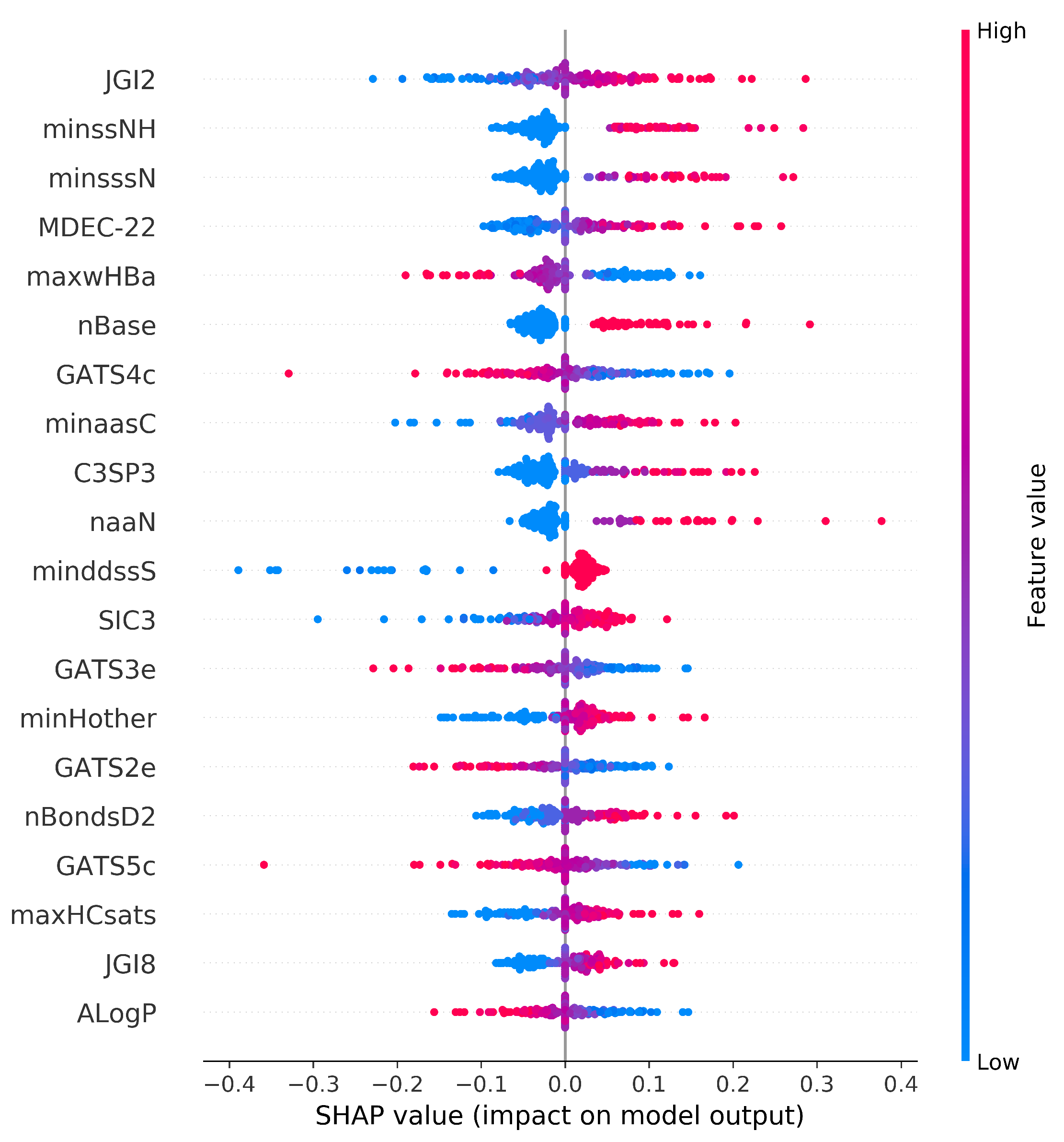
3.4.1. Global Feature Explanation
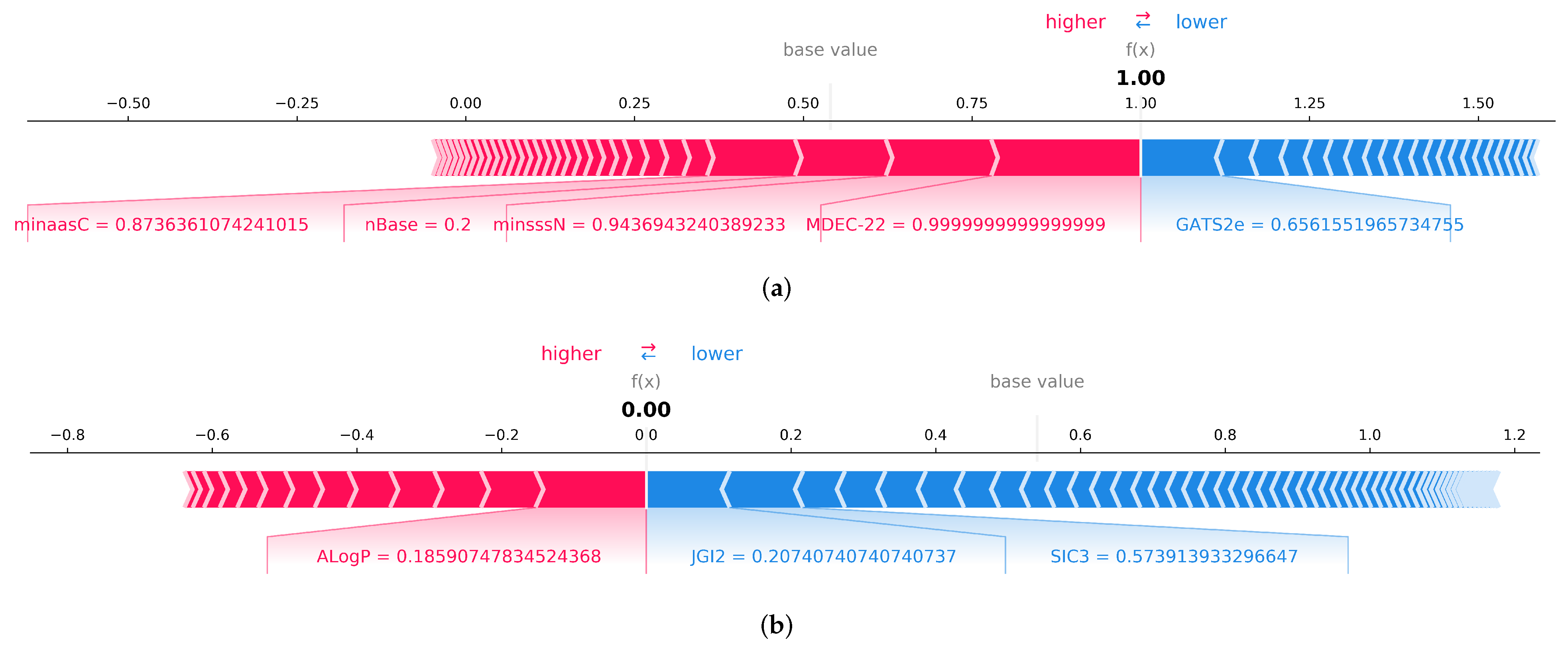
3.4.2. Local Feature Explanation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vo, A.H.; Van Vleet, T.R.; Gupta, R.R.; Liguori, M.J.; Rao, M.S. An overview of machine learning and big data for drug toxicity evaluation. Chem. Res. Toxicol. 2019, 33, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial intelligence for drug toxicity and safety. Trends Pharmacol. Sci. 2019, 40, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.C.; Xu, Q.; Pan, Y.J.; Pan, W.; Ji, N.; Li, Y.B.; Jin, H.J.; Liu, K.; Ji, Z.L. ADReCS: An ontology database for aiding standardization and hierarchical classification of adverse drug reaction terms. Nucleic Acids Res. 2015, 43, D907–D913. [Google Scholar] [CrossRef] [PubMed]
- Siramshetty, V.B.; Nickel, J.; Omieczynski, C.; Gohlke, B.O.; Drwal, M.N.; Preissner, R. WITHDRAWN—A resource for withdrawn and discontinued drugs. Nucleic Acids Res. 2016, 44, D1080–D1086. [Google Scholar] [CrossRef]
- Valverde-Monge, M.; Fernández-Nieto, M.; López, V.B.; Rodrigo-Muñoz, J.M.; Cañas, J.A.; Sastre, B.; Del Potro, M.G.; De las Heras, M.; Del Pozo, V.; Sastre, J. Novel causes of drug-induced occupational asthma. J. Allergy Clin. Immunol. Pract. 2019, 7, 740–742. [Google Scholar] [CrossRef]
- Skeoch, S.; Weatherley, N.; Swift, A.J.; Oldroyd, A.; Johns, C.; Hayton, C.; Giollo, A.; Wild, J.M.; Waterton, J.C.; Buch, M.; et al. Drug-induced interstitial lung disease: A systematic review. J. Clin. Med. 2018, 7, 356. [Google Scholar] [CrossRef]
- Bartal, C.; Sagy, I.; Barski, L. Drug-induced eosinophilic pneumonia: A review of 196 case reports. Medicine 2018, 97, e9688. [Google Scholar] [CrossRef]
- Varghese, M.; Glaum, M.; Lockey, R. Drug-induced rhinitis. Clin. Exp. Allergy 2010, 40, 381–384. [Google Scholar] [CrossRef]
- Schwaiblmair, M.; Behr, W.; Haeckel, T.; Märkl, B.; Foerg, W.; Berghaus, T. Drug induced interstitial lung disease. Open Respir. Med. J. 2012, 6, 63. [Google Scholar] [CrossRef]
- Chary, A.; Hennen, J.; Klein, S.G.; Serchi, T.; Gutleb, A.C.; Blömeke, B. Respiratory sensitization: Toxicological point of view on the available assays. Arch. Toxicol. 2018, 92, 803–822. [Google Scholar] [CrossRef]
- Matsuno, O. Drug-induced interstitial lung disease: Mechanisms and best diagnostic approaches. Respir. Res. 2012, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.D., Jr.; White, D.A.; Matthay, R.A. Drug-induced pulmonary disease: Part 1: Cytotoxic drugs. Am. Rev. Respir. Dis. 1986, 133, 321–340. [Google Scholar] [PubMed]
- Rossi, S.E.; Erasmus, J.J.; McAdams, H.P.; Sporn, T.A.; Goodman, P.C. Pulmonary drug toxicity: Radiologic and pathologic manifestations. Radiographics 2000, 20, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Baldotto, C.S.d.R.; Nunes, F.A.P.; Scheliga, A.A.d.S. Bleomycin-induced lung injury. J. Cancer Res. 2013, 2013, 480608. [Google Scholar] [CrossRef]
- De Jonge, M.E.; Huitema, A.D.; Rodenhuis, S.; Beijnen, J.H. Clinical pharmacokinetics of cyclophosphamide. Clin. Pharmacokinet. 2005, 44, 1135–1164. [Google Scholar] [CrossRef] [PubMed]
- Madani, Y.; Mann, B. Nitrofurantoin-induced lung disease and prophylaxis of urinary tract infections. Prim. Care Respir. J. 2012, 21, 337–341. [Google Scholar] [CrossRef]
- Casey, W.; Jacobs, A.; Maull, E.; Matheson, J.; Clarke, C.; Lowit, A. A new path forward: The interagency coordinating committee on the validation of alternative methods (ICCVAM) and national toxicology program’s interagency center for the evaluation of alternative toxicological methods (NICEATM). J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 170–173. [Google Scholar]
- Rácz, A.; Bajusz, D.; Miranda-Quintana, R.A.; Héberger, K. Machine learning models for classification tasks related to drug safety. Mol. Divers. 2021, 25, 1409–1424. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Li, W.; Liu, G.; Tang, Y. In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front. Chem. 2018, 6, 30. [Google Scholar] [CrossRef]
- Hua, Y.; Shi, Y.; Cui, X.; Li, X. In silico prediction of chemical-induced hematotoxicity with machine learning and deep learning methods. Mol. Divers. 2021, 25, 1585–1596. [Google Scholar] [CrossRef]
- Jiang, C.; Yang, H.; Di, P.; Li, W.; Tang, Y.; Liu, G. In silico prediction of chemical reproductive toxicity using machine learning. J. Appl. Toxicol. 2019, 39, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Tayara, H.; Chong, K.T. Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets. Int. J. Mol. Sci. 2021, 22, 8073. [Google Scholar] [CrossRef]
- Mekenyan, O.; Patlewicz, G.; Kuseva, C.; Popova, I.; Mehmed, A.; Kotov, S.; Zhechev, T.; Pavlov, T.; Temelkov, S.; Roberts, D.W. A mechanistic approach to modeling respiratory sensitization. Chem. Res. Toxicol. 2014, 27, 219–239. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, J.; Seed, M.; Stocks, S.; Agius, R. A refined QSAR model for prediction of chemical asthma hazard. Occup. Med. 2015, 65, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Seed, M.J.; Agius, R.M. Progress with Structure–Activity Relationship modelling of occupational chemical respiratory sensitizers. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Chen, F.; Liu, H.; Sun, H.; Kang, Y.; Li, D.; Li, Y.; Hou, T. ADMET evaluation in drug discovery. Part 17: Development of quantitative and qualitative prediction models for chemical-induced respiratory toxicity. Mol. Pharm. 2017, 14, 2407–2421. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, J.X.; Liu, C.T.; Ren, J.X.; Ding, L. Development and evaluation of in silico prediction model for drug-induced respiratory toxicity by using naïve Bayes classifier method. Food Chem. Toxicol. 2018, 121, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, P.; Zhang, X.; Xu, X.; Li, W.; Liu, G.; Tang, Y. In silico prediction of chemical respiratory toxicity via machine learning. Comput. Toxicol. 2021, 18, 100155. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Bannigan, P.; Aldeghi, M.; Bao, Z.; Häse, F.; Aspuru-Guzik, A.; Allen, C. Machine learning directed drug formulation development. Adv. Drug Deliv. Rev. 2021, 175, 113806. [Google Scholar] [CrossRef]
- Rehman, M.U.; Tayara, H.; Chong, K.T. DCNN-4mC: Densely connected neural network based N4-methylcytosine site prediction in multiple species. Comput. Struct. Biotechnol. J. 2021, 19, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Angelov, P.P.; Soares, E.A.; Jiang, R.; Arnold, N.I.; Atkinson, P.M. Explainable artificial intelligence: An analytical review. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2021, 11, e1424. [Google Scholar] [CrossRef]
- Rehman, M.U.; Akhtar, S.; Zakwan, M.; Mahmood, M.H. Novel architecture with selected feature vector for effective classification of mitotic and non-mitotic cells in breast cancer histology images. Biomed. Signal Process. Control 2022, 71, 103212. [Google Scholar] [CrossRef]
- Štrumbelj, E.; Kononenko, I. Explaining prediction models and individual predictions with feature contributions. Knowl. Inf. Syst. 2014, 41, 647–665. [Google Scholar] [CrossRef]
- Tjoa, E.; Guan, C. A survey on explainable artificial intelligence (xai): Toward medical xai. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4793–4813. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.I. A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 4768–4777. [Google Scholar]
- Rodríguez-Pérez, R.; Bajorath, J. Interpretation of machine learning models using shapley values: Application to compound potency and multi-target activity predictions. J. Comput.-Aided Mol. Des. 2020, 34, 1013–1026. [Google Scholar] [CrossRef]
- PNEUMOTOX. Available online: https://www.pneumotox.com/drug/index/ (accessed on 20 January 2022).
- Hazardous Chemical Information System. Available online: http://hcis.safeworkaustralia.gov.au/ (accessed on 20 January 2022).
- Dik, S.; Ezendam, J.; Cunningham, A.R.; Carrasquer, C.A.; van Loveren, H.; Rorije, E. Evaluation of in silico models for the identification of respiratory sensitizers. Toxicol. Sci. 2014, 142, 385–394. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Juan-Blanco, T.; Duran-Frigola, M.; Aloy, P. IntSide: A web server for the chemical and biological examination of drug side effects. Bioinformatics 2015, 31, 612–613. [Google Scholar] [CrossRef]
- Alves, V.M.; Capuzzi, S.J.; Braga, R.C.; Borba, J.V.; Silva, A.C.; Luechtefeld, T.; Hartung, T.; Andrade, C.H.; Muratov, E.N.; Tropsha, A. A perspective and a new integrated computational strategy for skin sensitization assessment. ACS Sustain. Chem. Eng. 2018, 6, 2845–2859. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 41. [Google Scholar]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Elisseeff, A. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar]
- Tax, D.; Duin, R. Feature scaling in support vector data descriptions. Learn. Imbalanced Datasets 2000, 25–30. [Google Scholar]
- Bollegala, D. Dynamic feature scaling for online learning of binary classifiers. Knowl.-Based Syst. 2017, 129, 97–105. [Google Scholar] [CrossRef][Green Version]
- Bommert, A.; Sun, X.; Bischl, B.; Rahnenführer, J.; Lang, M. Benchmark for filter methods for feature selection in high-dimensional classification data. Comput. Stat. Data Anal. 2020, 143, 106839. [Google Scholar] [CrossRef]
- Bolón-Canedo, V.; Sánchez-Marono, N.; Alonso-Betanzos, A.; Benítez, J.M.; Herrera, F. A review of microarray datasets and applied feature selection methods. Inf. Sci. 2014, 282, 111–135. [Google Scholar] [CrossRef]
- Bolboaca, S.D.; Jäntschi, L. Pearson versus Spearman, Kendall’s tau correlation analysis on structure-activity relationships of biologic active compounds. Leonardo J. Sci. 2006, 5, 179–200. [Google Scholar]
- Khanal, J.; Lim, D.Y.; Tayara, H.; Chong, K.T. i6ma-stack: A stacking ensemble-based computational prediction of dna n6-methyladenine (6ma) sites in the rosaceae genome. Genomics 2021, 113, 582–592. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Gao, Y. A hybrid short-term electricity price forecasting framework: Cuckoo search-based feature selection with singular spectrum analysis and SVM. Energy Econ. 2019, 81, 899–913. [Google Scholar] [CrossRef]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual Workshop on Computational Learning Theory, Pittsburgh, PA, USA, 27–29 July 1992; pp. 144–152. [Google Scholar]
- Murtagh, F. Multilayer perceptrons for classification and regression. Neurocomputing 1991, 2, 183–197. [Google Scholar] [CrossRef]
- Chen, T.; He, T.; Benesty, M.; Khotilovich, V.; Tang, Y.; Cho, H.; Chen, K. Xgboost: Extreme Gradient Boosting; R Package Version 0.4-2; 2015; pp. 1–4. Available online: https://cran.r-project.org/web/packages/xgboost/index.html (accessed on 20 January 2022).
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kleinbaum, D.G.; Dietz, K.; Gail, M.; Klein, M.; Klein, M. Logistic Regression; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Freund, Y.; Mason, L. The alternating decision tree learning algorithm. In Proceedings of the Sixteenth International Conference on Machine Learning (ICML 1999), Bled, Slovenia, 27–30 June 1999; Volume 99, pp. 124–133. [Google Scholar]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Rish, I. An empirical study of the naive Bayes classifier. In Proceedings of the IJCAI 2001 Workshop on Empirical Methods in Artificial Intelligence, Seattle, WA, USA, 4 August 2001; Volume 3, pp. 41–46. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Brownlee, J. XGBoost with Python. In Machine Learning Mastery; 2019; Available online: https://machinelearningmastery.com/xgboost-with-python/ (accessed on 20 January 2022).
- Czermiński, R.; Yasri, A.; Hartsough, D. Use of support vector machine in pattern classification: Application to QSAR studies. Quant. Struct.-Act. Relatsh. 2001, 20, 227–240. [Google Scholar] [CrossRef]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef]
- Arrieta, A.B.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; García, S.; Gil-López, S.; Molina, D.; Benjamins, R.; et al. Explainable Artificial Intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef]
- Shapley, L.S. A value for n-person games. In Contributions to the Theory of Games, 2; Princeton University Press: Princeton, NJ, USA, 1953; pp. 307–317. [Google Scholar]
- Hollas, B. An analysis of the autocorrelation descriptor for molecules. J. Math. Chem. 2003, 33, 91–101. [Google Scholar] [CrossRef]
- Broto, P.; Moreau, G.; Vandycke, C. Molecular structures: Perception, autocorrelation descriptor and sar studies: System of atomic contributions for the calculation of the n-octanol/water partition coefficients. Eur. J. Med. Chem. 1984, 19, 71–78. [Google Scholar]
- Hall, L.H.; Kier, L.B. Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Liu, S.; Cao, C.; Li, Z. Approach to estimation and prediction for normal boiling point (NBP) of alkanes based on a novel molecular distance-edge (MDE) vector, λ. J. Chem. Inf. Comput. Sci. 1998, 38, 387–394. [Google Scholar] [CrossRef]
- Galvez, J.; Garcia, R.; Salabert, M.; Soler, R. Charge indexes. New topological descriptors. J. Chem. Inf. Comput. Sci. 1994, 34, 520–525. [Google Scholar] [CrossRef]
- Chicco, D.; Tötsch, N.; Jurman, G. The Matthews correlation coefficient (MCC) is more reliable than balanced accuracy, bookmaker informedness, and markedness in two-class confusion matrix evaluation. BioData Min. 2021, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Abdelbaky, I.; Tayara, H.; Chong, K.T. Prediction of kinase inhibitors binding modes with machine learning and reduced descriptor sets. Sci. Rep. 2021, 11, 706. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Toxicants | Non-Toxicants | Total |
|---|---|---|---|
| Training set | 1043 | 826 | 1869 |
| Test set | 259 | 206 | 465 |
| External validation set | 136 | 57 | 193 |
| Total | 3772 | 1089 | 2527 |
| Descriptor Class | Dimension | Number of Descriptors |
|---|---|---|
| Constitutional descriptors | 1 | 120 |
| Autocorrelation descriptors | 2 | 346 |
| Basak descriptors | 2 | 42 |
| BCUT descriptors | 2 | 6 |
| Burden descriptors | 2 | 96 |
| Connectivity descriptors | 2 | 56 |
| E-state descriptors | 2 | 489 |
| Kappa descriptors | 2 | 3 |
| Molecular property descriptors | 2 | 15 |
| Quantum chemical descriptors | 2 | 6 |
| Topological descriptors | 2 | 265 |
| Descriptor Class | Count | Descriptor Category | Count |
|---|---|---|---|
| Autocorrelation | 75 | Geary Autocorrelation Descriptor | 25 |
| Moran Autocorrelation Descriptor | 13 | ||
| Centered Broto-Moreau Autocorrelation Descriptor | 13 | ||
| Average Broto-Moreau Autocorrelation Descriptor | 12 | ||
| Average centered Broto-Moreau Autocorrelation Descriptor | 12 | ||
| E-state | 33 | Electro topological State Atom Type Descriptor | 30 |
| Detour Matrix Descriptor | 3 | ||
| Topological | 24 | Extended Topo chemical Atom Descriptor | 8 |
| Molecular Distance-Edge Descriptor | 7 | ||
| Topological Charge Descriptor | 6 | ||
| Others | 3 |
| No. | Model Name | No. of Compounds | Test Method | ACC | SPE | SEN | MCC |
|---|---|---|---|---|---|---|---|
| 1 | XGBoost [26] | 468 | Test set | 0.826 | 0.832 | 0.822 | 0.644 |
| 2 | NB-2 [27] | 248 | Test set | 0.844 | 0.853 | 0.835 | 0.684 |
| 3 | PubchemFP-RF [28] | 1869 | 10-fold CV | 0.840 | 0.805 | 0.868 | 0.675 |
| 467 | Test set | 0.859 | 0.825 | 0.885 | 0.713 | ||
| 4 | Proposed Model | 1869 | 10-fold CV | 0.862 | 0.838 | 0.879 | 0.717 |
| 465 | Test set | 0.862 | 0.849 | 0.876 | 0.722 |
| Descriptor Name | Descriptor Class | Description | Impact |
|---|---|---|---|
| JGI2 | Topological descriptors | Mean topological charge index of order 2 | Positive |
| minssNH | E-state descriptors | Minimum atom-type E-State: -NH- | Positive |
| MDEC-22 | Topological descriptors | Molecular distance edge between all secondary carbons | Positive |
| minsssN | E-state descriptors | Minimum atom-type E-State: >N- | Positive |
| nBase | Constitutional descriptors | Number of basic groups. | Positive |
| maxwHBa | E-state descriptors | Maximum E-States for weak Hydrogen Bond acceptors | Negative |
| GATS4c | Autocorrelation descriptors | Geary autocorrelation - lag 4/weighted by charges | Negative |
| minaasC | E-state descriptors | Minimum atom-type E-State: :C:- | Positive |
| C3SP3 | Topological descriptors | Singly bound carbon bound to three other carbons | Positive |
| minddssS | E-state descriptors | Minimum atom-type E-State: >S== | Positive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaganathan, K.; Tayara, H.; Chong, K.T. An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors. Pharmaceutics 2022, 14, 832. https://doi.org/10.3390/pharmaceutics14040832
Jaganathan K, Tayara H, Chong KT. An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors. Pharmaceutics. 2022; 14(4):832. https://doi.org/10.3390/pharmaceutics14040832
Chicago/Turabian StyleJaganathan, Keerthana, Hilal Tayara, and Kil To Chong. 2022. "An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors" Pharmaceutics 14, no. 4: 832. https://doi.org/10.3390/pharmaceutics14040832
APA StyleJaganathan, K., Tayara, H., & Chong, K. T. (2022). An Explainable Supervised Machine Learning Model for Predicting Respiratory Toxicity of Chemicals Using Optimal Molecular Descriptors. Pharmaceutics, 14(4), 832. https://doi.org/10.3390/pharmaceutics14040832