
Acute Inflammation Is a Predisposing Factor for Weight Gain and Insulin Resistance

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Acute Endotoxemia Followed by Recovery Period under Chow Diet
2.3. Acute Endotoxemia Followed by High-Fat Diet (HFD)
2.4. Glucose and Insulin Tolerance Tests and Measurements of Serum Leptin, Adiponectin, Insulin, IGF-I, SAA and Endotoxin
2.5. Histological Analysis
2.6. In Vivo Peripheral Fat Area Quantification
2.7. Quantitative Real-Time PCR
2.8. Gene Set Enrichment Analysis of Publicly Available Microarray Data
2.9. Statistical Analysis
3. Results
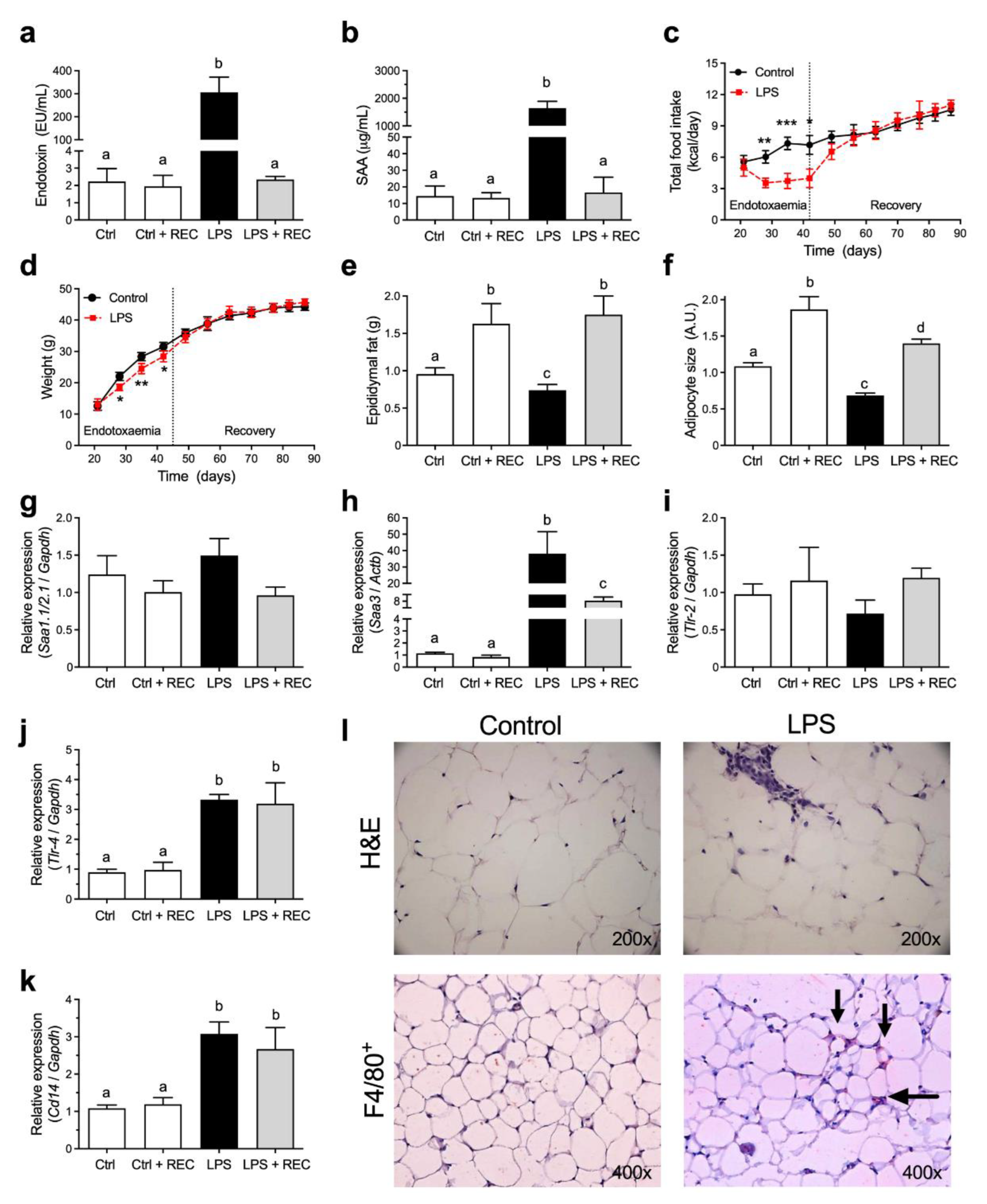
3.1. Acute Endotoxemia Affects Adipose Tissue but Does Not Lead to Weight Gain in Mice under Chow Diet
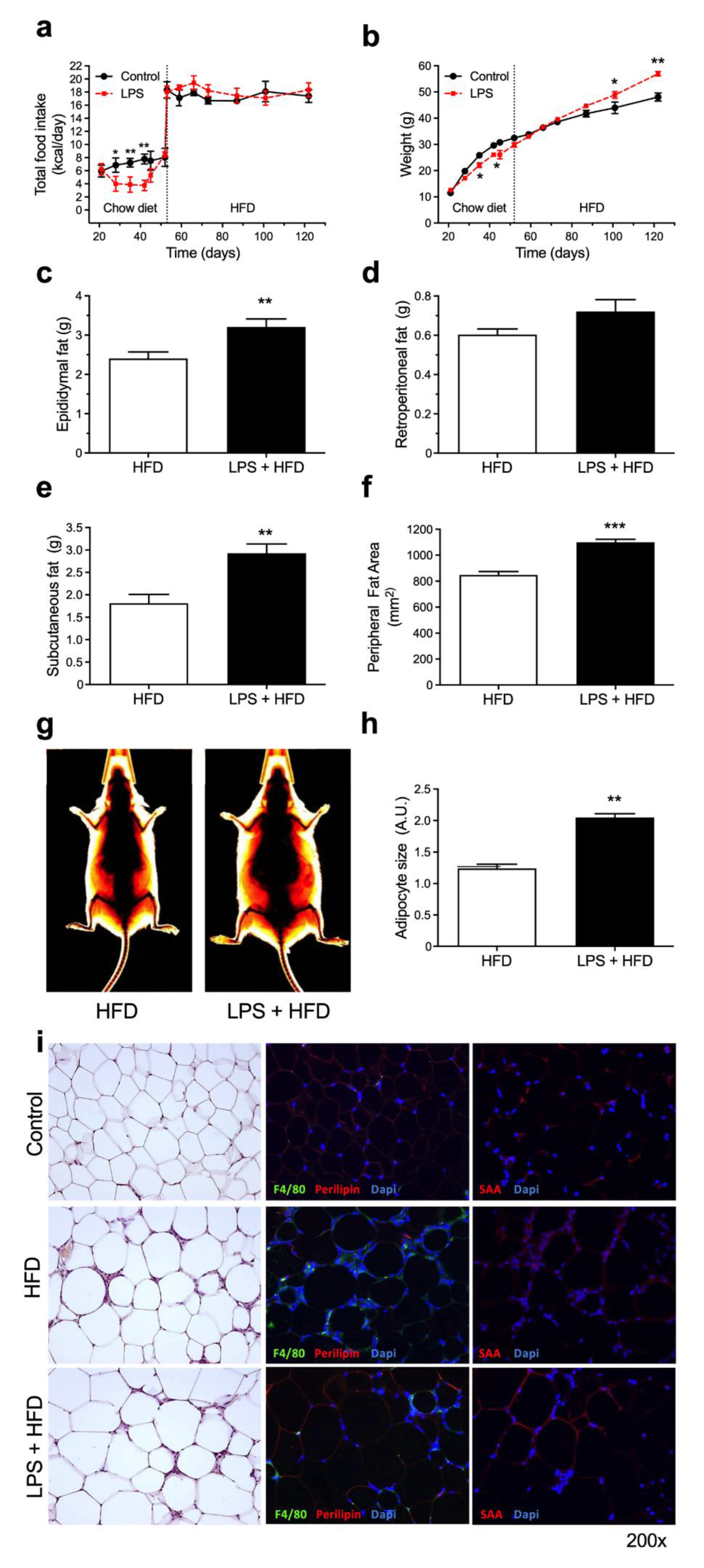
3.2. A Previous History of Acute Endotoxemia Exacerbates High-Fat Diet Complications
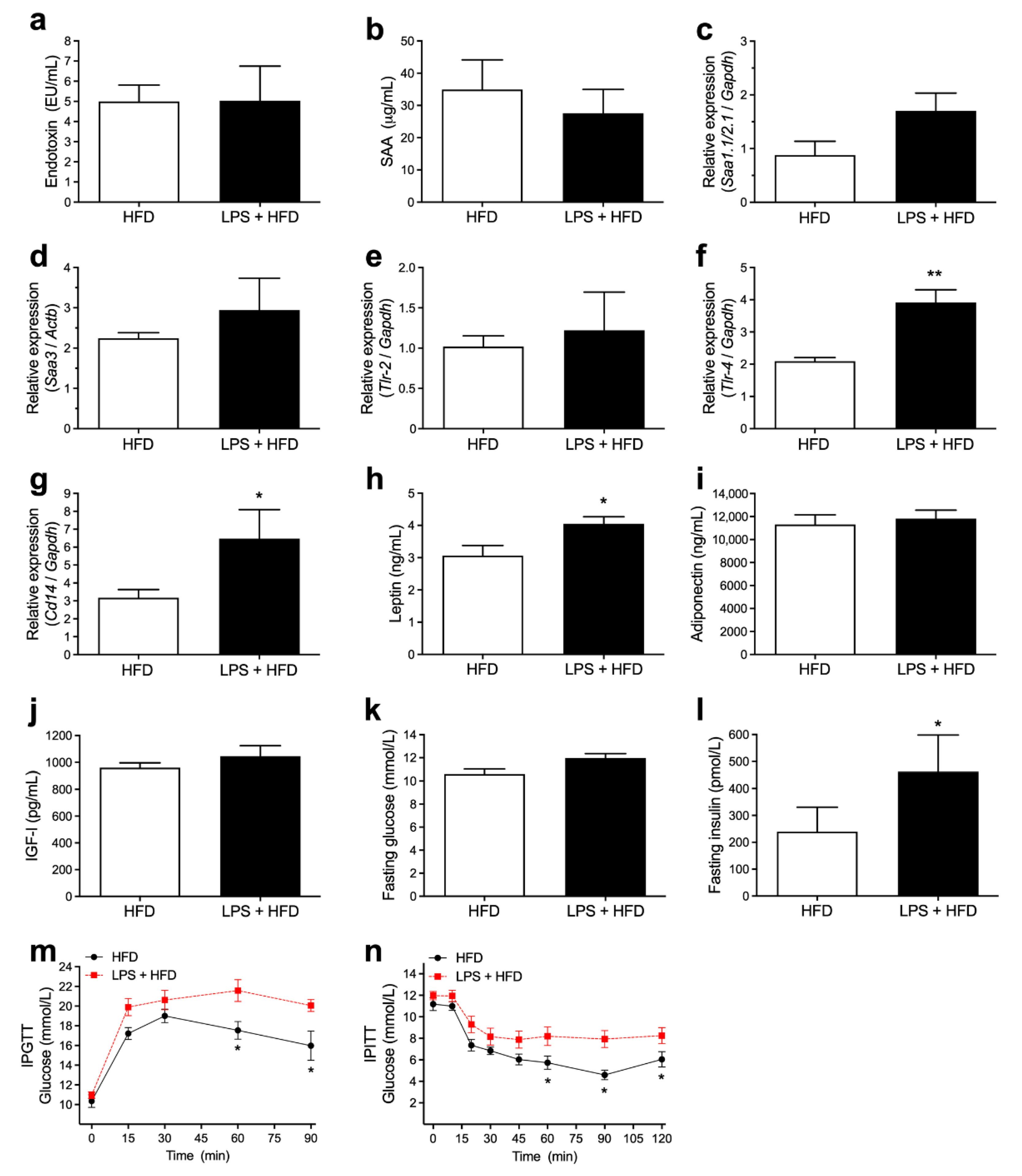
3.3. Acute Endotoxemia, Inflammatory Markers and Glucose Homeostasis
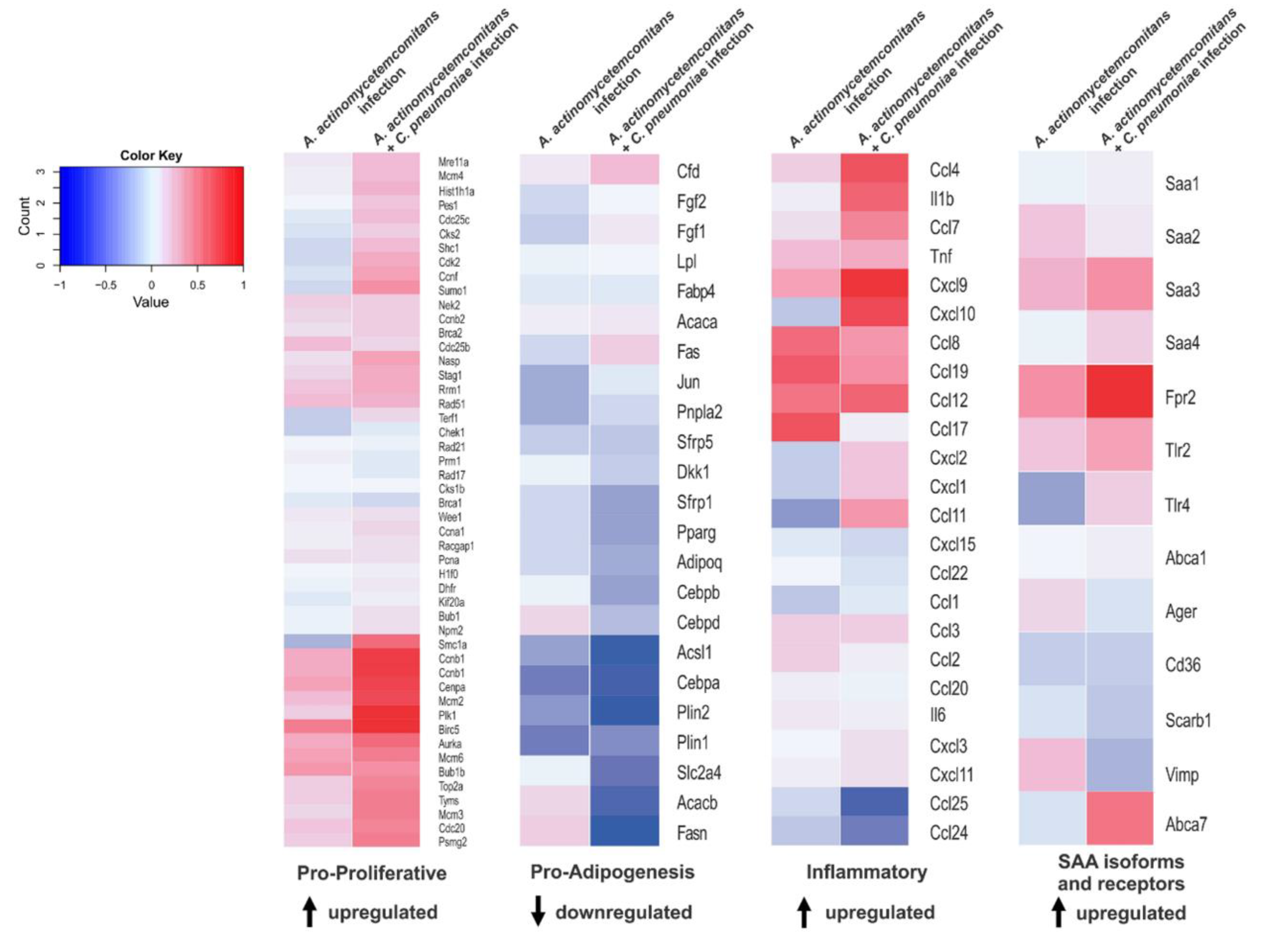
3.4. Recurrent Infection Upregulates Proliferative and Inflammatory Genes in Adipose Tissue
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Kushner, I. Mechanisms of disease: Acute-phase proteins and other systemic responses to inflammation. New Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Watkins, P.E. Experimental models of gram-negative sepsis. Br. J. Surg. 2001, 88, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Fock, R.A.; Vinolo, M.A.; de Moura Sá Rocha, V.; de Sá Rocha, L.C.; Borelli, P. Protein-energy malnutrition decreases the expression of TLR-4/MD-2 and CD14 receptors in peritoneal macrophages and reduces the synthesis of TNF-alpha in response to lipopolysaccharide (LPS) in mice. Cytokine 2007, 40, 105–114. [Google Scholar] [CrossRef]
- Tan, Y.; Kagan, J.C. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol. Cell 2014, 54, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.T. CRP as a mediator of disease. Circulation 2004, 109, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Bongers, S.H.; Chen, N.; van Grinsven, E.; van Staveren, S.; Hassani, M.; Spijkerman, R.; Hesselink, L.; Lo Tam Loi, A.T.; van Aalst, C.; Leijte, G.P.; et al. Kinetics of Neutrophil Subsets in Acute, Subacute, and Chronic Inflammation. Front. Immunol. 2021, 12, 674079. [Google Scholar] [CrossRef]
- Osrin, D.; Vergnano, S.; Costello, A. Serious bacterial infections in newborn infants in developing countries. Curr. Opin. Infect. Dis. 2004, 17, 217–224. [Google Scholar] [CrossRef]
- Di Cesare, M.; Sorić, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archibald, L.K.; Reller, L.B. Clinical microbiology in developing countries. Emerg. Infect. Dis. 2001, 7, 302–305. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.M.; Visniauskas, B.; Sandri, S.; Migliorini, S.; Andersen, M.L.; Tufik, S.; Fock, R.A.; Chagas, J.R.; Campa, A. Late effects of sleep restriction: Potentiating weight gain and insulin resistance arising from a high-fat diet in mice. Obesity 2015, 23, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyvarinen, K.; Tuomainen, A.M.; Laitinen, S.; Alfthan, G.; Salminen, I.; Leinonen, M.; Saikku, P.; Kovanen, P.T.; Jauhiainen, M.; Pussinen, P.J. The effect of proatherogenic pathogens on adipose tissue transcriptome and fatty acid distribution in apolipoprotein E-deficient mice. BMC Genom. 2013, 14, 709. [Google Scholar] [CrossRef] [Green Version]
- Van der Klaauw, A.A.; Farooqi, I.S. The Hunger Genes: Pathways to Obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Nymark, M.; Pussinen, P.J.; Tuomainen, A.M.; Forsblom, C.; Groop, P.-H.; Lehto, M.; Finndiane Study, G. Serum Lipopolysaccharide Activity Is Associated With the Progression of Kidney Disease in Finnish Patients With Type 1 Diabetes. Diabetes Care 2009, 32, 1689–1693. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.A. Drug therapy: Management of sepsis. N. Engl. J. Med. 2006, 355, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J. The immunopathogenesis of sepsis. Nat. 2002, 420, 885–891. [Google Scholar] [CrossRef]
- Sack, G.H. Serum amyloid A - a review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Song, M.J.; Kim, K.H.; Yoon, J.M.; Kim, J.B. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun. 2006, 346, 739–745. [Google Scholar] [CrossRef]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High Fat Diet-Induced Gut Microbiota Exacerbates Inflammation and Obesity in Mice via the TLR4 Signaling Pathway. PLoS ONE 2012, 7, 47713. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lee, H.; Berg, A.H.; Lisanti, M.P.; Shapiro, L.; Scherer, P.E. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J. Biol. Chem. 2000, 275, 24255–24263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehses, J.A.; Meier, D.T.; Wueest, S.; Rytka, J.; Boller, S.; Wielinga, P.Y.; Schraenen, A.; Lemaire, K.; Debray, S.; Van Lommel, L.; et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010, 53, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, N.; Echchannaoui, H.; Landmann, R.; Rivest, S. Cooperation between toll-like receptor 2 and 4 in the brain of mice challenged with cell wall components derived from gram-negative and gram-positive bacteria. Eur. J. Immunol. 2003, 33, 1127–1138. [Google Scholar] [CrossRef]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 2008, 181, 22–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, S.; Rodriguez, D.; Gomes, E.; Monteiro, H.P.; Russo, M.; Campa, A. Is serum amyloid A an endogenous TLR4 agonist? J. Leukoc. Biol. 2008, 83, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.L.; He, X.L.; Shi, X.G.; Huang, C.J.; Liu, J.; Zhou, S.L.; Heng, C.K. Association between serum amyloid A and obesity: A meta-analysis and systematic review. Inflamm. Res. 2010, 59, 323–334. [Google Scholar] [CrossRef]
- Eklund, K.K.; Niemi, K.; Kovanen, P.T. Immune Functions of Serum Amyloid, A. Crit. Rev. Immunol. 2012, 32, 335–348. [Google Scholar] [CrossRef]
- De Oliveira, E.M.; Sandri, S.; Knebel, F.H.; Iglesias Contesini, C.G.; Campa, A.; Filippin-Monteiro, F.B. Hypoxia Increases Serum Amyloid A3 (SAA3) in Differentiated 3T3-L1 Adipocytes. Inflammation 2013, 36, 1107–1110. [Google Scholar] [CrossRef]
- Filippin-Monteiro, F.B.; de Oliveira, E.M.; Sandri, S.; Knebel, F.H.; Albuquerque, R.C.; Campa, A. Serum amyloid A is a growth factor for 3T3-L1 adipocytes, inhibits differentiation and promotes insulin resistance. Int. J. Obes. 2012, 36, 1032–1039. [Google Scholar] [CrossRef] [Green Version]
- Furlaneto, C.J.; Campa, A. A novel function of serum amyloid A: A potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 by human blood neutrophil. Biochem. Biophys. Res. Commun. 2000, 268, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, E.; Ribeiro, F.P.; Campa, A. The acute phase protein serum amyloid A primes neutrophils. Fems Immunol. Med. Microbiol. 2003, 38, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, E.; Dermargos, A.; Armelin, H.A.; Curi, R.; Campa, A. Serum amyloid A induces reactive oxygen species (ROS) production and proliferation of fibroblast. Clin. Exp. Immunol. 2011, 163, 362–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knebel, F.H.; Albuquerque, R.C.; Massaro, R.R.; Maria-Engler, S.S.; Campa, A. Dual Effect of Serum Amyloid A on the Invasiveness of Glioma Cells. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemi, K.; Teirila, L.; Lappalainen, J.; Rajamaki, K.; Baumann, M.H.; Oorni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum Amyloid A Activates the NLRP3 Inflammasome via P2X(7) Receptor and a Cathepsin B-Sensitive Pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef] [Green Version]
- Ather, J.L.; Ckless, K.; Martin, R.; Foley, K.L.; Suratt, B.T.; Boyson, J.E.; Fitzgerald, K.A.; Flavell, R.A.; Eisenbarth, S.C.; Poynter, M.E. Serum Amyloid A Activates the NLRP3 Inflammasome and Promotes Th17 Allergic Asthma in Mice. J. Immunol. 2011, 187, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J. Serum Amyloid-A Is A Chemoattractant - Induction Of Migration, Adhesion, And Tissue Infiltration Of Monocytes And Polymorphonuclear Leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Faty, A.; Ferre, P.; Commans, S. The Acute Phase Protein Serum Amyloid A Induces Lipolysis and Inflammation in Human Adipocytes through Distinct Pathways. Plos One 2012, 7, 34031. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Wang, S.R.; Goodspeed, L.; Ding, Y.L.; Averill, M.; Subramanian, S.; Wietecha, T.; O'Brien, K.D.; Chait, A. Deletion of Serum Amyloid A3 Improves High Fat High Sucrose Diet-Induced Adipose Tissue Inflammation and Hyperlipidemia in Female Mice. Plos One 2014, 9, 108564. [Google Scholar] [CrossRef] [Green Version]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Ji, A.; Shridas, P.; Thompson, J.C.; van der Westhuyzen, D.R.; et al. Deficiency of Endogenous Acute Phase Serum Amyloid A Does Not Affect Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, E.M.; Visniauskas, B.; Tufik, S.; Andersen, M.L.; Chagas, J.R.; Campa, A. Serum Amyloid A Production Is Triggered by Sleep Deprivation in Mice and Humans: Is That the Link between Sleep Loss and Associated Comorbidities? Nutrients 2017, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.M.; Ascar, T.P.; Silva, J.C.; Sandri, S.; Migliorini, S.; Fock, R.A.; Campa, A. Serum amyloid A links endotoxaemia to weight gain and insulin resistance in mice. Diabetol. 2016, 59, 1760–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collaborators, G.D.D. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: A systematic analysis for the Global Burden of Disease Study 2015. Lancet. Infect. Dis. 2017, 17, 909–948. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.C.H.; Ezure, Y.; Rolley, L.; Spurling, G.; Lau, C.L.; Riaz, S.; Paterson, D.L.; Irwin, A.D. Gram-negative neonatal sepsis in low- and lower-middle-income countries and WHO empirical antibiotic recommendations: A systematic review and meta-analysis. PLoS Med 2021, 18, 1003787. [Google Scholar] [CrossRef]
- Huynh, B.T.; Kermorvant-Duchemin, E.; Chheang, R.; Randrianirina, F.; Seck, A.; Hariniaina Ratsima, E.; Andrianirina, Z.Z.; Diouf, J.B.; Abdou, A.Y.; Goyet, S.; et al. Severe bacterial neonatal infections in Madagascar, Senegal, and Cambodia: A multicentric community-based cohort study. PLoS Med 2021, 18, 1003681. [Google Scholar] [CrossRef]
- Singh, G.K.; Siahpush, M.; Kogan, M.D. Rising social inequalities in U.S. childhood obesity, 2003–2007. Ann. Epidemiol. 2010, 20, 40–52. [Google Scholar] [CrossRef]
- Singh, A.R.; Singh, S.A. Diseases of Poverty and Lifestyle, Well-Being and Human Development. Mens. Sana. Monogr. 2008, 6, 187–225. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.; Ruel, M.T.; Salm, L.; Sinclair, B.; Branca, F. Double-duty actions: Seizing programme and policy opportunities to address malnutrition in all its forms. Lancet 2020, 395, 142–155. [Google Scholar] [CrossRef]
- Nugent, R.; Levin, C.; Hale, J.; Hutchinson, B. Economic effects of the double burden of malnutrition. Lancet 2020, 395, 156–164. [Google Scholar] [CrossRef]
- Headey, D.D.; Alderman, H.H. The Relative Caloric Prices of Healthy and Unhealthy Foods Differ Systematically across Income Levels and Continents. J. Nutr. 2019, 149, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.C.; Forrest, C.B.; Zhang, P.; Richards, T.M.; Livshits, A.; DeRusso, P.A. Association of antibiotics in infancy with early childhood obesity. JAMA Pediatr. 2014, 168, 1063–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginneken, V.; Sitnyakowsky, L.; Jeffery, J.E. Infectobesity: Viral infections (especially with human adenovirus-36: Ad-36) may be a cause of obesity. Med. Hypotheses 2009, 72, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Dhurandhar, N.V.; Allison, D.B.; Bowen, R.L.; Israel, B.A.; Albu, J.B.; Augustus, A.S. Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids. Int. J. Obes. 2005, 29, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Miwata, H.; Yamada, T.; Okada, M.; Kudo, T.; Kimura, H.; Morishima, T. Serum Amyloid A-Protein In Acute Viral-Infections. Arch. Dis. Child. 1993, 68, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Machida, K.; Cheng, K.T.H.; Sung, V.M.H.; Levine, A.M.; Foung, S.; Lai, M.M.C. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J. Virol. 2006, 80, 866–874. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes de Oliveira, E.; Silva, J.C.; Ascar, T.P.; Sandri, S.; Marchi, A.F.; Migliorini, S.; Nakaya, H.T.I.; Fock, R.A.; Campa, A. Acute Inflammation Is a Predisposing Factor for Weight Gain and Insulin Resistance. Pharmaceutics 2022, 14, 623. https://doi.org/10.3390/pharmaceutics14030623
Mendes de Oliveira E, Silva JC, Ascar TP, Sandri S, Marchi AF, Migliorini S, Nakaya HTI, Fock RA, Campa A. Acute Inflammation Is a Predisposing Factor for Weight Gain and Insulin Resistance. Pharmaceutics. 2022; 14(3):623. https://doi.org/10.3390/pharmaceutics14030623
Chicago/Turabian StyleMendes de Oliveira, Edson, Jacqueline C. Silva, Thais P. Ascar, Silvana Sandri, Alexandre F. Marchi, Silene Migliorini, Helder T. I. Nakaya, Ricardo A. Fock, and Ana Campa. 2022. "Acute Inflammation Is a Predisposing Factor for Weight Gain and Insulin Resistance" Pharmaceutics 14, no. 3: 623. https://doi.org/10.3390/pharmaceutics14030623
APA StyleMendes de Oliveira, E., Silva, J. C., Ascar, T. P., Sandri, S., Marchi, A. F., Migliorini, S., Nakaya, H. T. I., Fock, R. A., & Campa, A. (2022). Acute Inflammation Is a Predisposing Factor for Weight Gain and Insulin Resistance. Pharmaceutics, 14(3), 623. https://doi.org/10.3390/pharmaceutics14030623