
Repurposing of Ciclopirox to Overcome the Limitations of Zidovudine (Azidothymidine) against Multidrug-Resistant Gram-Negative Bacteria



Abstract

1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Polymerase Chain Reaction (PCR) Amplification
2.2. Preparation of Bacterial Cells for Assays
2.3. Evaluation of Antibacterial Activity
2.4. Western Blotting Analysis
2.5. DNA Sequencing and Sequence Alignment
2.6. Cell Morphology Analysis
2.7. Motility Assay
2.8. Generation of Laboratory-Made Drug-Resistant E. coli Strains
2.9. Statistical Analysis
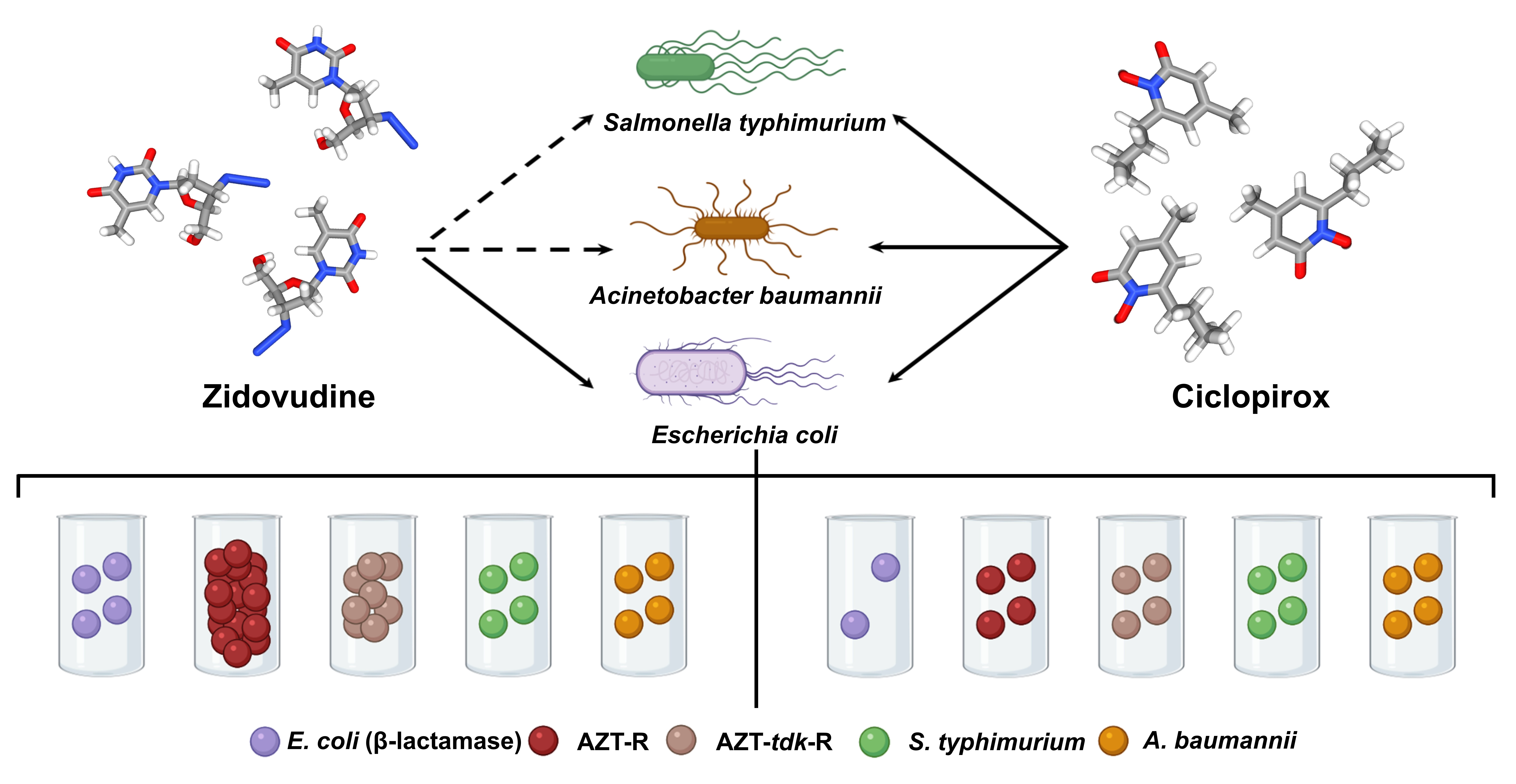
3. Results and Discussion
3.1. Activity of AZT and CPX against Gram-Negative Species with Different Antibiotic Resistance Status
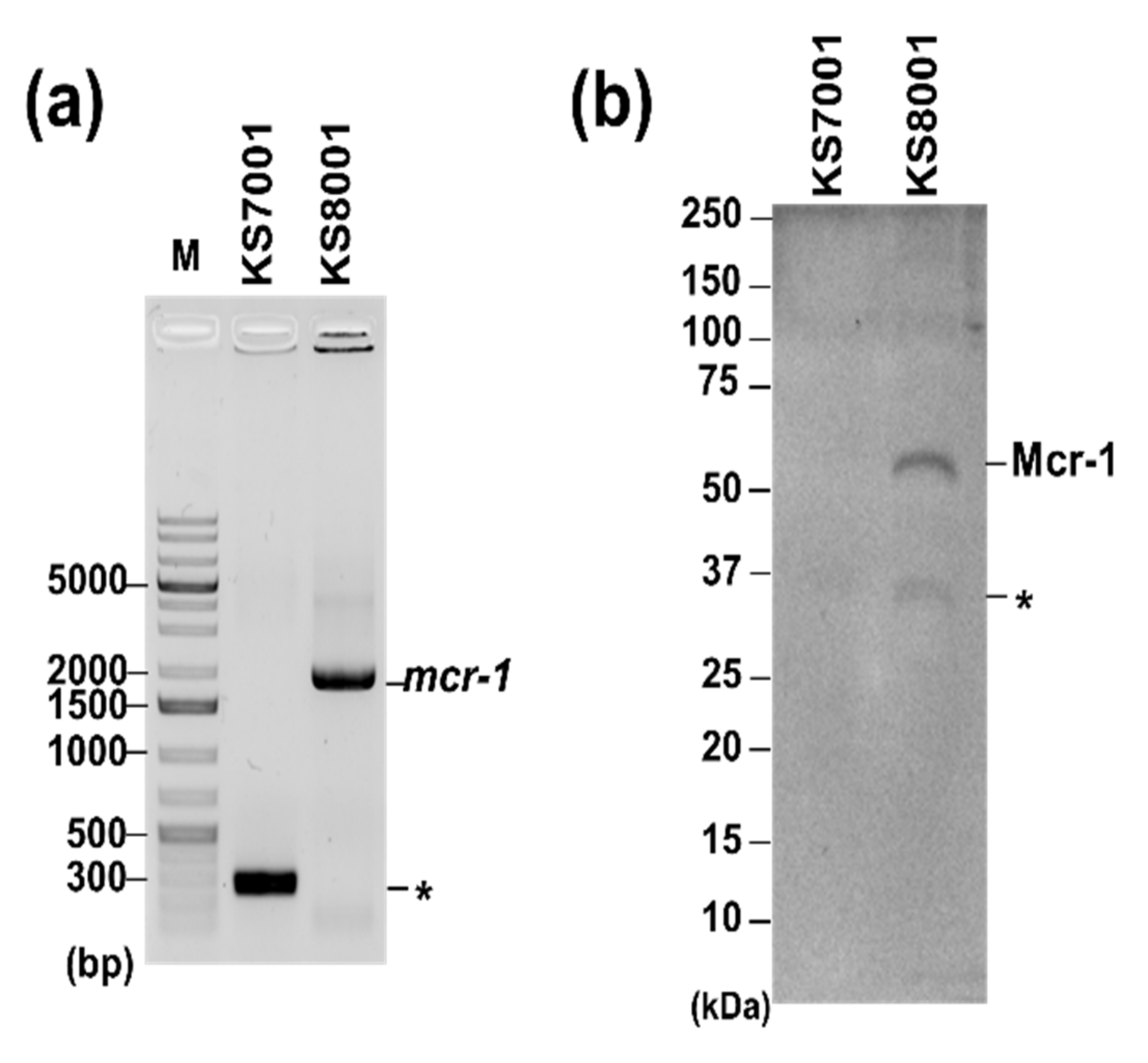
3.2. Phenotypic Studies of Increased CPX Action against β-Lactamase-Expressing Resistant E. coli Strains
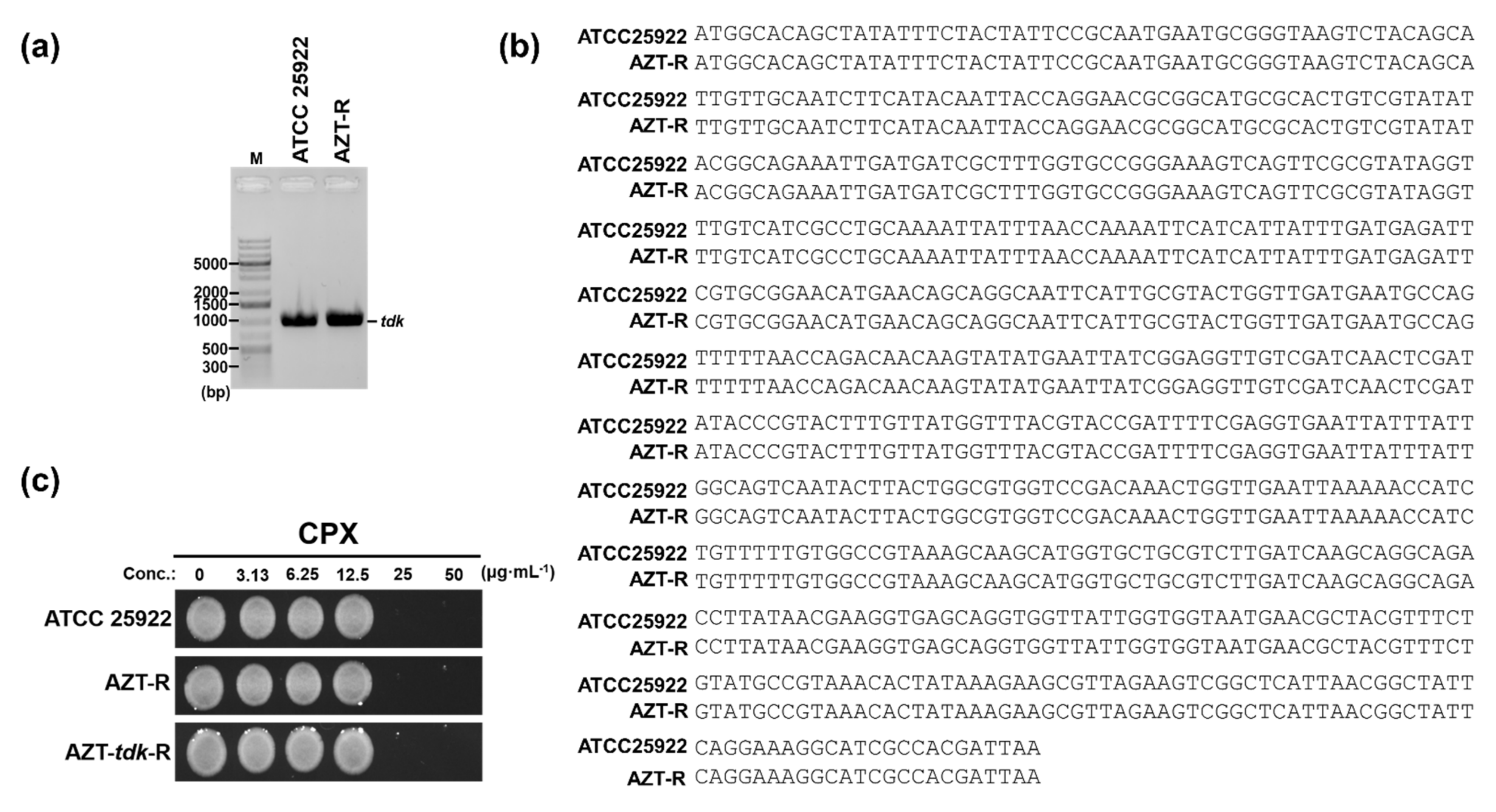
3.3. CPX Exhibits Bactericidal Activity against Tdk-Dependent AZT-Resistant E. coli
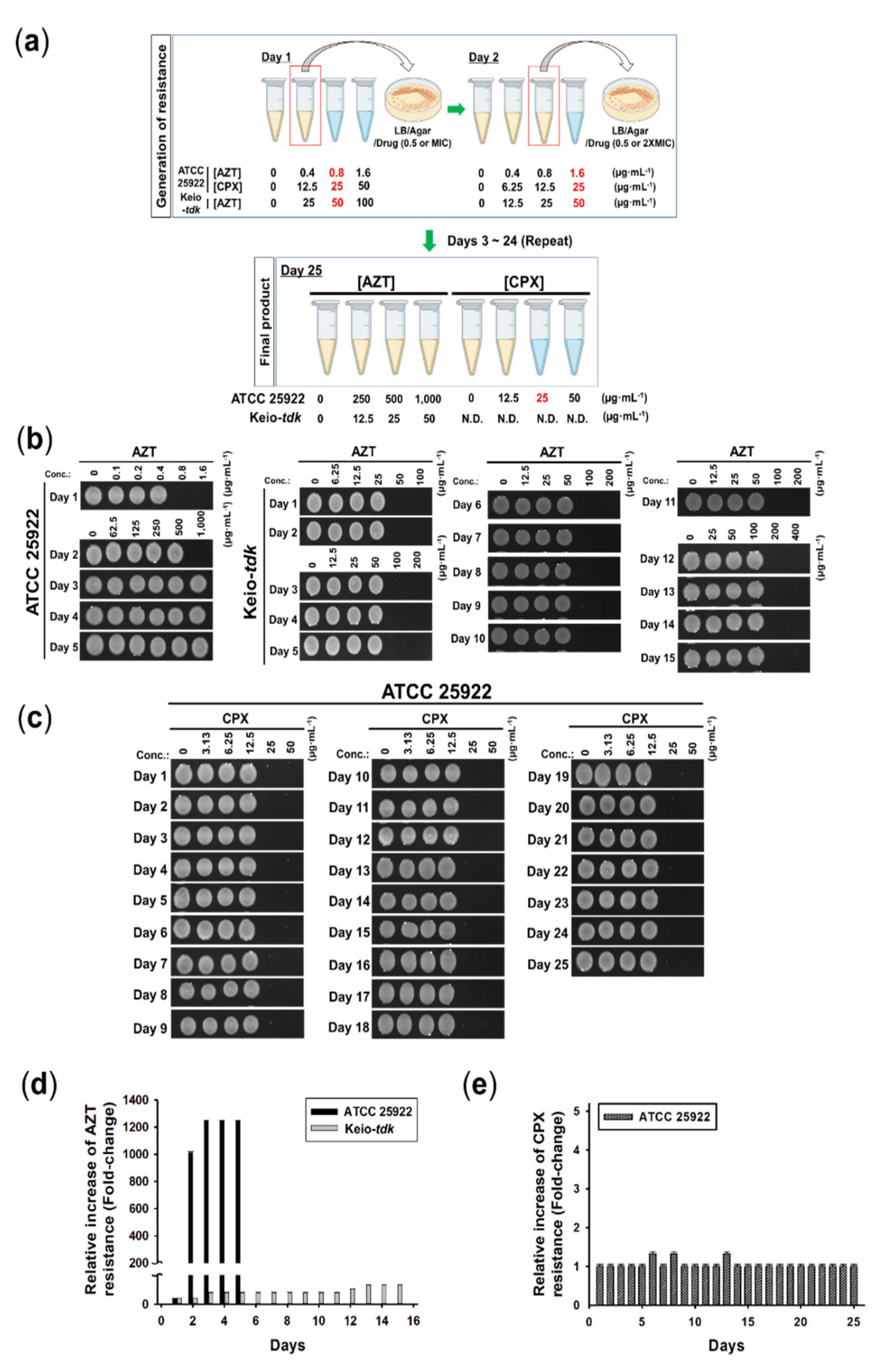
3.4. Induction of Resistance by AZT and CPX
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mercorelli, B.; Palù, G.; Loregian, A. Drug Repurposing for Viral Infectious Diseases: How Far Are We? Trends Microbiol. 2018, 26, 865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, F.; Zeng, M.; Mao, Y.; Song, Z. Drug repurposing strategies in the development of potential antifungal agents. Appl. Microbiol. Biotechnol. 2021, 105, 5259. [Google Scholar] [CrossRef] [PubMed]
- Miró-Canturri, A.; Ayerbe-Algaba, R.; Smani, Y. Drug Repurposing for the Treatment of Bacterial and Fungal Infections. Front. Microbiol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Farha, M.A.; Brown, E.D. Drug repurposing for antimicrobial discovery. Nat. Microbiol. 2019, 4, 565. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Williamson, P.R.; Zheng, W. Improving therapy of severe infections through drug repurposing of synergistic combinations. Curr. Opin. Pharmacol. 2019, 48, 92. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.K.; Teng, C.; Frei, C.R. Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus On Drug Repurposing. Front. Cell. Infect. Microbiol. 2021, 11, 684515. [Google Scholar] [CrossRef]
- Anderson, P.L.; Rower, J.E. Zidovudine and Lamivudine for HIV Infection. Clin. Med. Rev. Ther. 2010, 2, a2004. [Google Scholar]
- Elwell, L.P.; Ferone, R.; Freeman, G.A.; Fyfe, J.A.; Hill, J.A.; Ray, P.H.; Richards, C.A.; Singer, S.C.; Knick, V.B.; Rideout, J.L. Antibacterial activity and mechanism of action of 3′-azido-3′-deoxythymidine (BW A509U). Antimicrob. Agents Chemother. 1987, 31, 274. [Google Scholar] [CrossRef]
- Keith, B.R.; White, G.; Wilson, H.R. In vivo efficacy of zidovudine (3′-azido-3′-deoxythymidine) in experimental gram-negative-bacterial infections. Antimicrob. Agents Chemother. 1989, 33, 479. [Google Scholar] [CrossRef]
- Sandrini, M.P.; Clausen, A.R.; On, S.L.; Aarestrup, F.M.; Munch-Petersen, B.; Piskur, J. Nucleoside analogues are activated by bacterial deoxyribonucleoside kinases in a species-specific manner. J. Antimicrob. Chemother. 2007, 60, 510. [Google Scholar] [CrossRef]
- Doléans-Jordheim, A.; Bergeron, E.; Bereyziat, F.; Ben-Larbi, S.; Dumitrescu, O.; Mazoyer, M.A.; Morfin, F.; Dumontet, C.; Freney, J.; Jordheim, L.P. Zidovudine (AZT) has a bactericidal effect on enterobacteria and induces genetic modifications in resistant strains. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 1249. [Google Scholar] [CrossRef]
- Peyclit, L.; Baron, S.A.; Yousfi, H.; Rolain, J.M. Zidovudine: A salvage therapy for mcr-1 plasmid-mediated colistin-resistant bacterial infections? Int. J. Antimicrob. Agents 2018, 52, 11. [Google Scholar] [CrossRef]
- Ng, S.M.S.; Sioson, J.S.P.; Yap, J.M.; Ng, F.M.; Ching, H.S.V.; Teo, J.W.P.; Jureen, R.; Hill, J.; Chia, C.S.B. Repurposing Zidovudine in combination with Tigecycline for treating carbapenem-resistant Enterobacteriaceae infections. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 141. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Y.; Yang, K.; Li, R.; Xiao, X.; Wang, Z. Anti-HIV agent azidothymidine decreases Tet(X)-mediated bacterial resistance to tigecycline in Escherichia coli. Commun. Biol. 2020, 3, 162. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, Y.; Coates, A. Azidothymidine Produces Synergistic Activity in Combination with Colistin against Antibiotic-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 63, e01630. [Google Scholar] [CrossRef]
- Zeller, A.; Koenig, J.; Schmitt, G.; Singer, T.; Guérard, M. Genotoxicity profile of azidothymidine in vitro. Toxicol. Sci. 2013, 135, 317. [Google Scholar] [CrossRef] [PubMed]
- Lewin, C.S.; Allen, R.A.; Amyes, S.G. Mechanisms of zidovudine resistance in bacteria. J. Med. Microbiol. 1990, 33, 235. [Google Scholar] [CrossRef]
- Lawrence, E.O. Molecular Epidemiology of Escherichia coli in HIV-Positive Individuals in Southwest Nigeria. Int. Arch. Med. 2015, 8, 78. [Google Scholar] [CrossRef]
- Chatterjee, M.; Chakraborty, B.; Chatterjee, S.; Bose, M.; Mukherjee, K.; Basu, A.; Das, S.; Banerjee, M.; Ghosh, U. Enteric fever in an HIV/AIDS patient: Atypical manifestations. Iran. J. Microbiol. 2012, 4, 150. [Google Scholar] [PubMed]
- Manfredi, R.; Nanetti, A.; Valentini, R.; Chiodo, F. Acinetobacter infections in patients with human immunodeficiency virus infection: Microbiological and clinical epidemiology. Chemotherapy 2001, 47, 19. [Google Scholar] [CrossRef]
- Thi, P.L.N.; Yassibanda, S.; Aidara, A.; Le Bouguénec, C.; Germani, Y. Enteropathogenic Klebsiella pneumoniae HIV-infected adults, Africa. Emerg. Infect. Dis. 2003, 9, 135. [Google Scholar] [CrossRef]
- Salami, A.K.; Olatunji, P.O.; Oluboyo, P.O.; Akanbi, A.A., 2nd; Fawibe, E.A. Bacterial pneumonia in the AIDS patients. West Afr. J. Med. 2006, 25, 1. [Google Scholar]
- Furman, A.C.; Jacobs, J.; Sepkowitz, K.A. Lung abscess in patients with AIDS. Clin. Infect. Dis. 1996, 22, 81. [Google Scholar] [CrossRef] [PubMed]
- Abrams, B.B.; Hänel, H.; Hoehler, T. Ciclopirox olamine: A hydroxypyridone antifungal agent. Clin. Dermatol. 1991, 9, 471. [Google Scholar] [CrossRef]
- Bohn, M.; Kraemer, K.T. Dermatopharmacology of ciclopirox nail lacquer topical solution 8% in the treatment of onychomycosis. J. Am. Acad. Dermatol. 2000, 43, S57. [Google Scholar] [CrossRef]
- Subissi, A.; Monti, D.; Togni, G.; Mailland, F. Ciclopirox: Recent nonclinical and clinical data relevant to its use as a topical antimycotic agent. Drugs 2010, 70, 2133. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Kohli, Y. In vitro susceptibility testing of ciclopirox, terbinafine, ketoconazole and itraconazole against dermatophytes and nondermatophytes, and in vitro evaluation of combination antifungal activity. Br. J. Dermatol. 2003, 149, 296. [Google Scholar] [CrossRef]
- Carlson-Banning, K.M.; Chou, A.; Liu, Z.; Hamill, R.J.; Song, Y.; Zechiedrich, L. Toward repurposing ciclopirox as an antibiotic against drug-resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae. PLoS ONE 2013, 8, e69646. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Vega, A.; Bernstein, L.R.; Mandujano-Tinoco, E.A.; García-Contreras, S.J.; García-Contreras, R. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front. Microbiol. 2015, 6, 282. [Google Scholar] [CrossRef]
- Shin, J.; Cho, H.; Kim, S.; Kim, K.-s. Role of acid responsive genes in the susceptibility of Escherichia coli to ciclopirox. Biochem. Biophys. Res. Commun. 2018, 500, 296. [Google Scholar] [CrossRef] [PubMed]
- Coppi, G.; Silingardi, S.; Girardello, R.; De Aloysio, D.; Manzardo, S. Pharmacokinetics of ciclopirox olamine after vaginal application to rabbits and patients. J. Chemother. 1993, 5, 302. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K. Ciclopirox: An overview. Int. J. Dermatol. 2001, 40, 305. [Google Scholar] [CrossRef] [PubMed]
- Ceschin-Roques, C.G.; Hänel, H.; Pruja-Bougaret, S.M.; Lagarde, I.; Vandermander, J.; Michel, G. Ciclopiroxolamine cream 1%: In vitro and in vivo penetration into the stratum corneum. Skin Pharmacol. 1991, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Ji, W.; Xia, H.; Li, J. Combined analysis of gene expression and genome binding profiles identified potential therapeutic targets of ciclopirox in Ewing sarcoma. Mol. Med. Rep. 2018, 17, 4291. [Google Scholar]
- Shen, T.; Zhou, H.; Shang, C.; Luo, Y.; Wu, Y.; Huang, S. Ciclopirox activates ATR-Chk1 signaling pathway leading to Cdc25A protein degradation. Genes Cancer 2018, 9, 39. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mihailidou, C.; Papakotoulas, P.; Papavassiliou, A.G.; Karamouzis, M.V. Superior efficacy of the antifungal agent ciclopirox olamine over gemcitabine in pancreatic cancer models. Oncotarget 2017, 9, 10360. [Google Scholar] [CrossRef][Green Version]
- Mihailidou, C.; Chatzistamou, I.; Papavassiliou, A.G.; Kiaris, H. Modulation of Pancreatic Islets’ Function and Survival During Aging Involves the Differential Regulation of Endoplasmic Reticulum Stress by p21 and CHOP. Antioxid. Redox Signal. 2017, 27, 185. [Google Scholar] [CrossRef]
- Mihailidou, C.; Chatzistamou, I.; Papavassiliou, A.G.; Kiaris, H. Ciclopirox enhances pancreatic islet health by modulating the unfolded protein response in diabetes. Pflugers Arch. 2016, 468, 1957. [Google Scholar] [CrossRef]
- Bernier, K.M.; Morrison, L.A. Antifungal drug ciclopirox olamine reduces HSV-1 replication and disease in mice. Antivir. Res. 2018, 156, 102. [Google Scholar] [CrossRef]
- Cáceres, C.J.; Angulo, J.; Contreras, N.; Pino, K.; Vera-Otarola, J.; López-Lastra, M. Targeting deoxyhypusine hydroxylase activity impairs cap-independent translation initiation driven by the 5’untranslated region of the HIV-1, HTLV-1, and MMTV mRNAs. Antivir. Res. 2016, 134, 192. [Google Scholar] [CrossRef]
- Hanauske-Abel, H.M.; Saxena, D.; Palumbo, P.E.; Hanauske, A.R.; Luchessi, A.D.; Cambiaghi, T.D.; Hoque, M.; Spino, M.; Gandolfi, D.D.; Heller, D.S.; et al. Drug-induced reactivation of apoptosis abrogates HIV-1 infection. PLoS ONE 2013, 8, e74414. [Google Scholar] [CrossRef]
- Conley, Z.C.; Carlson-Banning, K.M.; Carter, A.G.; de la Cova, A.; Song, Y.; Zechiedrich, L. Sugar and iron: Toward understanding the antibacterial effect of ciclopirox in Escherichia coli. PLoS ONE 2019, 14, e0210547. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-s.; Kim, T.; Pan, J.G. In vitro evaluation of ciclopirox as an adjuvant for polymyxin B against gram-negative bacteria. J. Antibiot. 2015, 68, 395. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Cho, H.; Naskar, A.; Lee, S.; Kim, S.; Kim, K.-s. A New Surface Charge Neutralizing Nano-Adjuvant to Potentiate Polymyxins in Killing Mcr-1 Mediated Drug-Resistant Escherichia coli. Pharmaceutics 2021, 13, 250. [Google Scholar] [CrossRef]
- Kitagawa, M.; Ara, T.; Arifuzzaman, M.; Ioka-Nakamichi, T.; Inamoto, E.; Toyonaga, H.; Mori, H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res. 2005, 12, 291. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453. [Google Scholar] [CrossRef]
- Planesse, C.; Nativel, B.; Iwema, T.; Gasque, P.; Robert-Da Silva, C.; Viranaïcken, W. Recombinant human HSP60 produced in ClearColi™ BL21(DE3) does not activate the NFκB pathway. Cytokine 2015, 73, 190. [Google Scholar] [CrossRef]
- Jarvik, T.; Smillie, C.; Groisman, E.A.; Ochman, H. Short-term signatures of evolutionary change in the Salmonella enterica serovar typhimurium 14028 genome. J. Bacteriol. 2010, 192, 560. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threatsreport/2019-ar-threats-report-508.pdf (accessed on 10 January 2022).
- Duchesne, L.G.M.; Lam, J.S.; MacDonald, L.A. Effect of pH and acrylamide concentration on the separation of lipopolysaccharides in polyacrylamide gels. Curr. Microbiol. 1988, 16, 191. [Google Scholar] [CrossRef]
- Liu, D.; Reeves, P.R. Escherichia coli K12 regains its O antigen. Microbiology 1994, 140, 49. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.K.; Yasir, M.; Bastkowski, S.; Telatin, A.; Page, A.; Webber, M.; Charles, I. Chemical biology-whole genome engineering datasets predict new antibacterial combinations. Microb. Genom. 2021, 7, 718. [Google Scholar] [CrossRef] [PubMed]
- Peyclit, L.; Ben Khedher, M.; Zerrouki, L.; Diene, S.M.; Baron, S.A.; Rolain, J.M. Inactivation of thymidine kinase as a cause of resistance to zidovudine in clinical isolates of Escherichia coli: A phenotypic and genomic study. J. Antimicrob. Chemother. 2020, 75, 1410. [Google Scholar] [CrossRef] [PubMed]
- Anyanwu, M.U.; Jaja, I.F.; Nwobi, O.C. Occurrence and Characteristics of Mobile Colistin Resistance (mcr) Gene-Containing Isolates from the Environment: A Review. Int. J. Environ. Res. Public Health 2020, 17, 1028. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, L.; Wang, M.; Zhou, L.; Feng, X.; Yu, L.; Lan, J.; Gao, W.; Zhang, C.; Bu, Y.; et al. CPX Targeting DJ-1 Triggers ROS-induced Cell Death and Protective Autophagy in Colorectal Cancer. Theranostics 2019, 9, 5577. [Google Scholar] [CrossRef]
- Weir, S.J.; Patton, L.; Castle, K.; Rajewski, L.; Kasper, J.; Schimmer, A.D. The repositioning of the anti-fungal agent ciclopirox olamine as a novel therapeutic agent for the treatment of haematologic malignancy. J. Clin. Pharm. Ther. 2011, 36, 128. [Google Scholar] [CrossRef]
- Gupta, A.K.; Plott, T. Ciclopirox: A broad-spectrum antifungal with antibacterial and anti-inflammatory properties. Int. J. Dermatol. 2004, 43, 3. [Google Scholar] [CrossRef]
- Nyce, J.; Leonard, S.; Canupp, D.; Schulz, S.; Wong, S. Epigenetic mechanisms of drug resistance: Drug-induced DNA hypermethylation and drug resistance. Proc. Natl. Acad. Sci. USA 1993, 90, 2960. [Google Scholar] [CrossRef]
- Klein, R.J.; Czelusniak, S.M. Effect of a thymidine kinase inhibitor (L-653,180) on antiviral treatment of experimental herpes simplex virus infection in mice. Antivir. Res. 1990, 14, 207. [Google Scholar]
- Lin, J.; Zangi, M.; Kumar, T.V.N.H.; Shakar Reddy, M.; Reddy, L.V.R.; Sadhukhan, S.K.; Bradley, D.P.; Moreira-Walsh, B.; Edwards, T.C.; O’Dea, A.T.; et al. Synthetic Derivatives of Ciclopirox are Effective Inhibitors of Cryptococcus neoformans. ACS Omega 2021, 6, 8477. [Google Scholar] [CrossRef] [PubMed]
- Weir, S.J.; Wood, R.; Baltezor, M.J.; Reed, G.; Brinker, A.E.; Ham, T.; Schorno, K.; Toren, P.; Ramamoorthy, R.; Zhukova-Harrill, V.; et al. Pharmacokinetics of ciclopirox prodrug, a novel agent for the treatment of bladder cancer, in animals and humans. J. Clin. Oncol. 2019, 37, e14705. [Google Scholar] [CrossRef]
- Rangarajan, P.; Ramalingam, S.; Subramaniam, D.; Baltezor, M.J.; Wood, R.; Anant, S.; Weir, S. Clopirox prodrug for the prevention and therapy of non-muscle invasive bladder cancer. Cancer Res. 2015, 75, S1895. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Genetic Feature or Antibiotic Resistance | MIC (μg∙mL−1) 1 | Reference | |
|---|---|---|---|---|---|
| AZT | CPX | ||||
| A. baumannii | ATCC 17978 | Acinetobacter baumannii Bouvet and Grimont; Control | >12.8 | 25 | ATCC |
| ATCC 19606 | Acinetobacter baumannii Bouvet and Grimont; Control | >12.8 | 25 | ATCC | |
| TIG-R | TIGR ATCC 19606 | >12.8 | 25 | ATCC | |
| E. coli | BW25113 | K-12 F−Δ(araD-araB)567 ΔlacZ4787::rrnB-3 LAM− rph-1 Δ(rhaD-rhaB)568 hsdR514; O-antigen (−); control | 0.00625 | 25 | [44] |
| ATCC 25922 | Smooth LPS (serotype 6); control | 0.8 | 25 | ATCC | |
| MG1655 | K-12 F− λ−ilvG − rfb-50 rph-1; O-antigen (−); control | 0.00625 | 25 | [47] | |
| KS7001 | ATCC 25922 pQE-60 (AMPR) | 0.8 | 25 | This study | |
| KS8001 | ATCC 25922 pQE-60-mcr-1 (AMPR) | 0.8 | 25 | This study | |
| KS9000 | BW25113 pCA24N(-gfp) | 0.00625 | 25 | [46]; This study | |
| KS9001 | BW25113 ASKA-wbbL | 0.00625 | 25 | [46]; This study | |
| ClearColi® BL21 (DE3) | msbA148 ΔgutQ ΔkdsD ΔlpxL ΔlpxM ΔpagP ΔlpxP Δept; LPS-free (Lipid IVA) | 0.05 | 25 | [48] | |
| Keio-tdk | BW25113 tdk::KANR | 50 | 25 | [44] | |
| CCARM 0291 | NAL | 0.4 | 12.5 | CCARM | |
| CCARM 1013 | AMP, CEP, GM, NOR | 0.2 | 25 | CCARM | |
| CCARM 1120 | blaESBL; AMP, CEP, CIP, GM, NOR | 0.1 | 25 | CCARM | |
| CCARM 1368 | AMP, CEP, CTX, GM, NOR | 0.4 | 12.5 | CCARM | |
| ATCC BAA-2340 | blaNDM-1, AMC, AMP, TIC, PIP, CEP, CIP, CTX, FEP, FOX, DOR, MER, ETP, IMP, NAL, MOX, NOR, TOB, TET, TRI/SXT | 0.8 | 12.5 | ATCC | |
| ATCC BAA-2452 | blaNDM-1, ETP, IMP | 0.2 | 12.5 | ATCC | |
| ATCC BAA-2469 | blaNDM-1, ETP, IMP | 0.2 | 12.5 | ATCC | |
| ATCC BAA-2471 | blaNDM-1, ETP, IMP | 0.05 | 25 | ATCC | |
| NCCP 16283 | mcr-1, AMP, CAZ, CHL, CIP, COL, FEP, FOX, GEN, NAL, SXT, TET | 0.8–1.6 | 25 | NCCP | |
| NCCP 16284 | mcr-1, blaNDM-1; blaTEM-1, blaCTX-M-27, AMC, AMP, CAZ, CHL, CIP, COL, DOR, ETP, FEP, FOX, IMP, MEM, NAL, SXT, TET | 0.05 | 25 | NCCP | |
| AZT-R | Tdk-independent AZTR from ATCC 25922 | >1000 | 25 | This study | |
| AZT-tdk-R | Tdk-dependent AZTR from Keio-tdk | 200 | 25 | This study | |
| K. pneumoniae | KCTC 1726 | Klebsiella pneumoniae subsp. pneumoniae; Control | 1.6 | 100 | KCTC |
| KCTC 22057 | Klebsiella sp.; clinical isolate | 1.6 | 100 | KCTC | |
| KCTC 22058 | Klebsiella pneumoniae; clinical isolate | 1.6 | 100 | KCTC | |
| KCTC 22062 | Klebsiella sp.; clinical isolate | 1.6 | 100 | KCTC | |
| KCTC 32203 | Klebsiella pneumoniae; clinical isolate | 1.6 | 100 | KCTC | |
| S. typhimurium | 14028S | Salmonella enterica serovar typhimurium; Control | 0.4 | 25 | [49] |
| CCARM 0293 | blaAmpC; AMP, NAL | 0.4 | 25 | CCARM | |
| CCARM 8170 | blaAmpC; AMP, CHL, NAL, STR, TET | 0.1 | 50 | CCARM | |
| CCARM 8250 | blaAmpC; AMP, CHL, TET | 0.1 | 50 | CCARM | |
| CCARM 8254 | blaAmpC; AMP, CHL, TET | 0.1 | 50 | CCARM | |
| Strain | Diameter of Grown Bacterial Cells, mm | |
|---|---|---|
| CPX(−) 1 | CPX(+) 2 | |
| ATCC 25922 (Control) | 24.0 ± 1.4 | 15.3 ± 2.1 |
| ATCC BAA-2340 | 17.2 ± 2.1 | 1.33 ± 0.5 |
| ATCC BAA-2452 | 15.0 ± 1.6 | 2.00 ± 0.8 |
| ATCC BAA-2469 | 17.0 ± 2.2 | 2.67 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.; Kim, K.-s. Repurposing of Ciclopirox to Overcome the Limitations of Zidovudine (Azidothymidine) against Multidrug-Resistant Gram-Negative Bacteria. Pharmaceutics 2022, 14, 552. https://doi.org/10.3390/pharmaceutics14030552
Cho H, Kim K-s. Repurposing of Ciclopirox to Overcome the Limitations of Zidovudine (Azidothymidine) against Multidrug-Resistant Gram-Negative Bacteria. Pharmaceutics. 2022; 14(3):552. https://doi.org/10.3390/pharmaceutics14030552
Chicago/Turabian StyleCho, Hyejin, and Kwang-sun Kim. 2022. "Repurposing of Ciclopirox to Overcome the Limitations of Zidovudine (Azidothymidine) against Multidrug-Resistant Gram-Negative Bacteria" Pharmaceutics 14, no. 3: 552. https://doi.org/10.3390/pharmaceutics14030552
APA StyleCho, H., & Kim, K.-s. (2022). Repurposing of Ciclopirox to Overcome the Limitations of Zidovudine (Azidothymidine) against Multidrug-Resistant Gram-Negative Bacteria. Pharmaceutics, 14(3), 552. https://doi.org/10.3390/pharmaceutics14030552