
Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis

Abstract
:1. Introduction
2. Discussion
2.1. Alzheimer’s Disease
2.2. Amyloid Hypothesis
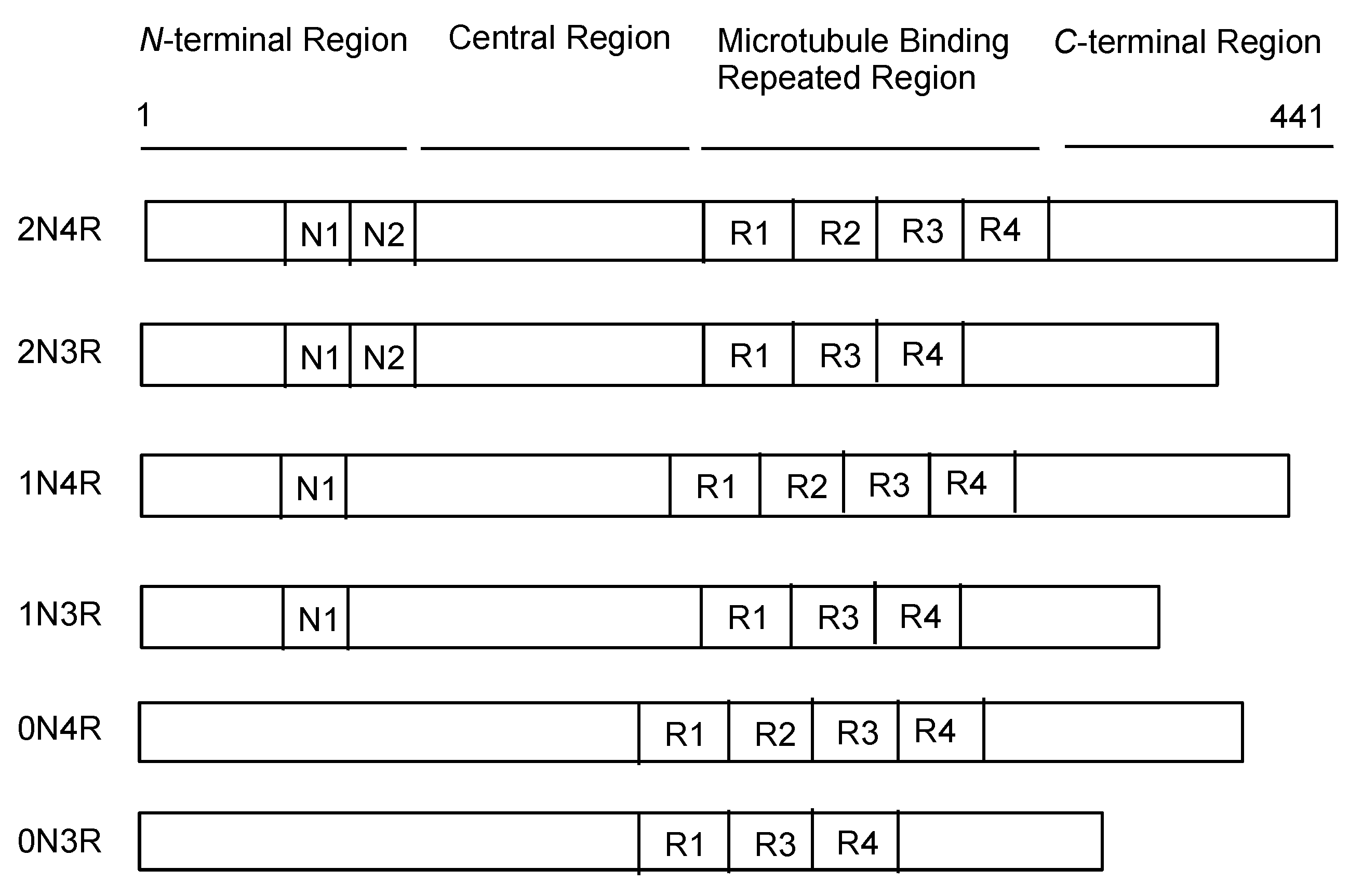
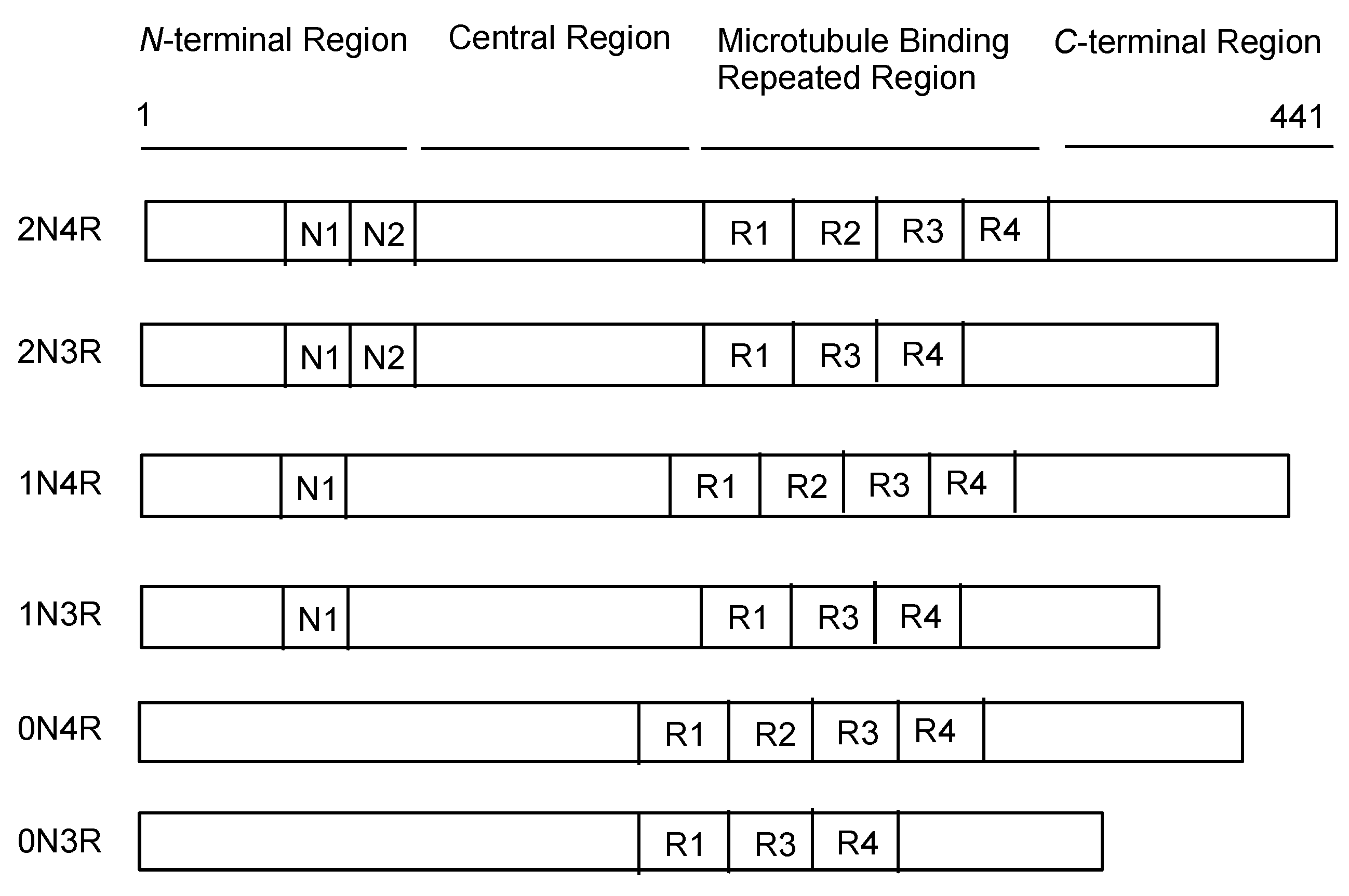
2.3. Tau Proteins
2.4. Aβ and Tau Pathologies
2.4.1. Temporal-Spatial Pathological Aβ and Tau Distribution in the Brain
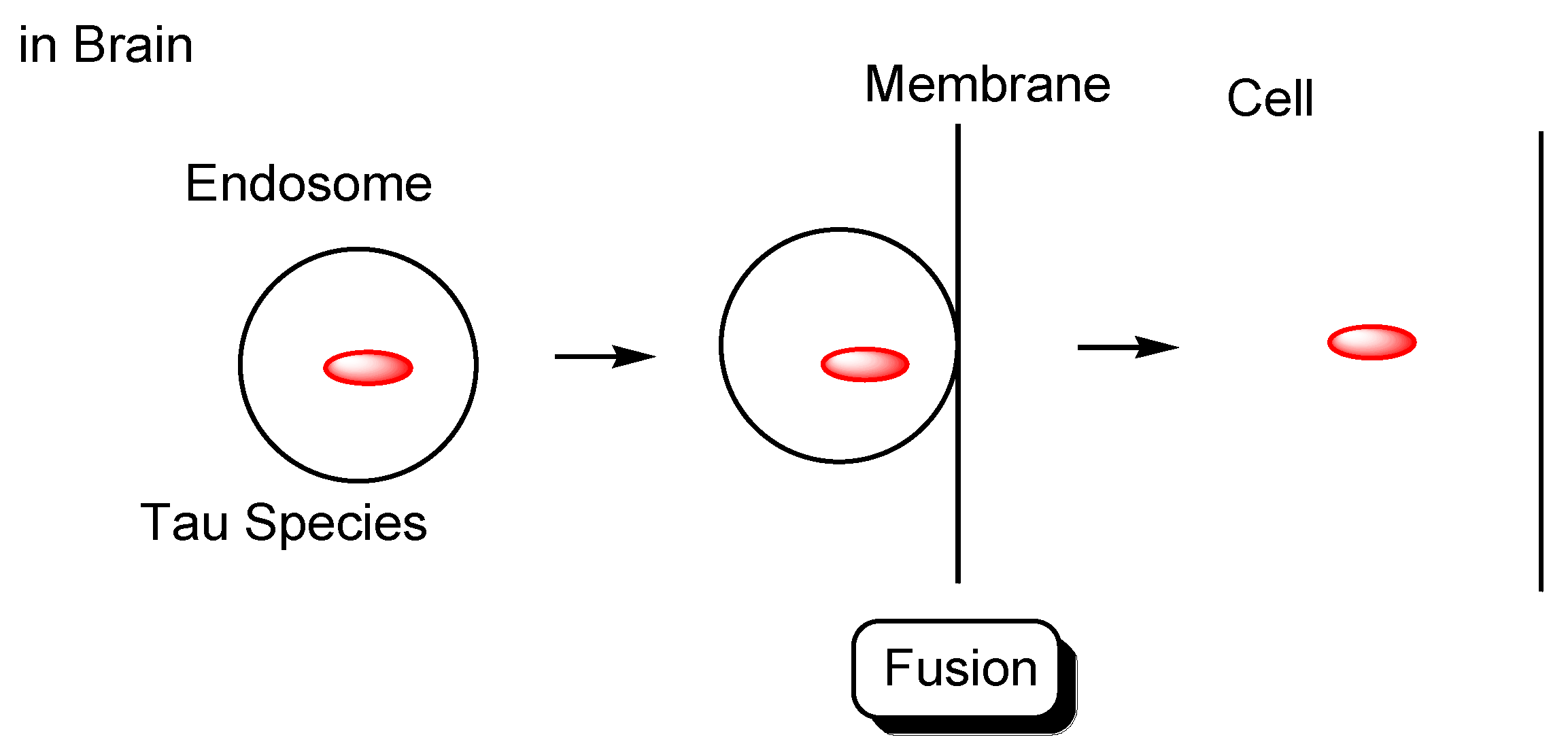
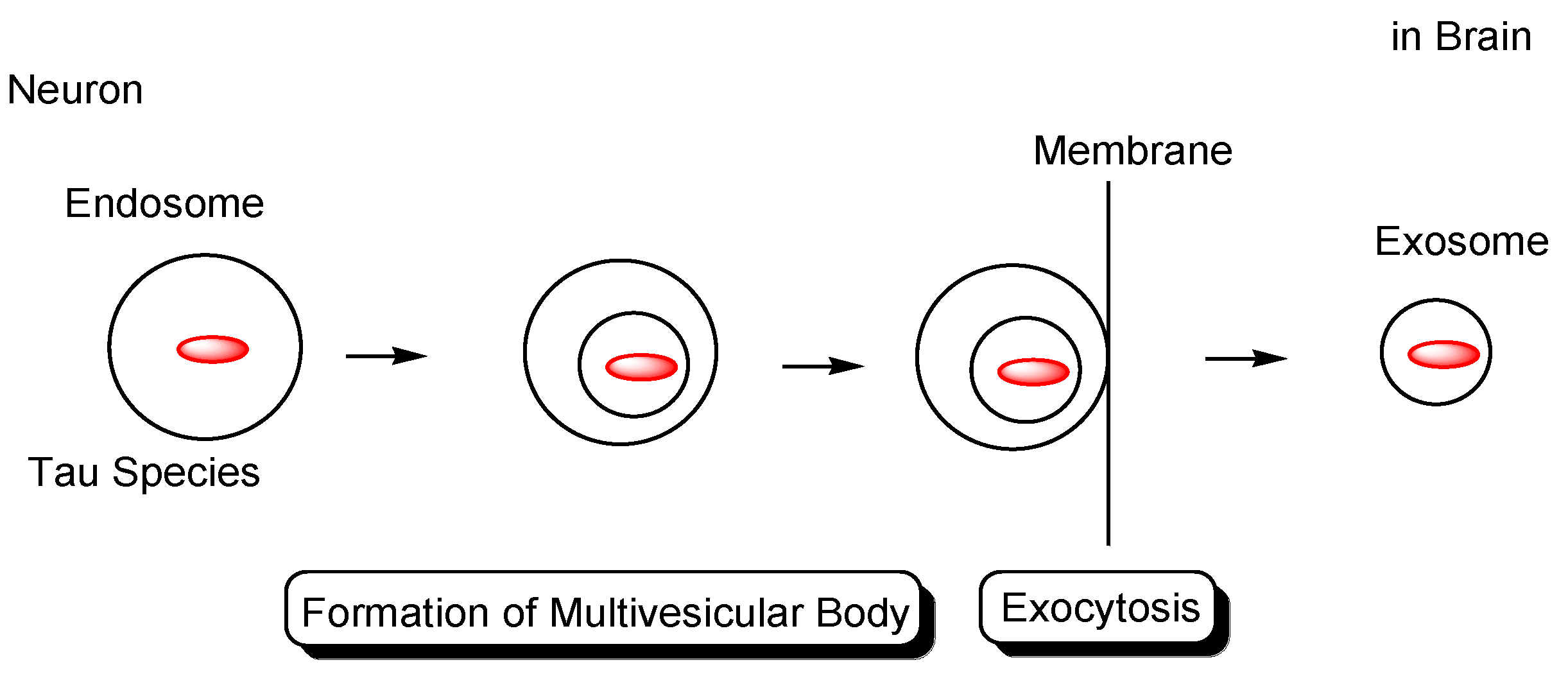
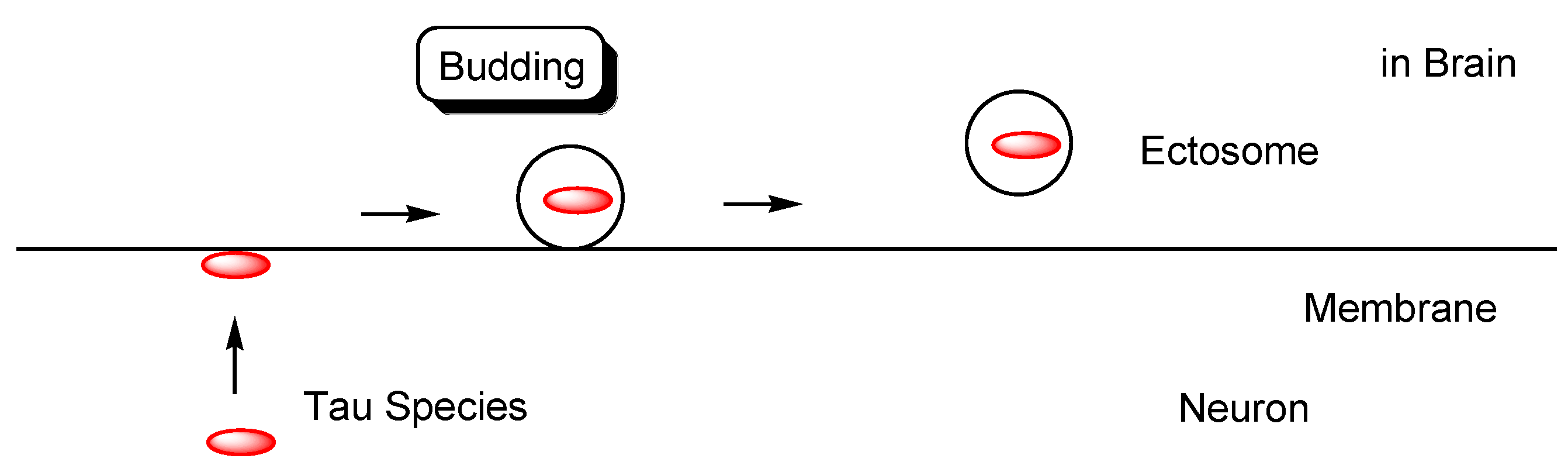
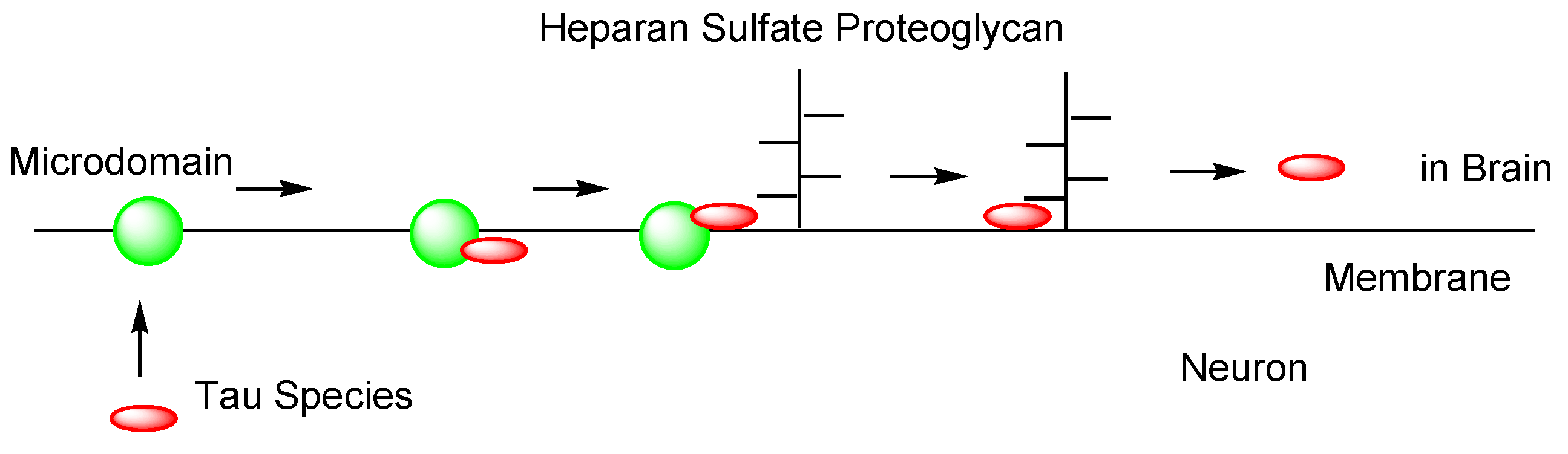
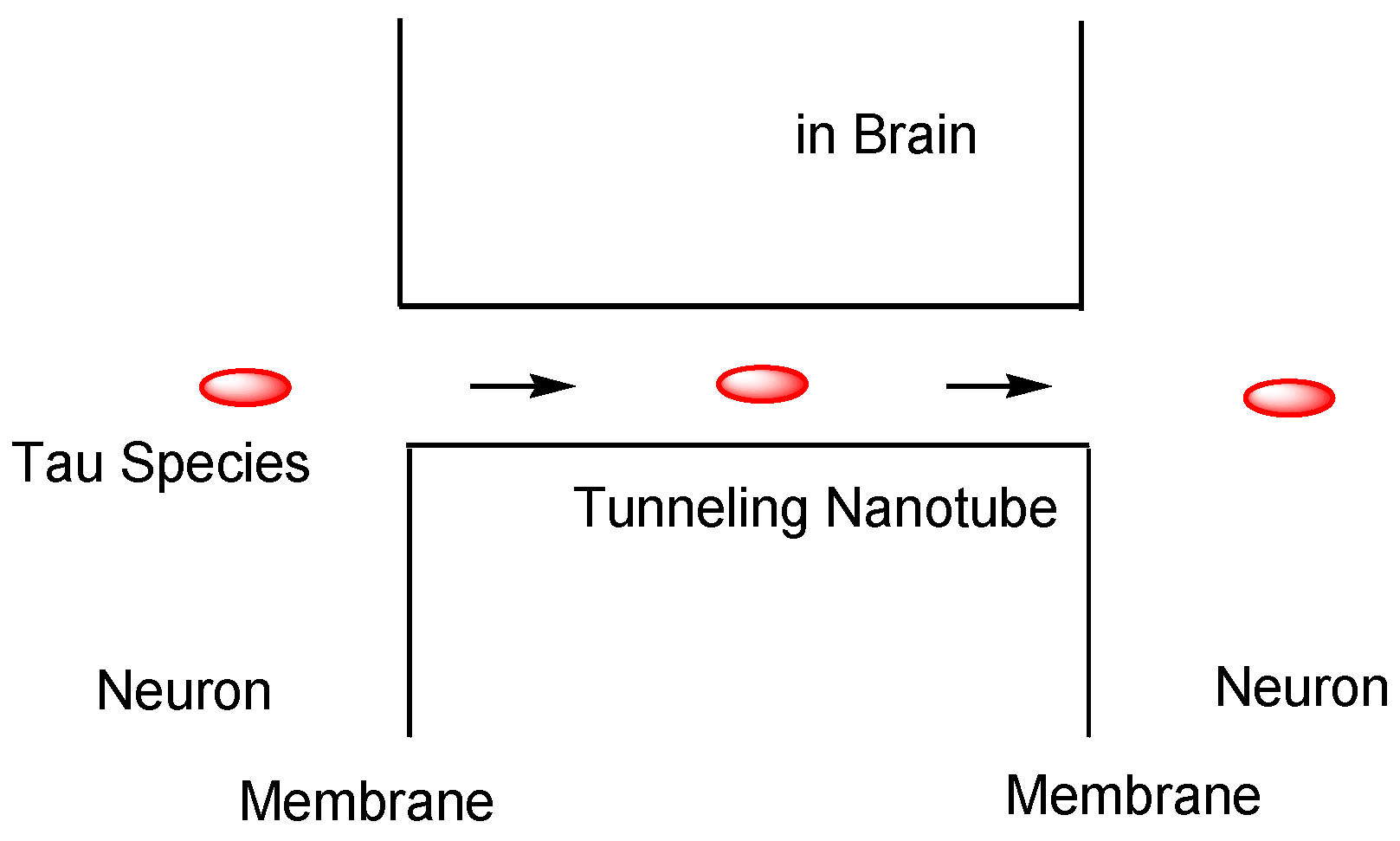
2.4.2. Cell-to-Cell Pathology Transmission
2.5. Tauopathies
2.6. Interactions between Aβ and Tau That Induce Neurotoxicity
2.6.1. Cyclin-Dependent Kinase 5 (CDK5) as a Matchmaker between Aβ and Tau
2.6.2. Fyn as a Matchmaker
2.6.3. Ca2+-Dependent Calmodulin Kinase IIα (CaMKIIα) as a Matchmaker
2.6.4. GSK3β as a Matchmaker
2.6.5. c-Jun N-Terminal Kinase (JNK) as a Matchmaker
2.7. Implementation of mAbs Targeting AD-Relevant Tau Species
2.7.1. Conformation-Selective Anti-Tau mAbs Block Cell-to-Cell Transmission of Tau Pathological Seeds
2.7.2. Clinical Trial for Anti-Tau mAbs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Administrated Drug | Formulation /Co-Administrated Drug | Disease | Sponsor | Phase | Study Start Date | Study Completion Date | ClinicalTrials.gov Identifier (Accessed on 25 December 2021) | Status | References |
|---|---|---|---|---|---|---|---|---|---|---|
| (i) | AADvac1 | An active tau vaccine (tau294–305) | AD | Axon Neuroscience SE | Phase 1 | January 2014 | December 2016 | NCT02031198 | Completed | [68] |
| (ii) | RG7345 | A humanized Ab against tau pS422 | Healthy Volunteer | Hoffmann-La Roche | Phase 1 | January 2015 | October 2015 | NCT02281786 | Completed | – |
| (iii) | RO7105705 (Semorinemab) | An IgG4 Ab against tau | Mild-to-Moderate AD | Genentech, Inc. | Phase 1 | June 2016 | June 2017 | NCT02820896 | Completed | – |
| (iv) | LY3303560 (Zagotenemab) | A humanized Ab against tau/Florbetapir F18 | AD | Eli Lilly and Company | Phase 1 | January 2017 | June 2019 | NCT03019536 | Completed | – |
| (v) | BIIB076 | A human IgG1 Ab against tau | AD | Biogen | Phase 1 | February 2017 | March 2020 | NCT03056729 | Completed | – |
| (vi) | Lu AF87908 | A humanized IgG1 Ab against tau | AD | H. Lundbeck A/S | Phase 1 | September 2019 | May 2021 | NCT04149860 | Recruiting | – |
| (vii) | AADvac1 | An active tau vaccine (tau294–305) | Mild AD | Axon Neuroscience SE | Phase 2 | March 2016 | June 2019 | NCT02579252 | Completed | [69] |
| (viii) | ACI-35 | An active tau vaccine (pSer396 and 404) | AD | AC Immune SA | Phase 1/2 | July 2019 | October 2023 | NCT04445831 | Recruiting | – |
| (ix) | E2814 | A humanized IgG1 Ab against tau | AD | Eisai Inc. | Phase 1/2 | June 2021 | April 2024 | NCT04971733 | Recruiting | – |
| (x) | RO7105705 (Semorinemab) | An IgG4 Ab against tau | Prodromal to Mild AD | Genentech, Inc. | Phase 2 | October 2017 | January 2021 | NCT03289143 | Completed | [70] |
| (xi) | BIIB092 (Gosuranemab) | A humanized IgG4 Ab against tau | AD | Biogen | Phase 2 | May 2018 | August 2021 | NCT03352557 | Active, not recruiting | – |
| (xii) | RO7105705 (Semorinemab) | An IgG4 Ab against tau | Moderate AD | Genentech, Inc. | Phase 2 | January 2019 | October 2023 | NCT03828747 | Active, not recruiting | – |
| (xiii) | ABBV-8E12 | A humanized IgG4 Ab against tau aggregates | Early AD | AbbVie | Phase 2 | March 2019 | July 2021 | NCT03712787 | Active, not recruiting | – |
| (xiv) | JNJ-63733657 | A humanized Ab against tau | Cognitive dysfunction | Janssen Research & Development, LLC | Phase 2 | January 2021 | March 2025 | NCT04619420 | Recruiting | – |
| (xv) | UCB0107 (Bepranemab) | A humanized IgG4 Ab against tau235–250 | AD | UCB Biopharma SRL | Phase 2 | June 2021 | November 2025 | NCT04867616 | Recruiting | – |
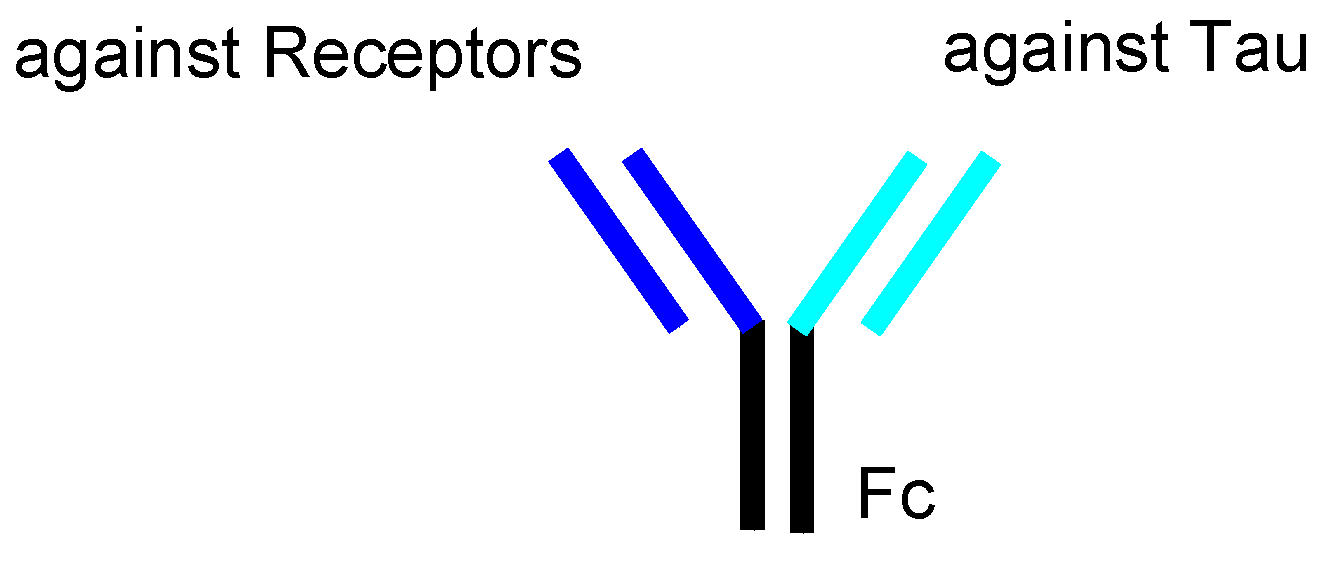
| (xvi) | intravenous mAbs | Bispecific mAbs against tau and TrR | AD | Under analysis in Tashima lab | – |
2.8. Possibility and Effective Use of mAbs Targeting AD-Relevant Tau Species
2.8.1. Lympatic and Immune System in the Brain
2.8.2. Transportation across the BBB
Role of FcRn of the Endothelium at the BBB
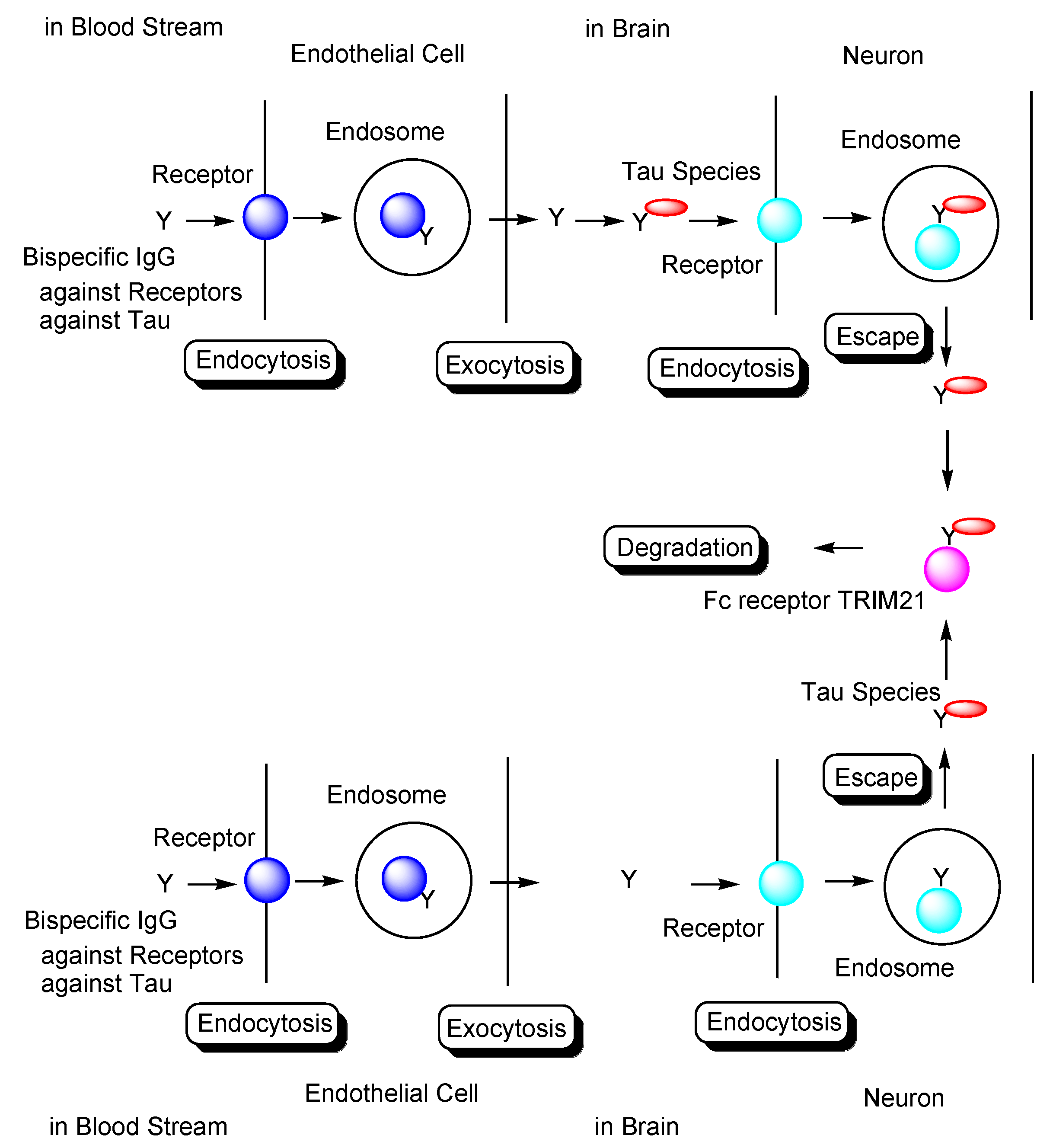
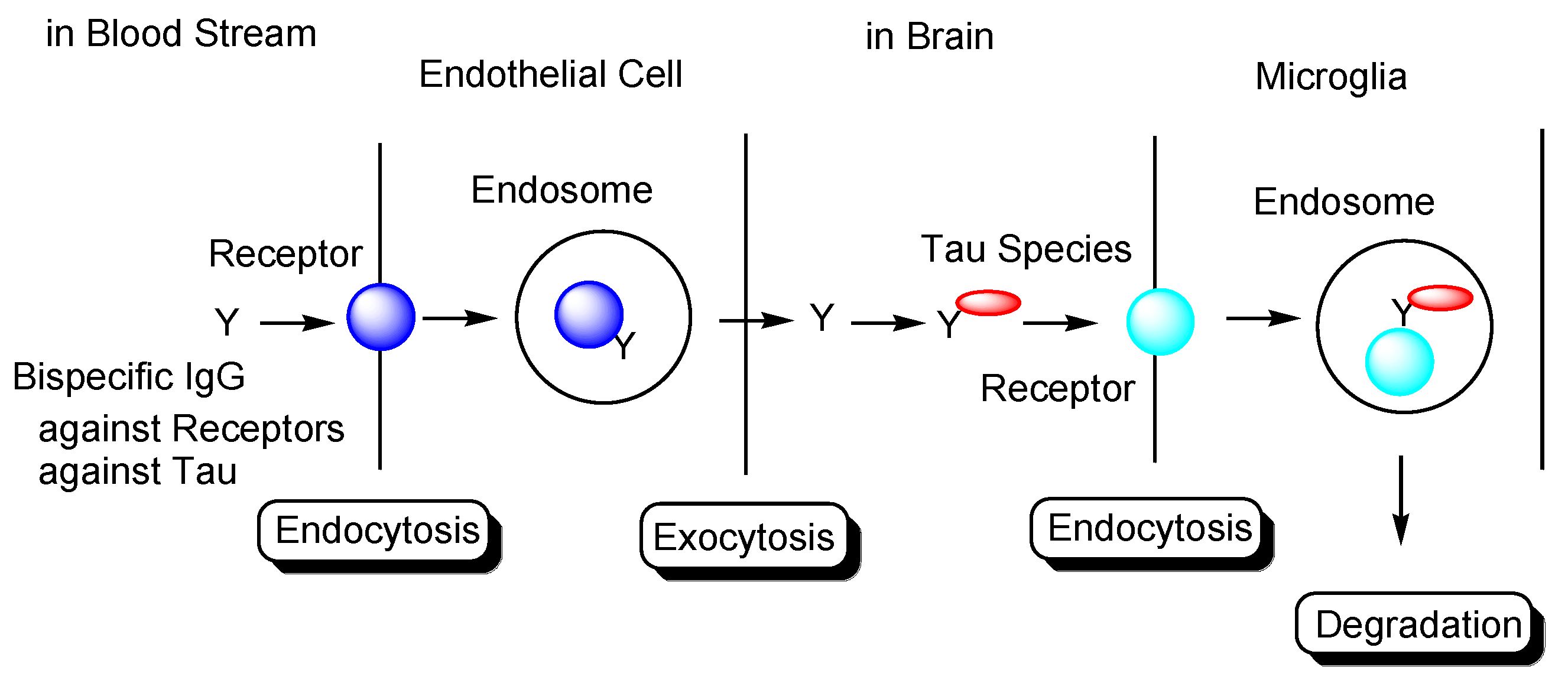
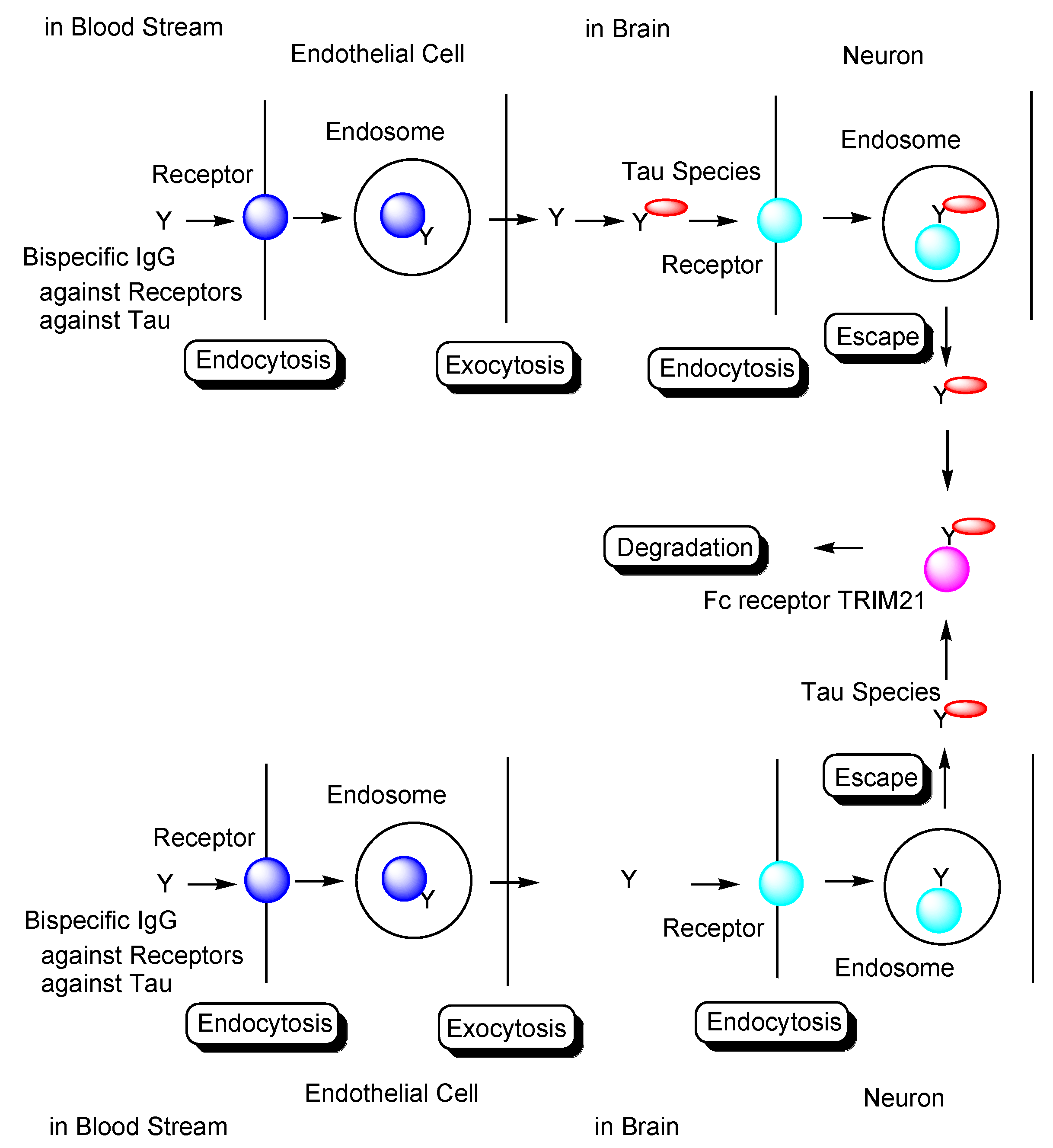
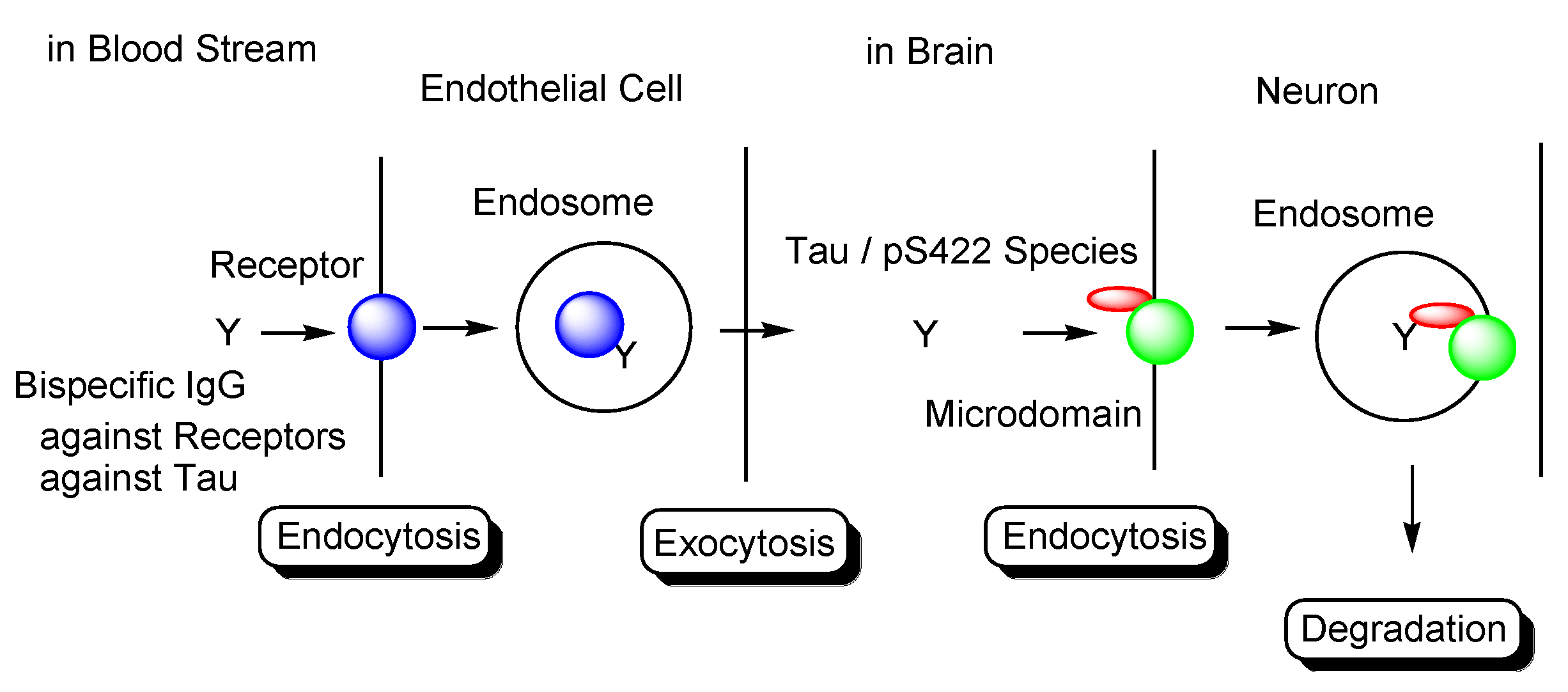
Transendothelium Based on Receptor-Mediated Transcytosis
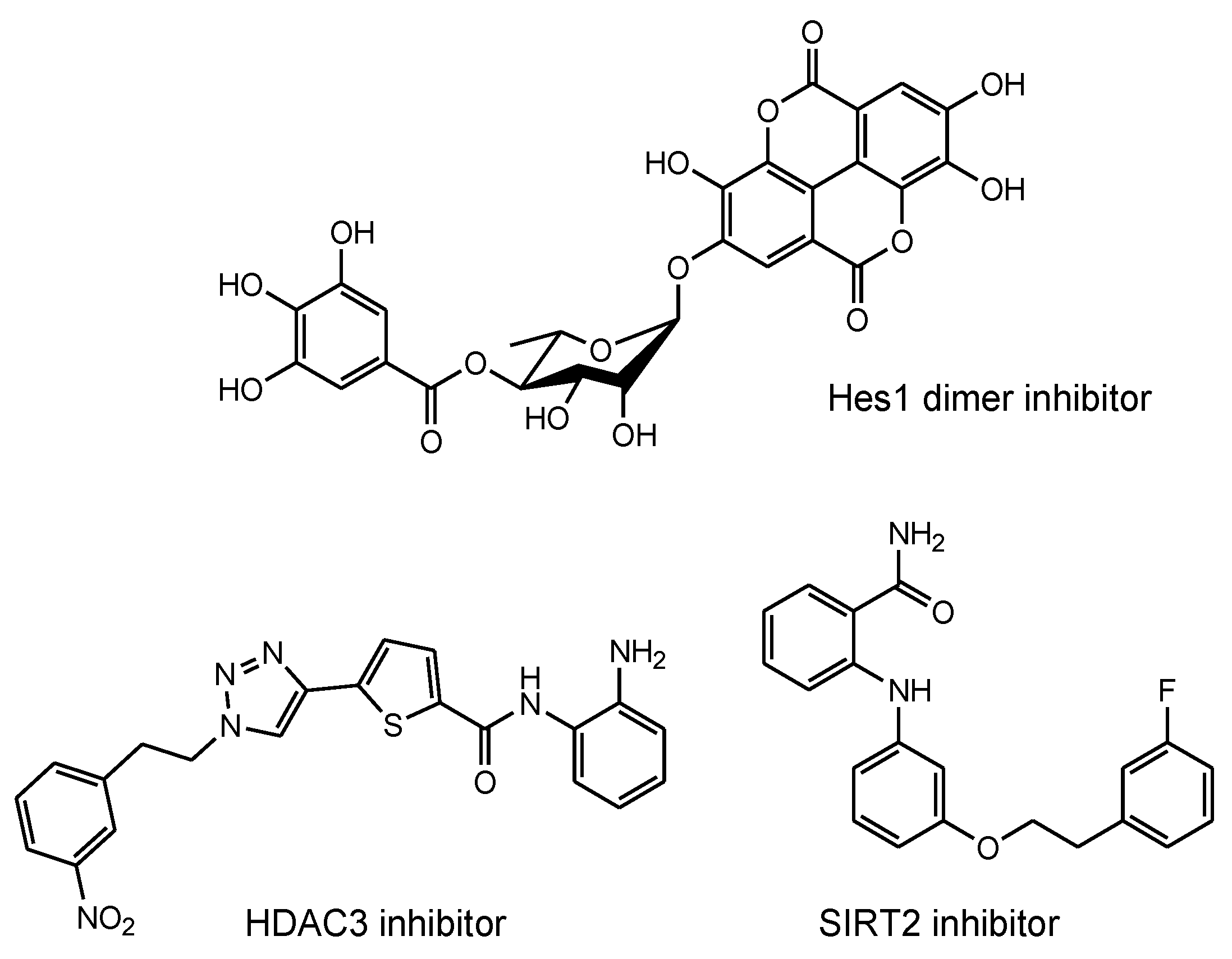
2.8.3. Plausible Design of mAbs to Clear Tau
2.8.4. Combination Therapies
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stimulus package. Nat. Med. 2018, 24, 247. [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2020, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T. Intriguing possibilities and beneficial aspects of transporter-conscious drug design. Bioorg. Med. Chem. 2015, 23, 4119–4131. [Google Scholar] [CrossRef]
- Tashima, T. Intelligent substance delivery into cells using cell-penetrating peptides. Bioorg. Med. Chem. Lett. 2017, 27, 121–130. [Google Scholar] [CrossRef]
- Tashima, T. Effective cancer therapy based on selective drug delivery into cells across their membrane using receptor-mediated endocytosis. Bioorg. Med. Chem. Lett. 2018, 28, 3015–3024. [Google Scholar] [CrossRef]
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Tashima, T. Shortcut Approaches to Substance Delivery into the Brain Based on Intranasal Administration Using Nanodelivery Strategies for Insulin. Molecules 2020, 25, 5188. [Google Scholar] [CrossRef]
- Tashima, T. Delivery of Orally Administered Digestible Antibodies Using Nanoparticles. Int. J. Mol. Sci. 2021, 22, 3349. [Google Scholar] [CrossRef] [PubMed]
- Joseph Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Weuve, J. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- WHO. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 10 November 2021).
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Maekawa, S.; Kimura, T.; Takashima, A.; Tomiyama, T.; Mori, H. Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice. Acta Neuropathol. 2014, 127, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Maarouf, C.L.; Daugs, I.D.; Kokjohn, T.A.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Masliah, E.; Nicoll, J.A.R.; Sabbagh, M.N.; Beach, T.G.; et al. The biochemical aftermath of anti-amyloid immunotherapy. Mol. Neurodegener. 2010, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Brunello, C.A.; Merezhko, M.; Uronen, R.-L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol. Life Sci. 2020, 77, 1721–1744. [Google Scholar] [CrossRef] [Green Version]
- Kopke, E.; Tung, Y.-C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; De Ture, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 510–514. [Google Scholar] [CrossRef] [Green Version]
- Bittar, A.; Bhatt, N.; Kayed, R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol. Dis. 2020, 134, 104707. [Google Scholar] [CrossRef] [PubMed]
- Kuzuhara, S.; Ihara, Y.; Toyokura, Y.; Shimada, H. A semiquantitative study on Alzheimer neurofibrillary tangles demonstrated immunohistochemically with anti-tau antibodies, in the brains of non-demented and demented old people. No Shinkei = Brain Nerve 1989, 41, 465–470. [Google Scholar]
- Braak, H.; Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef]
- Tarutani, A.; Miyata, H.; Nonaka, T.; Hasegawa, K.; Yoshida, M.; Saito, Y.; Murayama, S.; Robinson, A.C.; Mann, D.M.A.; Tomita, T.; et al. Human tauopathy-derived tau strains determine the substrates recruited for templated amplification. Brain 2021, 144, 2333–2348. [Google Scholar] [CrossRef] [PubMed]
- Pernègre, C.; Duquette, A.; Leclerc, N. Tau Secretion: Good and Bad for Neurons. Front. Neurosci. 2019, 13, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front. Mol. Neurosci. 2020, 13, 586731. [Google Scholar] [CrossRef] [PubMed]
- Barini, E.; Plotzky, G.; Mordashova, Y.; Hoppe, J.; Rodriguez-Correa, E.; Julier, S.; LePrieult, F.; Mairhofer, I.; Mezler, M.; Biesinger, S.; et al. Tau in the brain interstitial fluid is fragmented and seeding–competent. Neurobiol. Aging 2021, 109, 64–77. [Google Scholar] [CrossRef]
- Evans, L.D.; Wassmer, T.; Fraser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlapping but Distinct Pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef] [Green Version]
- Puangmalai, N.; Bhatt, N.; Montalbano, M.; Sengupta, U.; Gaikwad, S.; Ventura, F.; McAllen, S.; Ellsworth, A.; Garcia, S.; Kayed, R. Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer’s disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis. 2020, 11, 314. [Google Scholar] [CrossRef]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef]
- Zhu, H.-L.; Fernández, C.; Fan, J.-B.; Shewmaker, F.; Chen, J.; Minton, A.P.; Liang, Y. Quantitative characterization of heparin binding to Tau protein: Implication for inducer-mediated Tau filament formation. J. Biol. Chem. 2010, 5, 3592–3599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giamblanco, N.; Fichou, Y.; Janot, J.-M.; Balanzat, E.; Han, S.; Balme, S. Mechanisms of Heparin-Induced Tau Aggregation Revealed by a Single Nanopore. ACS Sens. 2020, 24, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Morozova, V.; Cohen, L.S.; Makki, A.E.-H.; Shur, A.; Pilar, G.; Idrissi, A.E.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell. Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [Green Version]
- La-Rocque, S.D.; Moretto, E.; Butnaru, I.; Schiavo, G. Knockin’ on heaven’s door: Molecular mechanisms of neuronal tau uptake. J. Neurochem. 2021, 156, 563–588. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.-C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.-L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Debaisieux, S.; Rayne, F.; Yezid, H.; Beaumelle, B. The Ins and Outs of HIV-1 Tat. Traffic 2012, 13, 355–363. [Google Scholar] [CrossRef]
- Steringer, J.P.; Nickel, W. A direct gateway into the extracellular space: Unconventional secretion of FGF2 through self-sustained plasma membrane pore. Cell Dev. Biol. 2018, 83, 3–7. [Google Scholar] [CrossRef]
- Zurzolo, C. Tunneling nanotubes: Reshaping connectivity. Curr. Opin. Cell Biol. 2021, 71, 139–147. [Google Scholar] [CrossRef]
- Gerson, J.E.; Sengupta, U.; Lasagna-Reeves, C.A.; Guerrero-Muñoz, M.J.; Troncoso, J.; Kayed, R. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol. Commun. 2014, 2, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampuscenko, K.; Morkuniene, R.; Krasauskas, L.; Smirnovas, V.; Tomita, T.; Borutaite, V. Distinct Neurotoxic Effects of Extracellular Tau Species in Primary Neuronal-Glial Cultures. Mol. Neurobiol. 2021, 58, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Rodríguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudy, C.C.; Hunsberger, H.C.; Weitzner, D.S.; Reed, M.N. The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology of Alzheimer’s Disease. Aging Dis. 2015, 6, 131–148. [Google Scholar] [CrossRef] [Green Version]
- Tovar, K.R.; Westbrook, G.L. The Incorporation of NMDA Receptors with a Distinct Subunit Composition at Nascent Hippocampal Synapses In Vitro. J. Neurosci. 1999, 19, 4180–4188. [Google Scholar] [CrossRef] [Green Version]
- Angulo, M.C.; Kozlov, A.S.; Charpak, S.; Audinat, E. Glutamate Released from Glial Cells Synchronizes Neuronal Activity in the Hippocampus. J. Neurosci. 2004, 2, 6920–6927. [Google Scholar] [CrossRef] [Green Version]
- Tovar, K.R.; McGinley, M.J.; Westbrook, G.L. Triheteromeric NMDA Receptors at Hippocampal Synapses. J. Neurosci. 2013, 33, 9150–9160. [Google Scholar] [CrossRef]
- Castro-Alvarez, J.F.; Uribe-Arias, S.A.; Mejía-Raigosa, D.; Cardona-Gómez, G.P. Cyclin-dependent kinase 5, a node protein in diminished tauopathy: A systems biology approach. Front. Aging Neurosci. 2014, 6, 232. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Nuvolone, M.; Lesné, S.E. The Complex PrPc-Fyn Couples Human Oligomeric Aβ with Pathological Tau Changes in Alzheimer’s Disease. J. Neurosci. 2012, 3, 16857–16871. [Google Scholar] [CrossRef] [Green Version]
- Goebel-Goody, S.M.; Davies, K.D.; Linger, R.M.A.; Freund, R.K.; Browning, M.D. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience 2009, 158, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; Eersel, J.V.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, S.; Vaz-Silva, J.; Pinto, V.; Dalla, C.; Kokras, N.; Bedenk, B.; Natalie, M.; Czisch, M.; Almeida, O.F.X.; Sousa, N. Tau protein is essential for stress-induced brain pathology. Proc. Natl. Acad. Sci. USA 2016, 113, E3755–E3763. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; Stein, L.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 41. [Google Scholar] [CrossRef]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Götz, J.; Buisson, A.; Lesné, S.E. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 2017, 10, eaal2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-R.; Liu, R.-T. The Toxicity and Polymorphism of β-Amyloid Oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 2020, 12, eaay1769. [Google Scholar] [CrossRef]
- Wu, H.; Wei, S.; Huang, Y.; Chen, L.; Wang, Y.; Wu, X.; Zhang, Z.; Pei, Y.; Wang, D. Aβ monomer induces phosphorylation of Tau at Ser-214 through β2AR-PKA-JNK signaling pathway. FASEB J. 2020, 34, 5092–5105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbons, G.S.; Kim, S.-J.; Wu, Q.; Riddle, D.M.; Leight, S.N.; Changolkar, L.; Xu, H.; Meymand, E.S.; O’Reilly, M.; Zhang, B.; et al. Conformation-selective tau monoclonal antibodies inhibit tau pathology in primary neurons and a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 64. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.R.; Falsig, J.; Stavenhagen, J.B.; Christensen, S.; Kartberg, F.; Rosenqvist, N.; Finsen, B.; Pedersen, J.T. Antibody-mediated clearance of tau in primary mouse microglial cultures requires Fcγ-receptor binding and functional lysosomes. Sci. Rep. 2019, 9, 4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, J.P.; Stavenhagen, J.B.; Teeling, J.L. New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer’s Disease. Front. Neurosci. 2014, 8, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilkova, M.; Nolle, A.; Kovacech, B.; Kontsekova, E.; Weisova, P.; Filipcik, P.; Skrabana, R.; Prcina, M.; Hromadka, T.; Cehlar, O.; et al. Humanized tau antibodies promote tau uptake by human microglia without any increase of inflammation. Acta Neuropathol. Commun. 2020, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Lubica Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P.; et al. Fundamant: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 24, 108. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Kovacech, B.; Katina, S.; Schmidt, R.; Scheltens, P.; Kontsekova, E.; Ropele, S.; Fialova, L.; Kramberger, M.; Paulenka-Ivanovova, N.; et al. Adamant: A placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nat. Aging 2021, 1, 521–534. [Google Scholar] [CrossRef]
- Mullard, A. Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat. Rev. Drug Discov. 2021, 20, 3–5. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.; Spires-Jones, T.L. Sleep well to slow Alzheimer’s progression? Science 2019, 363, 813–814. [Google Scholar] [CrossRef]
- Luo, W.; Liu, W.; Hu, X.; Hanna, M.; Caravaca, A.; Paul, S.M. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci. Rep. 2015, 5, 11161. [Google Scholar] [CrossRef] [Green Version]
- Collin, L.; Bohrmann, B.; Göpfert, U.; Oroszlan-Szovik, K.; Ozmen, L.; Grüninger, F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain 2014, 137, 2834–2846. [Google Scholar] [CrossRef] [Green Version]
- Congdon, E.E.; Gu, J.; Sait, H.B.R.; Sigurdsson, E.M. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγ receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 2013, 288, 35452–35465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwan, W.A.; Falcon, B.; Vaysburd, M.; Clift, D.; Oblak, A.L.; Ghetti, B.; Goedert, M.; James, L.C. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 74–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Caram-Salas, N.; Boileau, E.; Farrington, G.K.; Garber, E.; Brunette, E.; Abulrob, A.; Stanimirovic, D. In vitro and in vivo methods for assessing FcRn-mediated reverse transcytosis across the blood-brain barrier. Methods Mol. Biol. 2011, 763, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, J.M.; Shusta, E.V. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haqqani, A.S.; Thom, G.; Burrell, M.; Delaney, C.E.; Brunette, E.; Baumann, E.; Sodja, C.; Jezierski, A.; Webster, C.; Stanimirovic, D.B. Intracellular sorting and transcytosis of the rat transferrin receptor antibody OX26 across the blood-brain barrier in vitro is dependent on its binding affinity. J. Neurochem. 2018, 146, 735–752. [Google Scholar] [CrossRef] [Green Version]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef]
- Lindvall, O.; Rehncrona, S.; Gustavii, B.; Brundin, P.; Astedt, B.; Widner, H.; Lindholm, T.; Björklund, A.; Leenders, K.L.; Rothwell, J.C.; et al. Fetal dopamine-rich mesencephalic grafts in Parkinson’s disease. Lancet 1988, 2, 1483–1484. [Google Scholar] [CrossRef]
- Weiss, M.L.; Medicetty, S.; Bledsoe, A.R.; Rachakatla, R.S.; Choi, M.; Merchav, S.; Luo, Y.; Rao, M.S.; Velagaleti, G.; Troyer, D. Human umbilical cord matrix stem cells: Preliminary characterization and effect of transplantation in a rodent model of Parkinson’s disease. Stem Cells 2006, 24, 781–792. [Google Scholar] [CrossRef]
- Liesveld, J.L.; Sharma, N.; Aljitawi, O.S. Stem cell homing: From physiology to therapeutics. Stem Cells 2020, 38, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
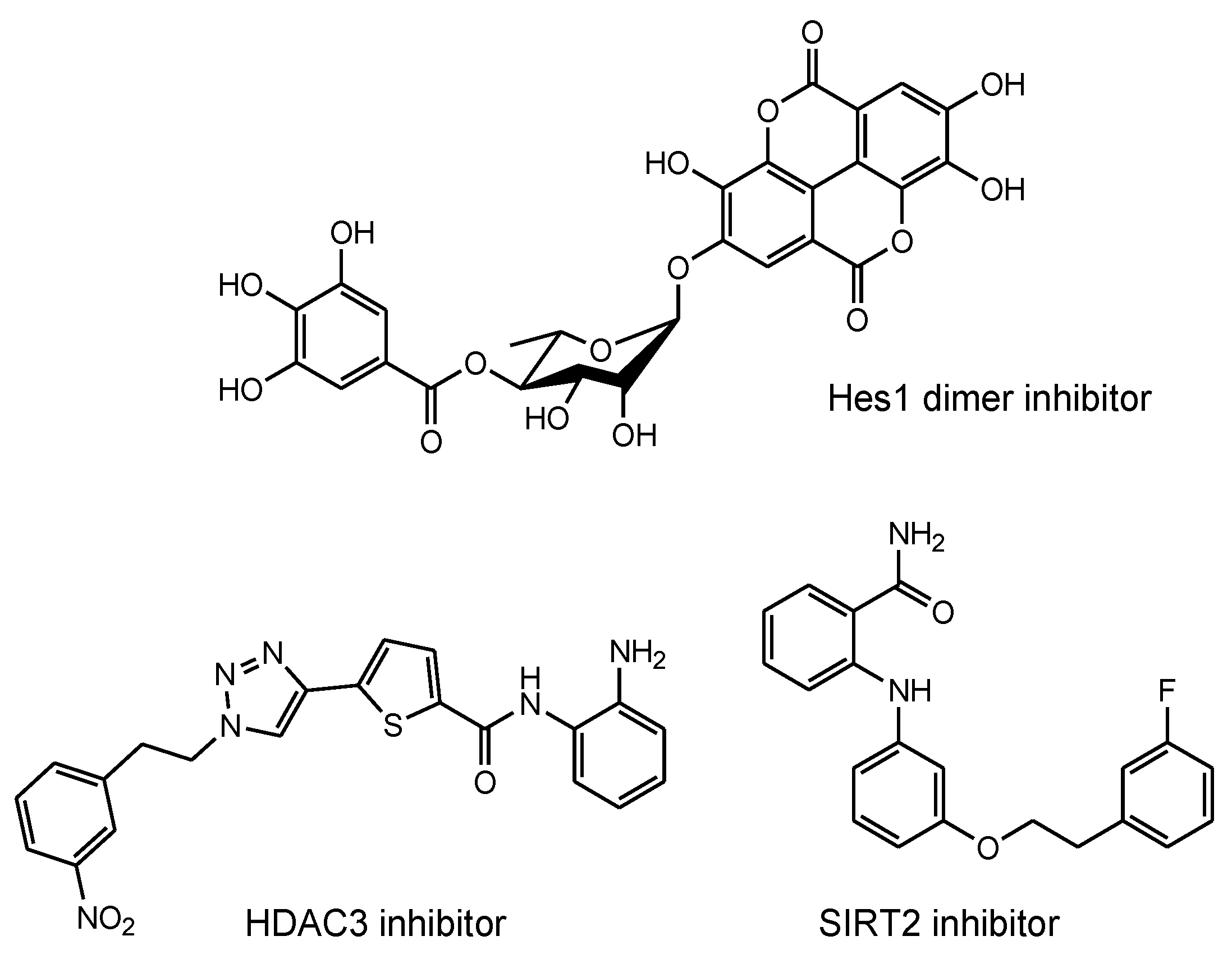
- Arai, M.A. Target Protein-Oriented Isolations for Bioactive Natural Products. Chem. Pharm. Bull. 2021, 69, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kasuya, Y.; Itoh, Y.; Ota, Y.; Zhan, P.; Asamitsu, K.; Nakagawa, H.; Okamoto, T.; Miyata, N. Identification of Highly Selective and Potent Histone Deacetylase 3 Inhibitors Using Click Chemistry-Based Combinatorial Fragment Assembly. PLoS ONE 2013, 14, e68669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Khan, M.N.A.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashima, T. Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics 2022, 14, 411. https://doi.org/10.3390/pharmaceutics14020411
Tashima T. Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics. 2022; 14(2):411. https://doi.org/10.3390/pharmaceutics14020411
Chicago/Turabian StyleTashima, Toshihiko. 2022. "Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis" Pharmaceutics 14, no. 2: 411. https://doi.org/10.3390/pharmaceutics14020411
APA StyleTashima, T. (2022). Delivery of Intravenously Administered Antibodies Targeting Alzheimer’s Disease-Relevant Tau Species into the Brain Based on Receptor-Mediated Transcytosis. Pharmaceutics, 14(2), 411. https://doi.org/10.3390/pharmaceutics14020411