

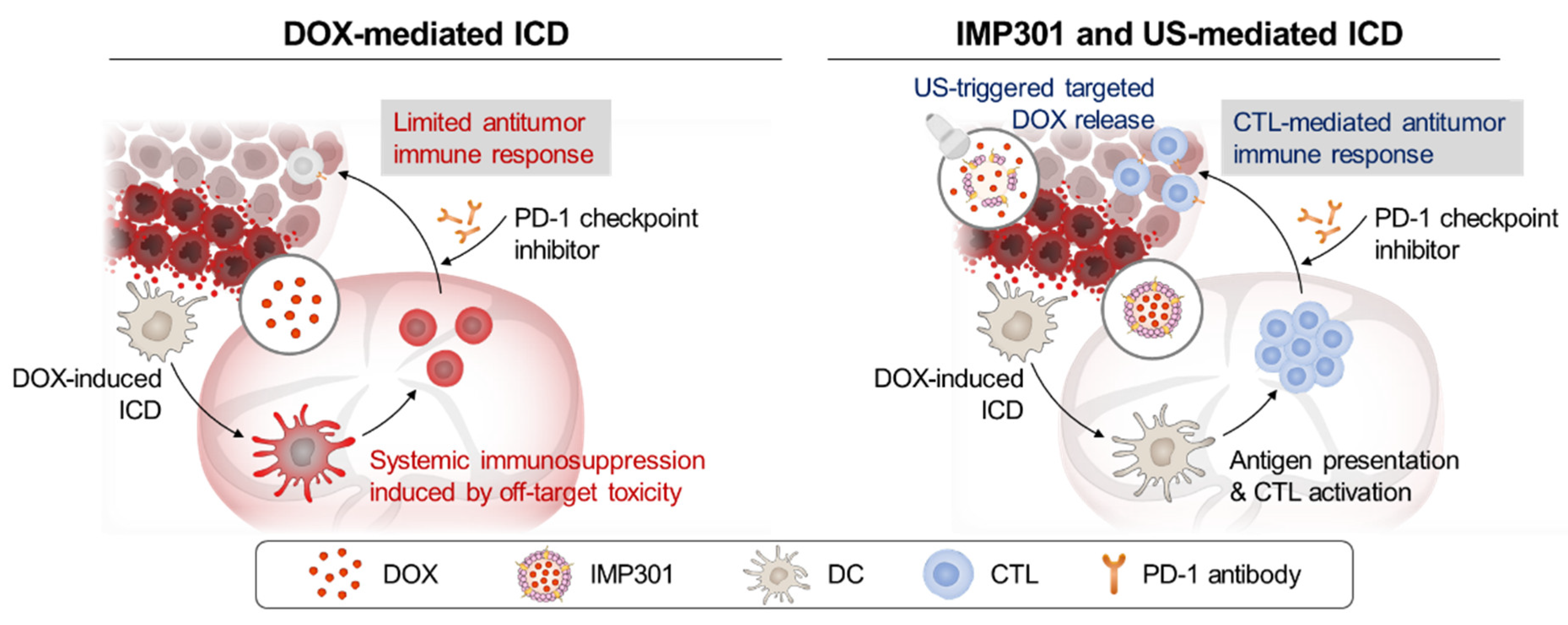
Evading Doxorubicin-Induced Systemic Immunosuppression Using Ultrasound-Responsive Liposomes Combined with Focused Ultrasound

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of IMP301
2.3. In Vitro Release Behavior of IMP301
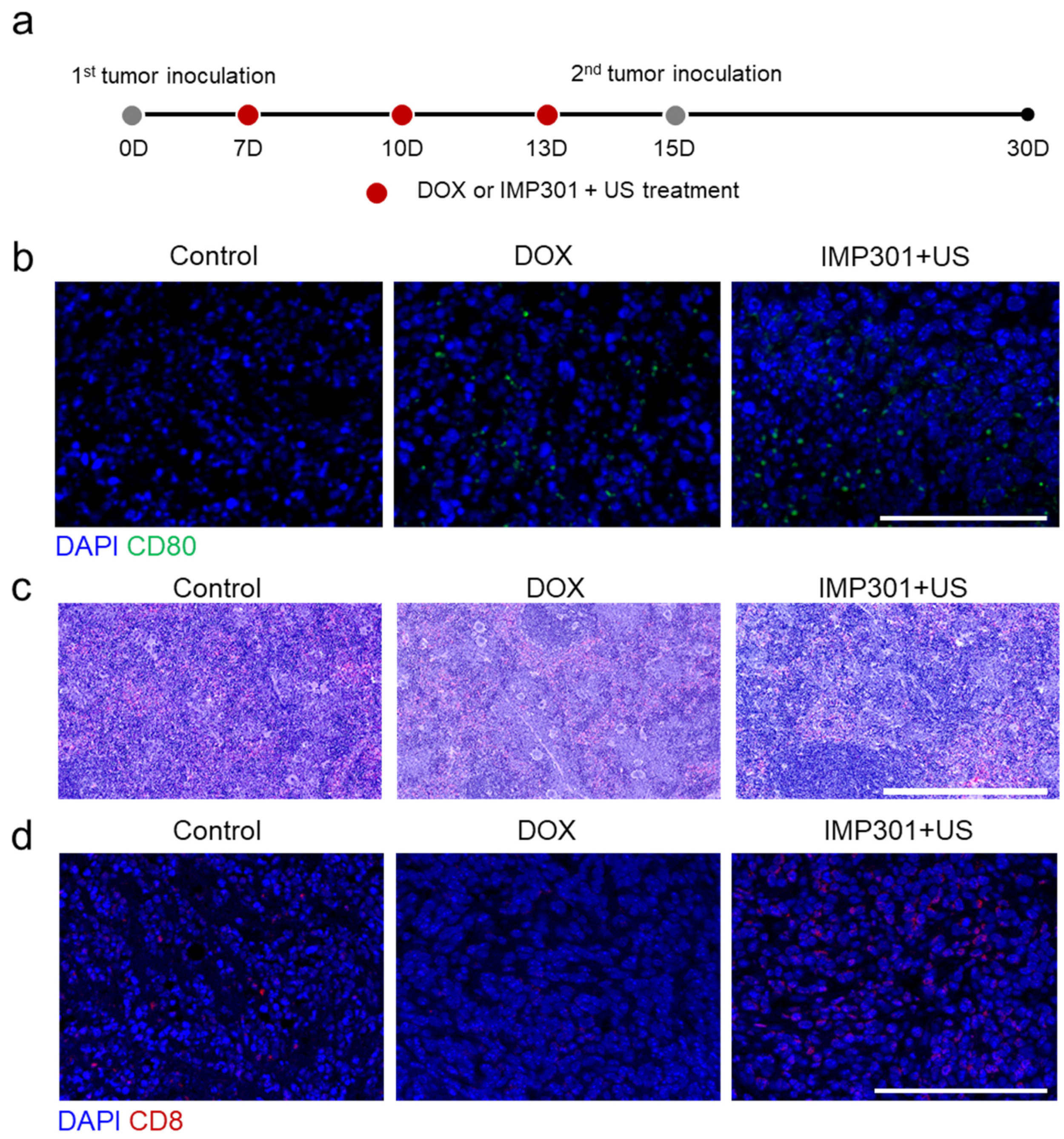
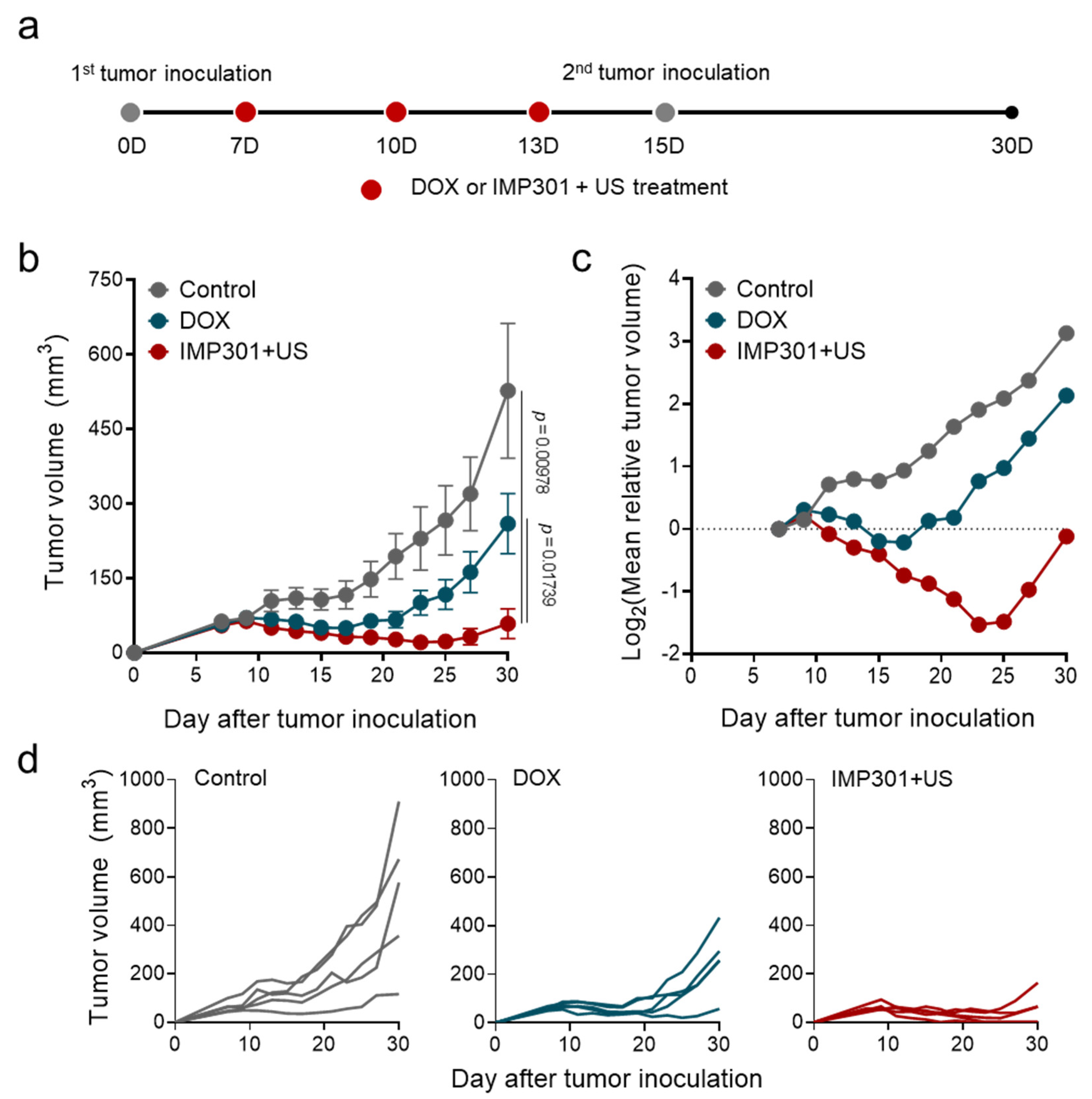
2.4. Animal Models
2.5. In Vivo Antitumor Therapy
2.6. Immunohistochemistry
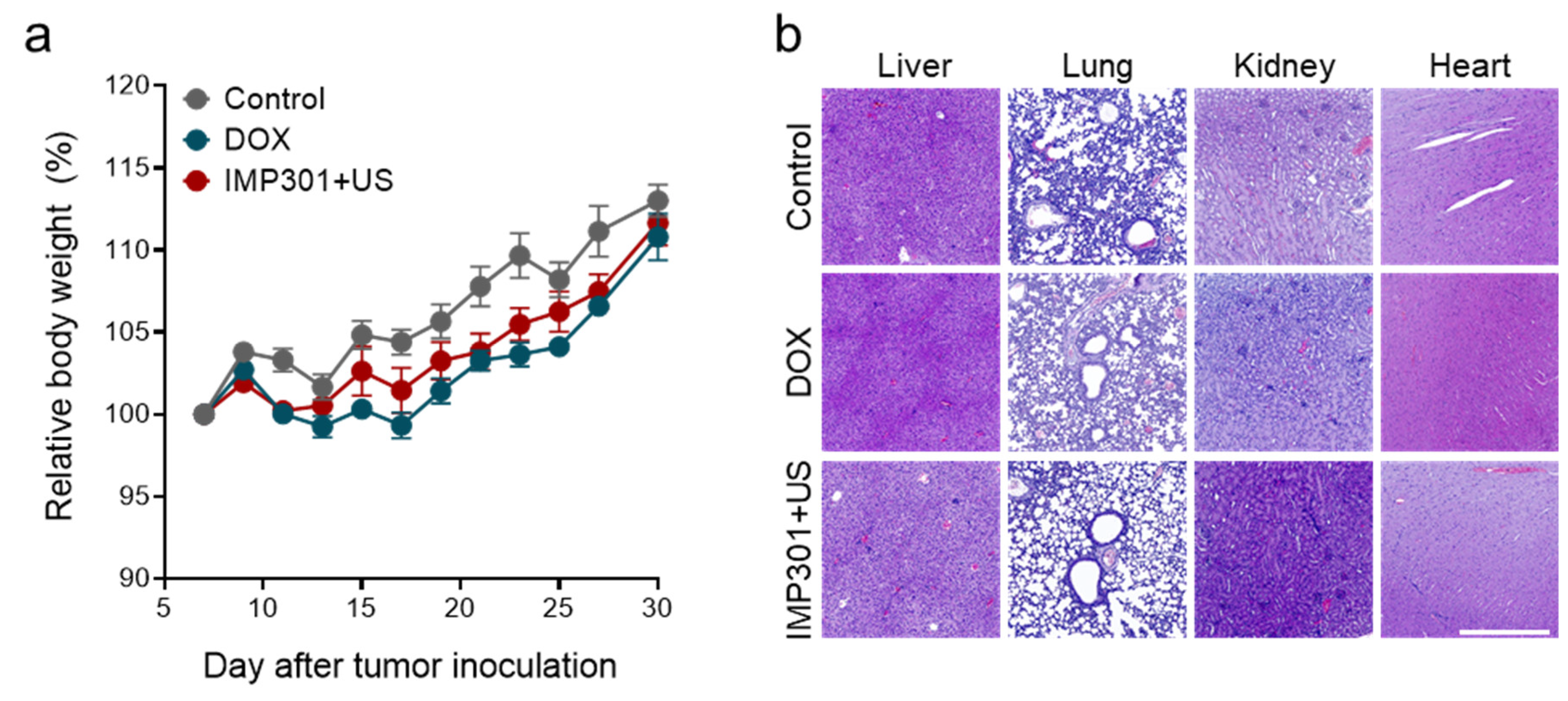
2.7. Ex Vivo Histology
2.8. In Vivo Anti-Tumor Efficacy Test with Immune Checkpoint Blockade
2.9. Statistics
3. Results and Discussion
3.1. In Vivo Immunogenicity and Systemic Toxicity of the Combination of IMP301 and US Treatment
3.2. In Vivo Therapeutic Efficacy of the Combination of IMP301 and US in 4T1 Tumor-Bearing Mice
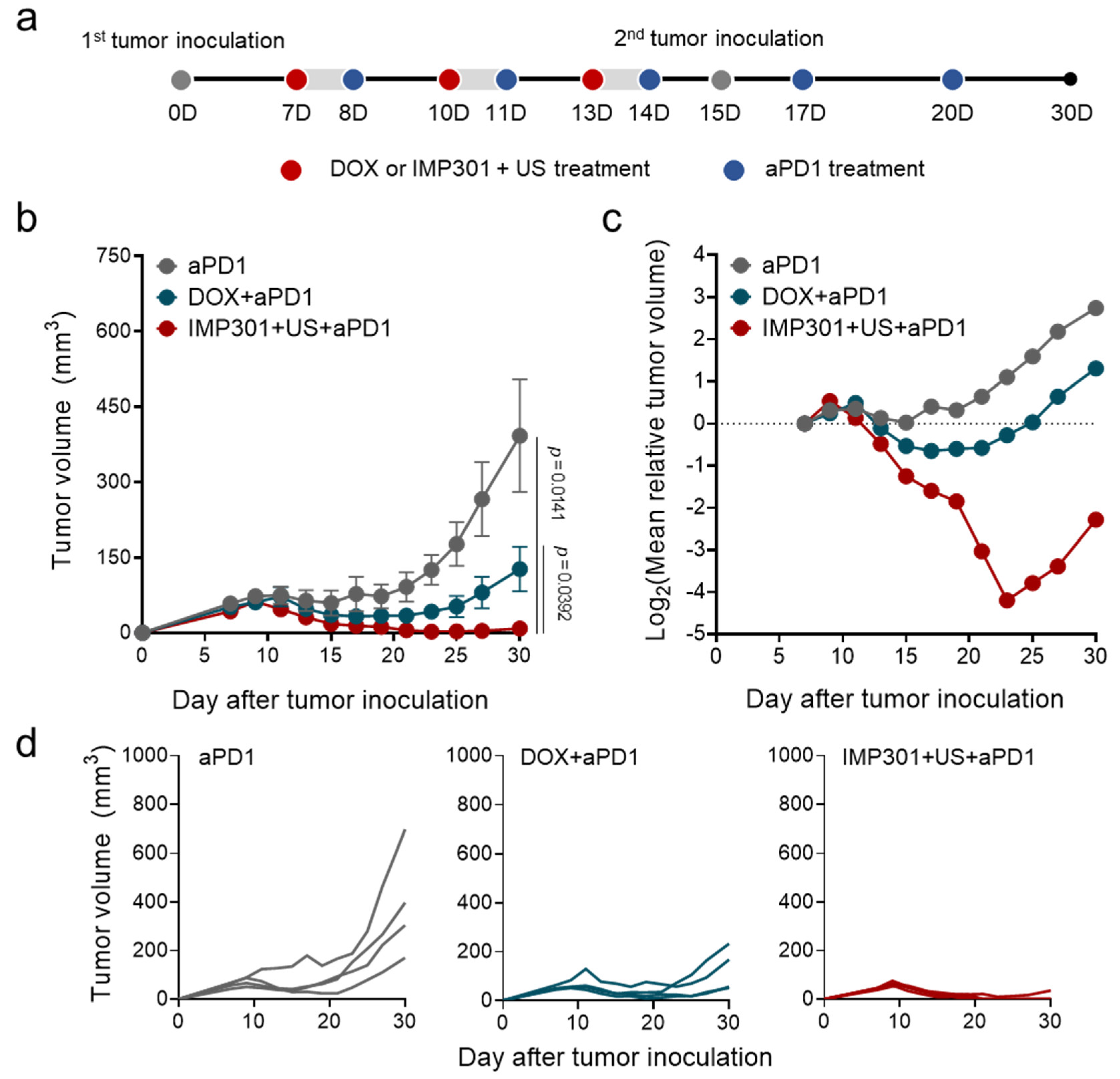
3.3. In Vivo Therapeutic Efficacy of the Combination of aPD1, IMP301, and US Treatment in 4T1 Tumor-Bearing Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Casares, N.; Pequignot, M.O.; Chaput, N.; Albert, M.L.; Kroemer, G. Immune response against dying tumor cells. Adv. Immunol. 2004, 84, 131–179. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Rufo, N.; Garg, A.D.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Q.; Qi, H.; Wang, C.; Wang, C.; Zhang, J.; Dong, L. Doxorubicin-Induced Systemic Inflammation Is Driven by Upregulation of Toll-Like Receptor TLR4 and Endotoxin Leakage. Cancer Res. 2016, 76, 6631–6642. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [PubMed]
- Riad, A.; Bien, S.; Gratz, M.; Escher, F.; Westermann, D.; Heimesaat, M.M.; Bereswill, S.; Krieg, T.; Felix, S.B.; Schultheiss, H.P.; et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur. J. Heart Fail. 2008, 10, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.D.; Lyon, P.C.; Mannaris, C.; Folkes, L.K.; Stratford, M.; Campo, L.; Chung, D.Y.F.; Scott, S.; Anderson, M.; Goldin, R.; et al. Focused Ultrasound Hyperthermia for Targeted Drug Release from Thermosensitive Liposomes: Results from a Phase I Trial. Radiology 2019, 291, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Lyon, P.C.; Griffiths, L.F.; Lee, J.; Chung, D.; Carlisle, R.; Wu, F.; Middleton, M.R.; Gleeson, F.V.; Coussios, C.C. Clinical trial protocol for TARDOX: A phase I study to investigate the feasibility of targeted release of lyso-thermosensitive liposomal doxorubicin (ThermoDox(R)) using focused ultrasound in patients with liver tumours. J. Ther. Ultrasound 2017, 5, 28. [Google Scholar] [CrossRef]
- Escoffre, J.M.; Novell, A.; de Smet, M.; Bouakaz, A. Focused ultrasound mediated drug delivery from temperature-sensitive liposomes: In-vitro characterization and validation. Phys. Med. Biol. 2013, 58, 8135–8151. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ko, M.J.; Moon, H.; Sim, W.; Cho, A.S.; Gil, G.; Kim, H.R. Ultrasound-Responsive Liposomes for Targeted Drug Delivery Combined with Focused Ultrasound. Pharmaceutics 2022, 14, 1314. [Google Scholar] [CrossRef]
- Lee, H.; Moon, H.; Kim, H.R. Effects of Lipid Shape and Interactions on the Conformation, Dynamics, and Curvature of Ultrasound-Responsive Liposomes. Pharmaceutics 2022, 14, 1512. [Google Scholar] [CrossRef]
- Minotti, G.; Recalcati, S.; Menna, P.; Salvatorelli, E.; Corna, G.; Cairo, G. Doxorubicin cardiotoxicity and the control of iron metabolism: Quinone-dependent and independent mechanisms. Methods Enzymol. 2004, 378, 340–361. [Google Scholar] [CrossRef]
- Jadapalli, J.K.; Wright, G.W.; Kain, V.; Sherwani, M.A.; Sonkar, R.; Yusuf, N.; Halade, G.V. Doxorubicin triggers splenic contraction and irreversible dysregulation of COX and LOX that alters the inflammation-resolution program in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1091–H1100. [Google Scholar] [CrossRef]
- De Sousa, M. Immune cell functions in iron overload. Clin. Exp. Immunol. 1989, 75, 1–6. [Google Scholar]
- Um, W.; Ko, H.; You, D.G.; Lim, S.; Kwak, G.; Shim, M.K.; Yang, S.; Lee, J.; Song, Y.; Kim, K.; et al. Necroptosis-Inducible Polymeric Nanobubbles for Enhanced Cancer Sonoimmunotherapy. Adv. Mater. 2020, 32, 1907953. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Yoon, B.; Song, S.H.; Um, W.; Song, Y.; Lee, J.; You, D.G.; An, J.Y.; Park, J.H. Chemiluminescence resonance energy transfer-based immunostimulatory nanoparticles for sonoimmunotherapy. Biomaterials 2022, 283, 121466. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zhang, C.; He, S.; Li, J.; Pu, K. Activatable Cancer Sono-Immunotherapy using Semiconducting Polymer Nanobodies. Adv. Mater. 2022, 34, e2203246. [Google Scholar] [CrossRef] [PubMed]
- Um, W.; Kumar, E.K.P.; Song, Y.; Lee, J.; An, J.Y.; Joo, H.; You, D.G.; Park, J.H. Carboxymethyl dextran-based nanocomposites for enhanced chemo-sonodynamic therapy of cancer. Carbohydr. Polym. 2021, 273, 118488. [Google Scholar] [CrossRef]
- Yu, T.H.; Wang, Z.B.; Mason, T.J. A review of research into the uses of low level ultrasound in cancer therapy. Ultrason. Sonochem. 2004, 11, 95–103. [Google Scholar] [CrossRef]
- Mitragotri, S. Innovation-Healing sound: The use of ultrasound in drug delivery and other therapeutic applications. Nat. Rev. Drug Discov. 2005, 4, 255–260. [Google Scholar] [CrossRef]
- Eisenbrey, J.R.; Huang, P.; Hsu, J.; Wheatley, M.A. Ultrasound triggered cell death in vitro with doxorubicin loaded poly lactic-acid contrast agents. Ultrasonics 2009, 49, 628–633. [Google Scholar] [CrossRef][Green Version]
- Aryal, M.; Vykhodtseva, N.; Zhang, Y.Z.; McDannold, N. Multiple sessions of liposomal doxorubicin delivery via focused ultrasound mediated blood-brain barrier disruption: A safety study. J. Control. Release 2015, 204, 60–69. [Google Scholar] [CrossRef]
- Yang, S.; Shim, M.K.; Song, S.; Cho, H.; Choi, J.; Jeon, S.I.; Kim, W.J.; Um, W.; Park, J.H.; Yoon, H.Y.; et al. Liposome-mediated PD-L1 multivalent binding promotes the lysosomal degradation of PD-L1 for T cell-mediated antitumor immunity. Biomaterials 2022, 290, 121841. [Google Scholar] [CrossRef]
- Abdalkader, R.; Kawakami, S.; Unga, J.; Suzuki, R.; Maruyama, K.; Yamashita, F.; Hashida, M. Evaluation of the potential of doxorubicin loaded microbubbles as a theranostic modality using a murine tumor model. Acta Biomater. 2015, 19, 112–118. [Google Scholar] [CrossRef]
- Escoffre, J.M.; Mannaris, C.; Geers, B.; Novell, A.; Lentacker, I.; Averkiou, M.; Bouakaz, A. Doxorubicin liposome-loaded microbubbles for contrast imaging and ultrasound-triggered drug delivery. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2013, 60, 78–87. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Um, W.; Moon, H.; Joo, H.; Song, Y.; Park, M.; Yoon, B.; Kim, H.-R.; Park, J.H. Evading Doxorubicin-Induced Systemic Immunosuppression Using Ultrasound-Responsive Liposomes Combined with Focused Ultrasound. Pharmaceutics 2022, 14, 2603. https://doi.org/10.3390/pharmaceutics14122603
Lee J, Um W, Moon H, Joo H, Song Y, Park M, Yoon B, Kim H-R, Park JH. Evading Doxorubicin-Induced Systemic Immunosuppression Using Ultrasound-Responsive Liposomes Combined with Focused Ultrasound. Pharmaceutics. 2022; 14(12):2603. https://doi.org/10.3390/pharmaceutics14122603
Chicago/Turabian StyleLee, Jeongjin, Wooram Um, Hyungwon Moon, Hyeyeon Joo, Yeari Song, Minsung Park, Been Yoon, Hyun-Ryoung Kim, and Jae Hyung Park. 2022. "Evading Doxorubicin-Induced Systemic Immunosuppression Using Ultrasound-Responsive Liposomes Combined with Focused Ultrasound" Pharmaceutics 14, no. 12: 2603. https://doi.org/10.3390/pharmaceutics14122603
APA StyleLee, J., Um, W., Moon, H., Joo, H., Song, Y., Park, M., Yoon, B., Kim, H.-R., & Park, J. H. (2022). Evading Doxorubicin-Induced Systemic Immunosuppression Using Ultrasound-Responsive Liposomes Combined with Focused Ultrasound. Pharmaceutics, 14(12), 2603. https://doi.org/10.3390/pharmaceutics14122603