
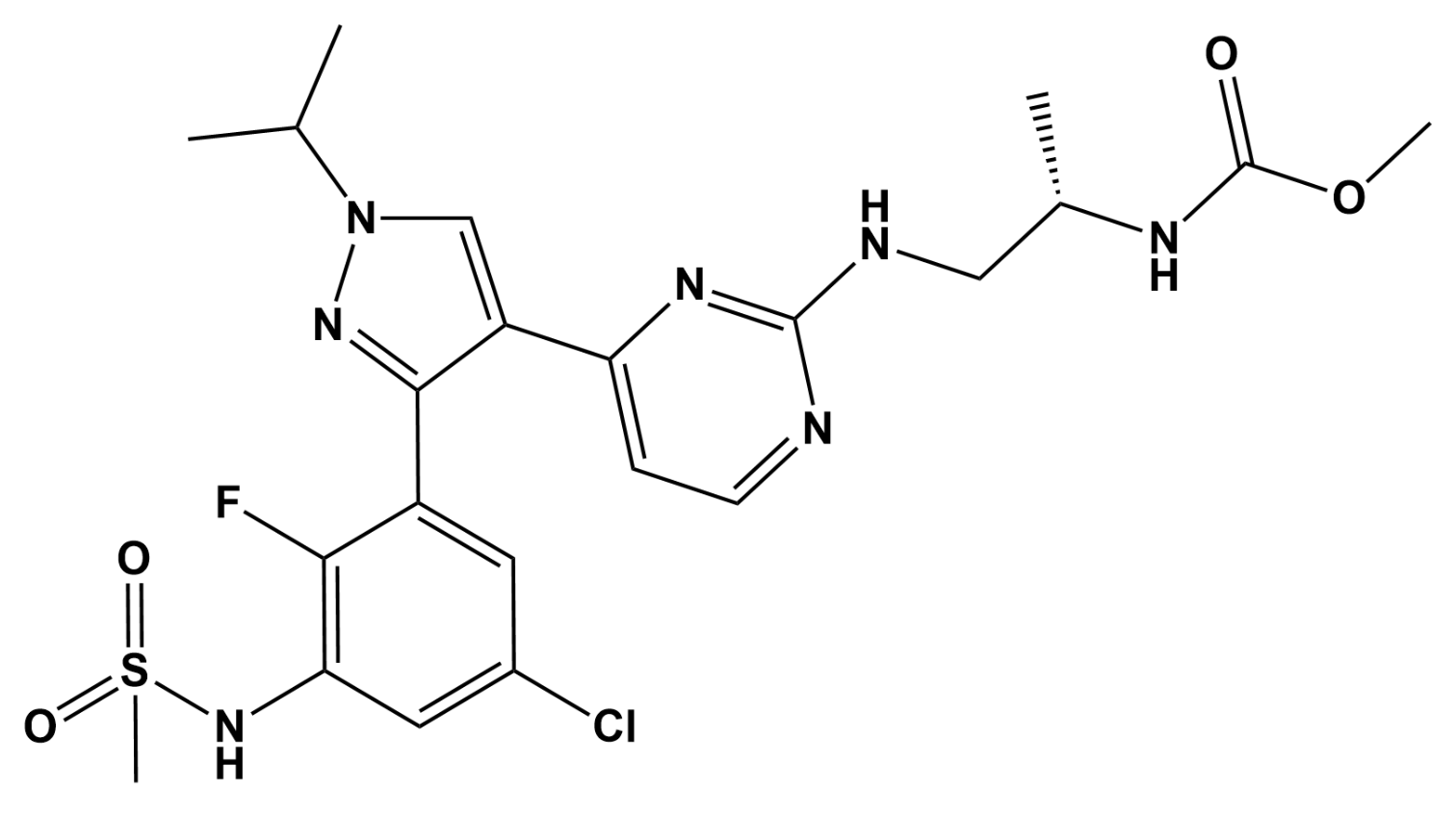
Encorafenib Acts as a Dual-Activity Chemosensitizer through Its Inhibitory Effect on ABCC1 Transporter In Vitro and Ex Vivo
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagent and Chemicals
2.2. Cell Culture
2.3. Isolation and Purification of NSCLC Patient-Derived Primary Cells
2.4. Drug Accumulation Study for ABC Transporters
2.5. MTT Proliferation Assay
2.6. Drug Combination Study
2.7. Western Blotting
2.8. Detection of Gene Expression
2.9. Human Recombinant CYP3A4 Enzyme Inhibition Study
2.10. Statistical Analysis
3. Results
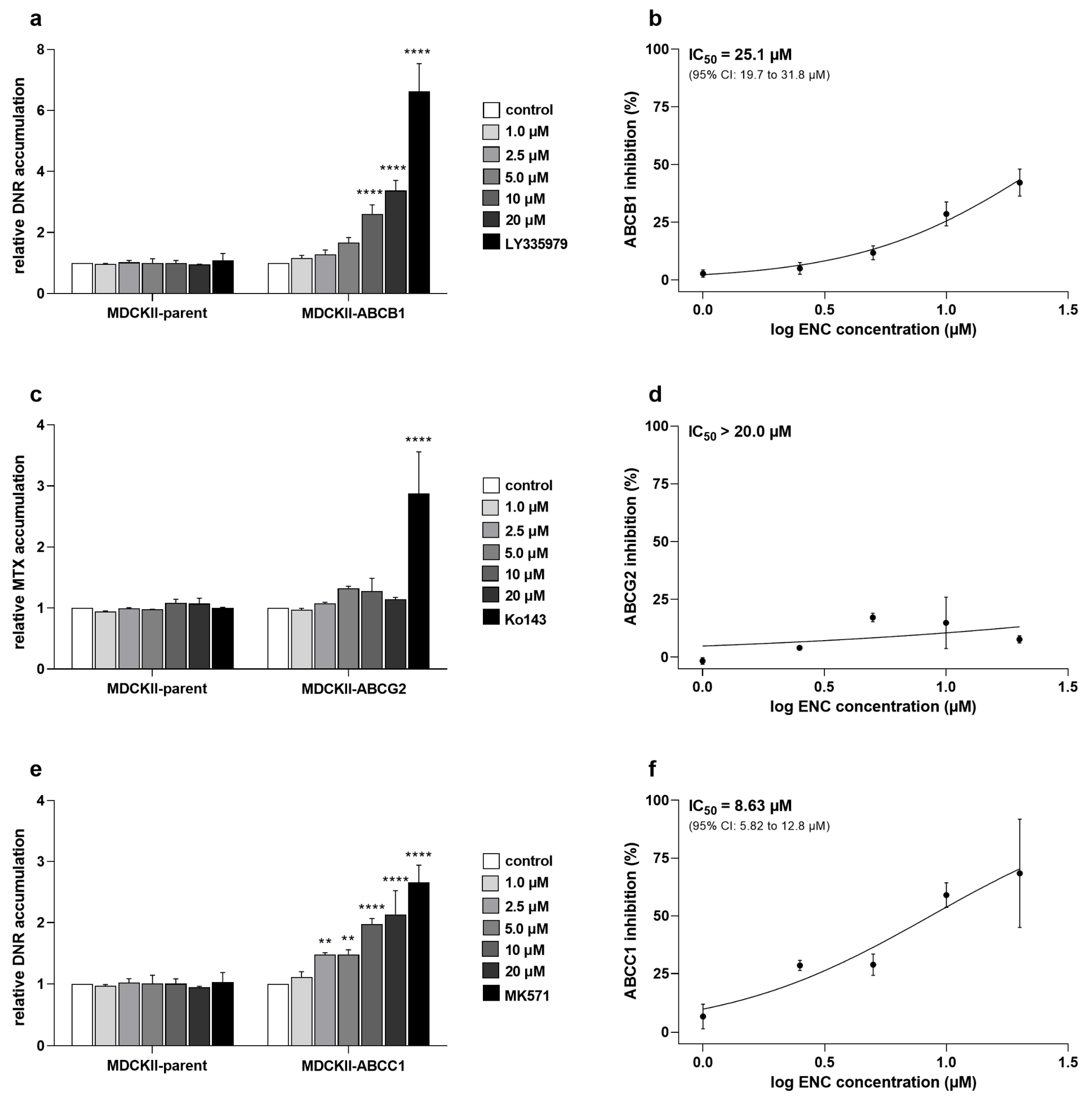
3.1. Encorafenib Shows Functional Inhibitory Effect on ABCC1 in MDCKII Cells
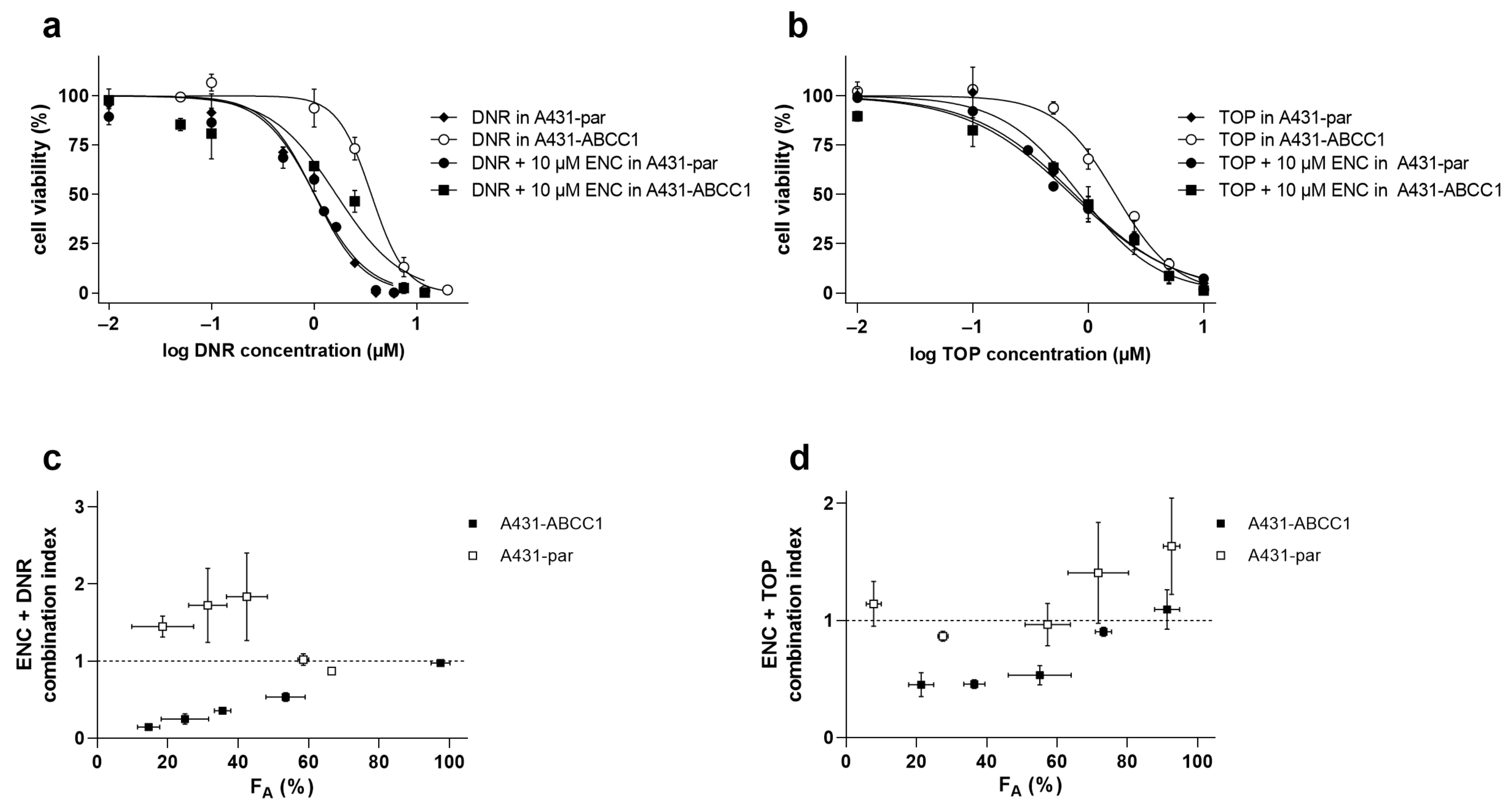
3.2. Encorafenib Decreases a Cytostatic Resistance Driven by the ABCC1 Overexpression In Vitro
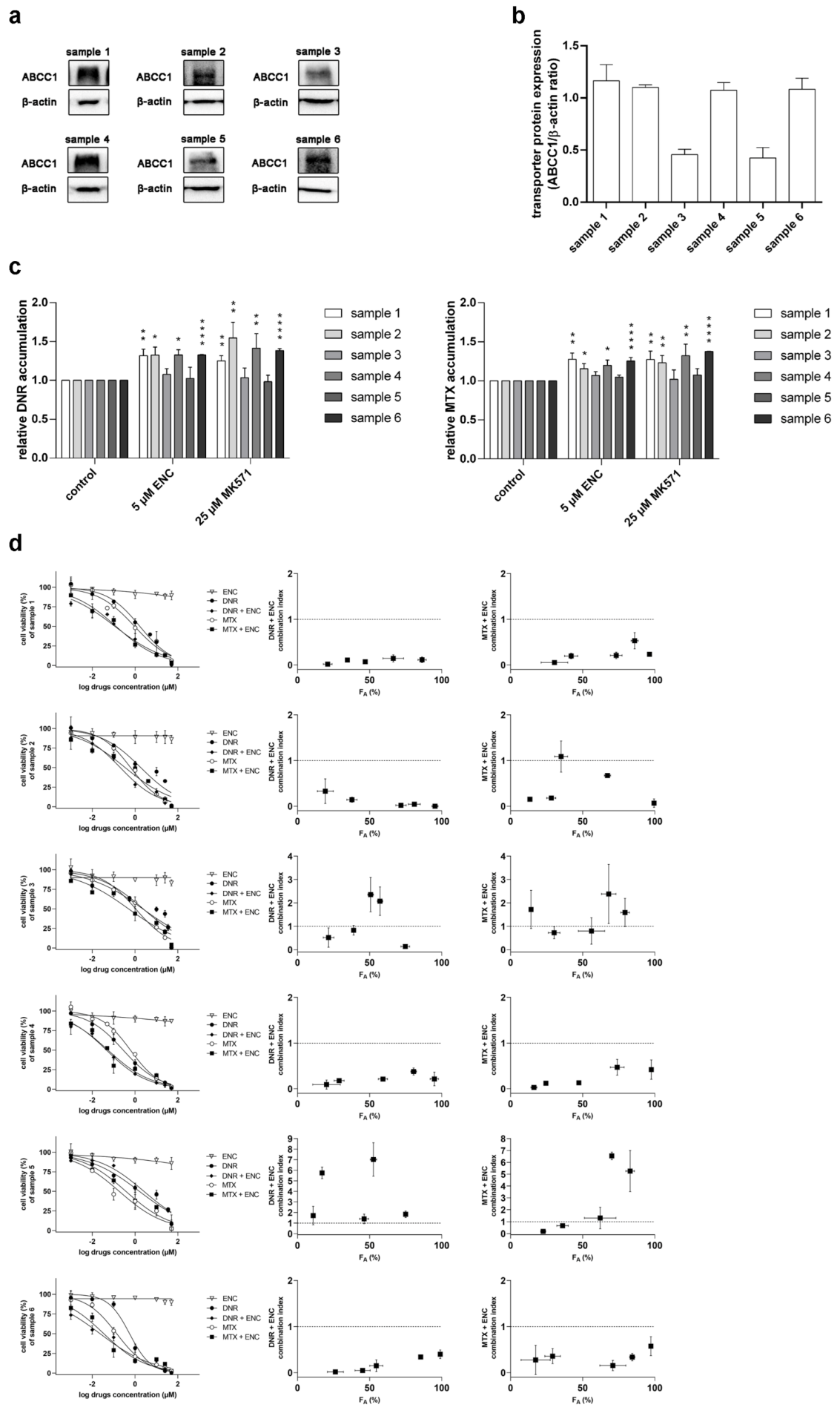
3.3. Encorafenib Is Effective against MDR in NSCLC Primary Explants
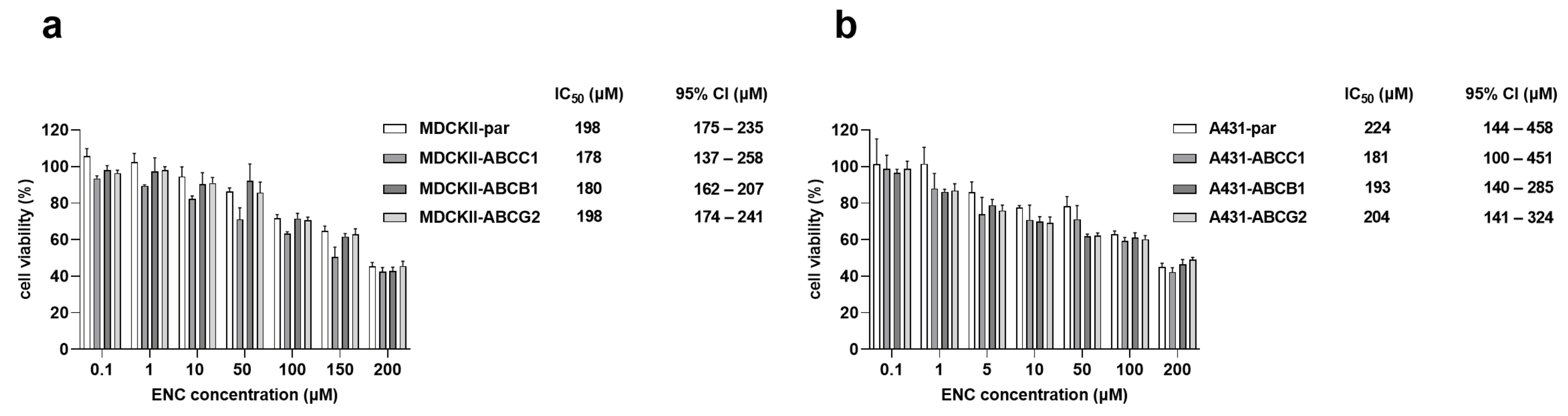
3.4. The Overexpression of ABCB1, ABCG2 and ABCC1 Fails to Establish Resistance to Encorafenib
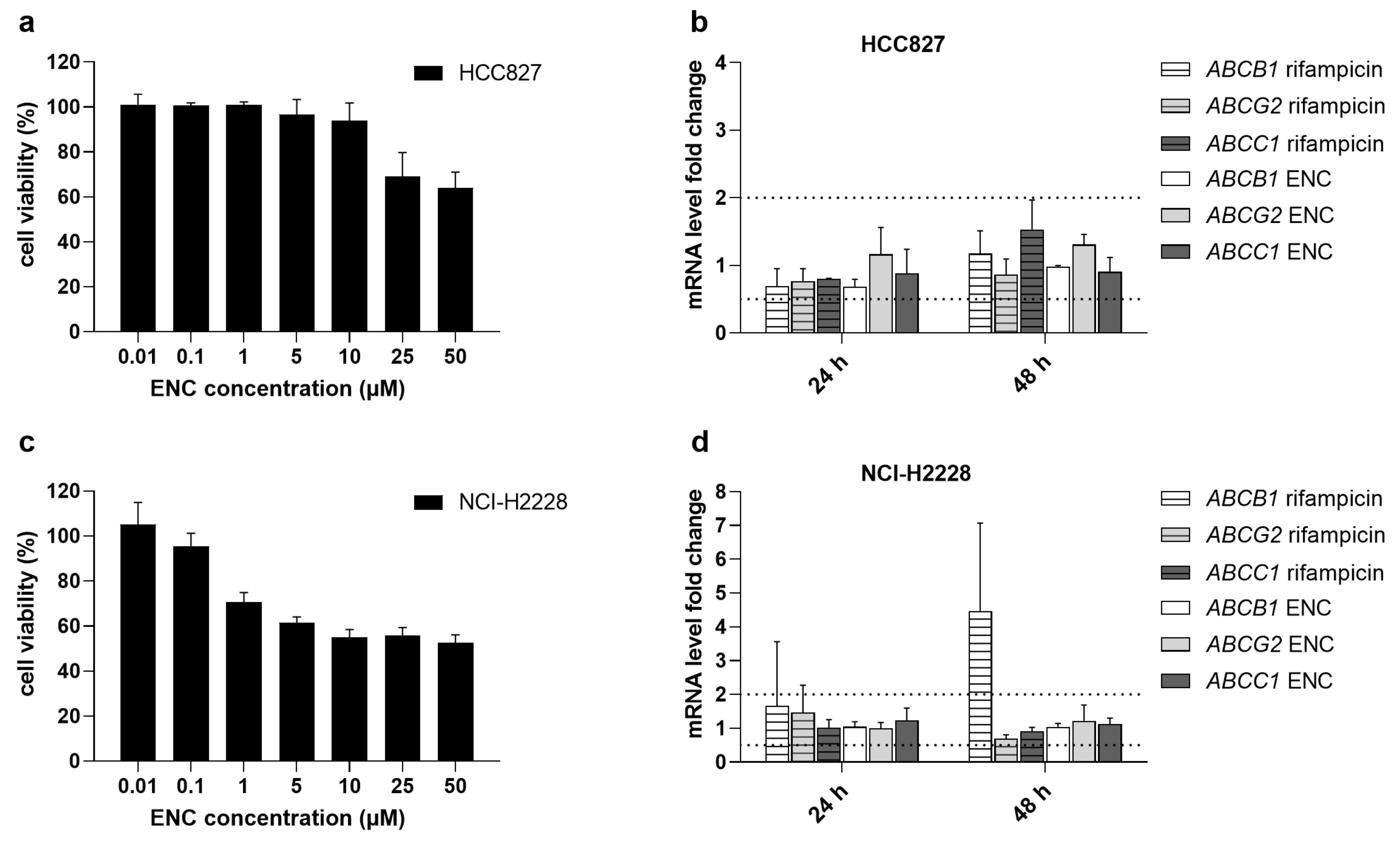
3.5. Encorafenib Does Not Alter the Gene Expression Levels of MDR-Causing ABC Transporters
3.6. Encorafenib Does Not Suppress the CYP3A4 Enzyme’s Activity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Kaur, G.; Gupta, S.K.; Singh, P.; Ali, V.; Kumar, V.; Verma, M. Drug-metabolizing enzymes: Role in drug resistance in cancer. Clin. Transl. Oncol. 2020, 22, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Juan-Carlos, P.M.; Perla-Lidia, P.P.; Stephanie-Talia, M.M.; Monica-Griselda, A.M.; Luz-Maria, T.E. ABC transporter superfamily. An updated overview, relevance in cancer multidrug resistance and perspectives with personalized medicine. Mol. Biol. Rep. 2021, 48, 1883–1901. [Google Scholar] [CrossRef] [PubMed]
- Amawi, H.; Sim, H.M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef]
- Wang, J.Q.; Wu, Z.X.; Yang, Y.; Teng, Q.X.; Li, Y.D.; Lei, Z.N.; Jani, K.A.; Kaushal, N.; Chen, Z.S. ATP-binding cassette (ABC) transporters in cancer: A review of recent updates. J. Evid. Based. Med. 2021, 14, 232–256. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Hofman, J.; Vagiannis, D.; Chen, S.; Guo, L. Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance. Chem. Biol. Interact. 2021, 340, 109448. [Google Scholar] [CrossRef]
- van Eijk, M.; Boosman, R.J.; Schinkel, A.H.; Huitema, A.D.R.; Beijnen, J.H. Cytochrome P450 3A4, 3A5, and 2C8 expression in breast, prostate, lung, endometrial, and ovarian tumors: Relevance for resistance to taxanes. Cancer Chemother. Pharmacol. 2019, 84, 487–499. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Limon, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Encorafenib and Binimetinib: First Global Approvals. Drugs 2018, 78, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Boileve, A.; Samalin, E. New drug approval: Encorafenib-metastatic colorectal cancers with BRAF V600E mutation after systemic chemotherapy. Bull Cancer 2020, 107, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X.; et al. FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist 2018, 23, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Wu, R. Growth of human lung tumor cells in culture. In Culture of Human Tumor Cells; Freshney, R.P.a.R.I., Ed.; Wiley-Liss, Inc.: Hoboken, NJ, USA, 2004; pp. 1–21. [Google Scholar]
- Vagiannis, D.; Budagaga, Y.; Morell, A.; Zhang, Y.; Novotna, E.; Skarka, A.; Kammerer, S.; Kupper, J.H.; Hanke, I.; Rozkos, T.; et al. Tepotinib Inhibits Several Drug Efflux Transporters and Biotransformation Enzymes: The Role in Drug-Drug Interactions and Targeting Cytostatic Resistance In Vitro and Ex Vivo. Int. J. Mol. Sci. 2021, 22, 11936. [Google Scholar] [CrossRef]
- Zhang, Y.; Vagiannis, D.; Budagaga, Y.; Sabet, Z.; Hanke, I.; Rozkos, T.; Hofman, J. Sonidegib potentiates the cancer cells’ sensitivity to cytostatic agents by functional inhibition of ABCB1 and ABCG2 in vitro and ex vivo. Biochem. Pharmacol. 2022, 199, 115009. [Google Scholar] [CrossRef]
- Vagiannis, D.; Yu, Z.; Novotna, E.; Morell, A.; Hofman, J. Entrectinib reverses cytostatic resistance through the inhibition of ABCB1 efflux transporter, but not the CYP3A4 drug-metabolizing enzyme. Biochem. Pharmacol. 2020, 178, 114061. [Google Scholar] [CrossRef] [PubMed]
- Vagiannis, D.; Zhang, Y.; Budagaga, Y.; Novotna, E.; Skarka, A.; Kammerer, S.; Kupper, J.H.; Hofman, J. Alisertib shows negligible potential for perpetrating pharmacokinetic drug-drug interactions on ABCB1, ABCG2 and cytochromes P450, but acts as dual-activity resistance modulator through the inhibition of ABCC1 transporter. Toxicol. Appl. Pharmacol. 2022, 434, 115823. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Agency, E.M. Guideline on the investigation of drug interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2**. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf (accessed on 21 June 2022).
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- Liu, X.D.; Pan, G.Y. Drug Transporters in Drug Disposition, Effects and Toxicity Preface. Drug Transp. Drug Dispos. Eff. Toxic. 2019, 1141, V. [Google Scholar] [CrossRef]
- Trojaniello, C.; Festino, L.; Vanella, V.; Ascierto, P.A. Encorafenib in combination with binimetinib for unresectable or metastatic melanoma with BRAF mutations. Expert. Rev. Clin. Pharmacol. 2019, 12, 259–266. [Google Scholar] [CrossRef]
- Trullas, A.; Delgado, J.; Koenig, J.; Fuerstenau, U.; Dedorath, J.; Hausmann, S.; Stock, T.; Enzmann, H.; Pignatti, F. The EMA assessment of encorafenib in combination with cetuximab for the treatment of adult patients with metastatic colorectal carcinoma harbouring the BRAFV600E mutation who have received prior therapy. ESMO Open. 2021, 6, 100031. [Google Scholar] [CrossRef] [PubMed]
- Ross, K.C.; Chin, K.F.; Kim, D.; Marion, C.D.; Yen, T.J.; Bhattacharjee, V. Methotrexate sensitizes drug-resistant metastatic melanoma cells to BRAF V600E inhibitors dabrafenib and encorafenib. Oncotarget 2018, 9, 13324–13336. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van der Deen, M.; de Vries, E.G.; Timens, W.; Scheper, R.J.; Timmer-Bosscha, H.; Postma, D.S. ATP-binding cassette (ABC) transporters in normal and pathological lung. Respir. Res. 2005, 6, 59. [Google Scholar] [CrossRef]
- Hlavata, I.; Mohelnikova-Duchonova, B.; Vaclavikova, R.; Liska, V.; Pitule, P.; Novak, P.; Bruha, J.; Vycital, O.; Holubec, L.; Treska, V.; et al. The role of ABC transporters in progression and clinical outcome of colorectal cancer. Mutagenesis 2012, 27, 187–196. [Google Scholar] [CrossRef]
- Hofman, J.; Sorf, A.; Vagiannis, D.; Sucha, S.; Kammerer, S.; Kupper, J.H.; Chen, S.; Guo, L.; Ceckova, M.; Staud, F. Brivanib Exhibits Potential for Pharmacokinetic Drug-Drug Interactions and the Modulation of Multidrug Resistance through the Inhibition of Human ABCG2 Drug Efflux Transporter and CYP450 Biotransformation Enzymes. Mol. Pharm. 2019, 16, 4436–4450. [Google Scholar] [CrossRef]
- Cihalova, D.; Ceckova, M.; Kucera, R.; Klimes, J.; Staud, F. Dinaciclib, a cyclin-dependent kinase inhibitor, is a substrate of human ABCB1 and ABCG2 and an inhibitor of human ABCC1 in vitro. Biochem. Pharmacol. 2015, 98, 465–472. [Google Scholar] [CrossRef]
- Peterson, B.G.; Tan, K.W.; Osa-Andrews, B.; Iram, S.H. High-content screening of clinically tested anticancer drugs identifies novel inhibitors of human MRP1 (ABCC1). Pharmacol. Res. 2017, 119, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Patel, A.; Ma, S.L.; Li, X.J.; Zhang, Y.K.; Yang, P.Q.; Kathawala, R.J.; Wang, Y.J.; Anreddy, N.; Fu, L.W.; et al. In vitro, in vivo and ex vivo characterization of ibrutinib: A potent inhibitor of the efflux function of the transporter MRP1. Br. J. Pharmacol. 2014, 171, 5845–5857. [Google Scholar] [CrossRef]
- Ma, S.L.; Hu, Y.P.; Wang, F.; Huang, Z.C.; Chen, Y.F.; Wang, X.K.; Fu, L.W. Lapatinib antagonizes multidrug resistance-associated protein 1-mediated multidrug resistance by inhibiting its transport function. Mol. Med. 2014, 20, 390–399. [Google Scholar] [CrossRef]
- Zheng, L.S.; Wang, F.; Li, Y.H.; Zhang, X.; Chen, L.M.; Liang, Y.J.; Dai, C.L.; Yan, Y.Y.; Tao, L.Y.; Mi, Y.J.; et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS ONE 2009, 4, e5172. [Google Scholar] [CrossRef]
- Wang, J.; Gan, C.; Sparidans, R.W.; Wagenaar, E.; van Hoppe, S.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (MDR1/ABCB1) and Breast Cancer Resistance Protein (BCRP/ABCG2) affect brain accumulation and intestinal disposition of encorafenib in mice. Pharmacol. Res. 2018, 129, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Al-Salama, Z.T. Encorafenib: A Review in Metastatic Colorectal Cancer with a BRAF V600E Mutation. Drugs 2021, 81, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.R.; Potukuchi, P.K.; Schuetz, E.G.; Schuetz, J.D. Beyond Competitive Inhibition: Regulation of ABC Transporters by Kinases and Protein-Protein Interactions as Potential Mechanisms of Drug-Drug Interactions. Drug. Metab. Dispos. 2018, 46, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Vagiannis, D.; Novotna, E.; Skarka, A.; Kammerer, S.; Kupper, J.H.; Chen, S.; Guo, L.; Staud, F.; Hofman, J. Ensartinib (X-396) Effectively Modulates Pharmacokinetic Resistance Mediated by ABCB1 and ABCG2 Drug Efflux Transporters and CYP3A4 Biotransformation Enzyme. Cancers 2020, 12, 813. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Darwish, H.W.; Al-Shakliah, N.S.; Kadi, A.A. A Validated LC-MS/MS Assay for the Simultaneous Quantification of the FDA-Approved Anticancer Mixture (Encorafenib and Binimetinib): Metabolic Stability Estimation. Molecules 2021, 26, 2717. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Drug (s) | IC50 (μM) | 95% CI (μM) | RR |
|---|---|---|---|---|
| A431-par | ||||
| DNR | 1.01 | 0.864–1.18 | ||
| TOP | 0.832 | 0.707–1.01 | ||
| DNR + ENC | 1.01 ns | 0.900–1.15 | 1.00 | |
| TOP + ENC | 0.764 ns | 0.663–0.879 | 1.09 | |
| A431-ABCC1 | ||||
| DNR | 3.76 | 3.27–4.00 | ||
| TOP | 1.66 | 1.56–1.90 | ||
| DNR + ENC | 1.45 ** | 1.27–2.13 | 2.59 | |
| TOP + ENC | 0.662 ** | 0.560–0.911 | 2.51 |
| Tyrosine Kinase Inhibitor | ABCC1 Inhibition Model | ABCC1 IC50(µM) | MDR-Reversal Model | MDR-Victim Cytotoxic Drug | Combination Outcome |
|---|---|---|---|---|---|
| encorafenib | accumulation assays in MDCKII cells with daunorubicin | 8.63 | A431-par and A431-ABCC1 cells | daunorubicin and topotecan | additive to antagonistic effects in A431-par; synergistic effects in A431-ABCC1 |
| alisertib [22] | accumulation assays in MDCKII cells with calcein AM and daunorubicin | 19.9 and 2.59, respectively | MDCKII-par and MDCKII-ABCC1 cells | daunorubicin | additivity to antagonism in parent cells; synergism in ABCC1-overexpressing cells |
| brivanib [34] | accumulation assays in MDCKII cells with calcein AM and daunorubicin | >50.0 and 25.3, respectively | MDCKII-par, MDCKII-ABCC1, A431-par and A431-ABCC1 cells | daunorubicin | additivity to antagonism in all variants |
| dinaciclib [35] | accumulation assays in MDCKII cells with daunorubicin | 18.0 | MDCKII-par, MDCKII-ABCC1 and ABCC1-expressing T47D cells | daunorubicin and topotecan | additive to antagonistic effects in MDCKII-par; synergistic effects in MDCKII-ABCC1; weak synergism in T47D |
| sirolimus [36] | imaging-based accumulation assays in H69 and H69AR cells with calcein AM | 2.80 | H69AR cells | vincristine | potentiation in resistant H69AR cells |
| ridaforolimus [36] | imaging-based accumulation assays in H69 and H69AR cells with calcein AM | 4.90 | H69AR cells | vincristine | potentiation in resistant H69AR cell line |
| everolimus [36] | imaging-based accumulation assays in H69 and H69AR cells with calcein AM | 2.60 | H69AR cells | vincristine | potentiation in resistant H69AR cell line |
| temsirolimus [36] | imaging-based accumulation assays in H69 and H69AR cells with calcein AM | 5.60 | H69AR cells | vincristine | potentiation in resistant cell line H69AR |
| ibrutinib [37] | vinblastine accumulation assays in HEK293 cells; doxorubicin accumulation in HL60 cells | NA | HEK293/pcDNA 3.1, HEK293/MRP1, HL60 and HL60/Adr cells | vincristine and doxorobicin | potentiation in resistant cell lines; no effect in parental cells |
| lapatinib [38] | doxorubicin and rhodamine 123 accumulation assays in KB-3-1 (drug-sensitive) and C-A120 (MRP1 overexpressing) cells | NA | KB-3-1 and C-A120 cells | vincristine and doxorobicin | potentiation in resistant C-A120 cell line; no effect in KB-3-1 drug-sensitive cell line |
| vandetanib [39] | doxorubicin and rhodamine 123 accumulation assays in KB-3-1 and C-A120 | NA | KB-3-1 and C-A120 cells | vincristine | significant IC50 shift in resistant C-A120 cell line; non-significant IC50 shift in drug-sensitive KB-3-1 cell line |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Vagiannis, D.; Budagaga, Y.; Sabet, Z.; Hanke, I.; Rozkoš, T.; Hofman, J. Encorafenib Acts as a Dual-Activity Chemosensitizer through Its Inhibitory Effect on ABCC1 Transporter In Vitro and Ex Vivo. Pharmaceutics 2022, 14, 2595. https://doi.org/10.3390/pharmaceutics14122595
Zhang Y, Vagiannis D, Budagaga Y, Sabet Z, Hanke I, Rozkoš T, Hofman J. Encorafenib Acts as a Dual-Activity Chemosensitizer through Its Inhibitory Effect on ABCC1 Transporter In Vitro and Ex Vivo. Pharmaceutics. 2022; 14(12):2595. https://doi.org/10.3390/pharmaceutics14122595
Chicago/Turabian StyleZhang, Yu, Dimitrios Vagiannis, Youssif Budagaga, Ziba Sabet, Ivo Hanke, Tomáš Rozkoš, and Jakub Hofman. 2022. "Encorafenib Acts as a Dual-Activity Chemosensitizer through Its Inhibitory Effect on ABCC1 Transporter In Vitro and Ex Vivo" Pharmaceutics 14, no. 12: 2595. https://doi.org/10.3390/pharmaceutics14122595
APA StyleZhang, Y., Vagiannis, D., Budagaga, Y., Sabet, Z., Hanke, I., Rozkoš, T., & Hofman, J. (2022). Encorafenib Acts as a Dual-Activity Chemosensitizer through Its Inhibitory Effect on ABCC1 Transporter In Vitro and Ex Vivo. Pharmaceutics, 14(12), 2595. https://doi.org/10.3390/pharmaceutics14122595