

Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease


 ,
,  and
and
Abstract
1. Introduction
2. General Mechanism of Mitophagy
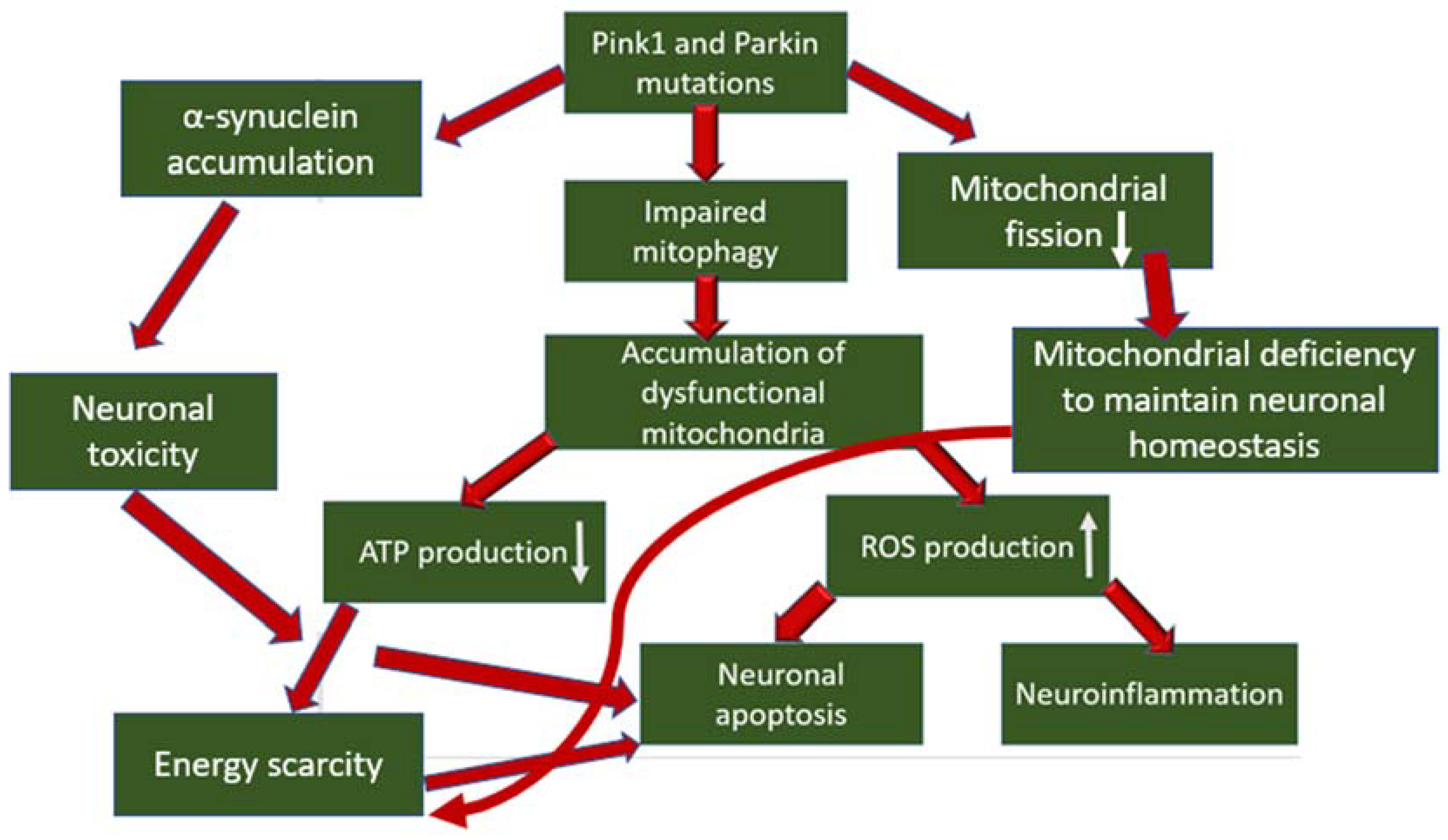
3. The Role of Mitophagy Disorders in the Development of Parkinson’s Disease
4. Features of Pink1 and Parkin as Targets for the Therapy of Parkinson’s Disease
4.1. Characterized Mutant Forms of Mitophagy Proteins
4.2. Level of Expression of Mitophagy Proteins in Neurons
4.3. Labeled Cellular Localization of Mitophagy Proteins
5. Current State of Development of Pink1 and Parkin Activators
6. Evaluation of the Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- What Is Parkinson’s? Date Views. Available online: www.parkinson.org/understanding-parkinsons/what-is-parkinsons (accessed on 10 April 2021).
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 1. [Google Scholar] [CrossRef]
- Chen, R.C.; Chang, S.F.; Su, C.L.; Chen, T.H.H.; Yen, M.F.; Wu, H.M.; Chen, Z.Y.; Liou, H.H. Prevalence, incidence, and mortality of PD: A door-to-door survey in Ilan County, Taiwan. Neurology 2001, 57, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Cubo, E.; Goetz, C.G. Parkinson’s Disease. In Encyclopedia of the Neurological Sciences, 2nd ed.; Michael, J., Aminoff, R., Daroff, B., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 828–832. ISBN 9780123851581. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Steece-Collier, K.; Maries, E.; Kordower, J.H. Etiology of Parkinson’s disease: Genetics and environment revisited. Proc. Natl. Acad. Sci. USA 2002, 99, 13972–13974. [Google Scholar] [CrossRef] [PubMed]
- Hernán, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Ascherio, A.; Checkoway, H.; Marder, K.S.; Nelson, L.M.; Rocca, W.A.; Ross, G.W.; Strickland, D.; Van Den Eeden, S.K.; Gorel, J. Pooled analysis of tobacco use and risk of Parkinson disease. Arch. Neurol. 2007, 64, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, C.B.; Berry, C.; Chang, E.T.; Sielken, R.L., Jr.; Mandel, J.S. Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: Systematic review and meta-analysis. PLoS ONE 2016, 11, e0151841. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.-H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Parkinson’s Disease. Date Views. Available online: www.mayoclinic.org/diseases-conditions/parkinsons-disease/diagnosis-treatment/drc-20376062 (accessed on 10 April 2021).
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The Role of Mitophagy in Innate Immunity. Front. Immunol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway: A mitochondrial quality control system? J. Bioenerg. Biomembr. 2009, 41, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.-F. Mechanisms and Functions of Mitophagy and Potential Roles in Renal Disease. Front. Physiol. 2020, 11, 935. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Arano, T.; Imai, Y. Mitophagy Regulated by the PINK1-Parkin Pathway. In Cell Death—Autophagy, Apoptosis and Necrosis; IntechOpen: London, UK, 2015. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell. Dev. Biol. 2021, 8, 624216. [Google Scholar] [CrossRef]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Model. Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yang, J.; Wang, D.; Li, C.; Fu, Y.; Wang, H.; He, W.; Zhang, J. Mitophagy in Parkinson’s Disease: Pathogenic and Therapeutic Implications. Front. Neurol. 2017, 8, 527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Lu, Y.; Duan, C.; Gao, G.; Lu, L.; Yang, H. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017, 8, e3056. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Fiesel, F.C.; Hudec, R.; Caulfield, T.R.; Ogaki, K.; Gorka-Skoczylas, P.; Koziorowski, D.; Friedman, A.; Chen, L.; Dawson, V.L.; et al. The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 2017, 12, 32. [Google Scholar] [CrossRef]
- Trempe, J.F.; Sauve, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Menade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Siuda, J.; Jasinska-Myga, B.; Boczarska-Jedynak, M.; Opala, G.; Fiesel, F.C.; Moussaud-Lamodiere, E.L.; Scarffe, L.A.; Dawson, V.L.; Ross, O.A.; Springer, W.; et al. Early-onset Parkinson’s disease due to PINK1 p.Q456X mutation--clinical and functional study. Park. Relat. Disord. 2014, 20, 1274–1278. [Google Scholar] [CrossRef]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Aguirre, J.D.; Dunkerley, K.M.; Lam, R.; Rusal, M.; Shaw, G.S. Impact of altered phosphorylation on loss of function of juvenile Parkinsonism-associated genetic variants of the E3 ligase parkin. J. Biol. Chem. 2018, 293, 6337–6348. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Sultan, A.; Platzer, J.; Dolatabadi, N.; Soldner, F.; McClatchy, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Ambasudhan, R.; Nakamura, T.; et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017, 21, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sun, Y.; Guo, S.; Lu, B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 2011, 20, 3227–3240. [Google Scholar] [CrossRef]
- Gómez-Sánchez, R.; Gegg, M.; Pedro, J.M.B.-S.; Niso-Santano, M.; Alvarez-Erviti, L.; Pizarro-Estrella, E.; Gutiérrez-Martín, Y.; Alvarez-Barrientos, A.; Fuentes, J.M.; González-Polo, R.A.; et al. Mitochondrial impairment increases FL-PINK1 levels by calcium-dependent gene expression. Neurobiol. Dis. 2014, 62, 426–440. [Google Scholar] [CrossRef]
- Sainger, R.; Grau, J.B.; Poggio, P.; Branchetti, E.; Bavaria, J.E.; Gorman, J.H.; Gorman, R.C.; Ferrari, G. Dephosphorylation of circulating human osteopontin correlates with severe valvular calcification in patients with calcific aortic valve disease. Biomarkers 2012, 17, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Lupoli, R.; Calcaterra, I.; Poggio, P.; Forte, F.; Spadarella, G.; Ambrosino, P.; Iannuzzo, G.; Di Minno, M.N.D. Efficacy and Safety of Bempedoic Acid in Patients with Hypercholesterolemia: Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2020, 9, e016262. [Google Scholar] [CrossRef]
- Okatsu, K.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015, 128, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef]
- Perrot, N.; Valerio, V.; Moschetta, D.; Boekholdt, S.M.; Dina, C.; Chen, H.Y.; Abner, E.; Martinsson, A.; Manikpurage, H.D.; Rigade, S.; et al. Genetic and In Vitro Inhibition of PCSK9 and Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2020, 5, 649–661. [Google Scholar] [CrossRef]
- Fallaize, D.; Chin, L.S.; Li, L. Differential submitochondrial localization of PINK1 as a molecular switch for mediating distinct mitochondrial signaling pathways. Cell. Signal. 2015, 27, 2543–2554. [Google Scholar] [CrossRef]
- Shires, S.E.; Quiles, J.M.; Najor, R.H.; Leon, L.J.; Cortez, M.Q.; Lampert, M.A.; Mark, A.; Gustafsson, B. Nuclear Parkin Activates the ERRα Transcriptional Program and Drives Widespread Changes in Gene Expression Following Hypoxia. Sci. Rep. 2020, 10, 8499. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, F.B.; Liebes, L.; Simson, G.G.; Mendoza, S.; Mull, J.; Leyne, M.; Norcliffe-Kaufmann, L.; Kaufmann, H.; Slaugenhaupt, S.A. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr. Res. 2011, 70, 480–483. [Google Scholar] [CrossRef]
- Orr, A.L.; Rutaganira, F.U.; de Roulet, D.; Huang, E.J.; Hertz, N.T.; Shokat, K.M.; Nakamura, K. Long-term oral kinetin does not protect against α-synuclein-induced neurodegeneration in rodent models of Parkinson’s disease. Neurochem. Int. 2017, 109, 106–116. [Google Scholar] [CrossRef]
- Lin, M.W.; Lin, C.C.; Chen, Y.H.; Yang, H.B.; Hung, S.Y. Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’s Disease through Activating Mitophagy. Antioxidants 2019, 9, 37. [Google Scholar] [CrossRef]
- Li, R.; Chen, J. Salidroside Protects Dopaminergic Neurons by Enhancing PINK1/Parkin-Mediated Mitophagy. Oxid. Med. Cell. Longev. 2019, 2019, 9341018. [Google Scholar] [CrossRef]
- Clark, E.H.; Vázquez de la Torre, A.; Hoshikawa, T.; Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 2021, 296, 100209. [Google Scholar] [CrossRef]
- Parolari, A.; Poggio, P.; Myasoedova, V.; Songia, P.; Bonalumi, G.; Pilozzi, A.; Pacini, D.; Alamanni, F.; Tremoli, E. Biomarkers in Coronary Artery Bypass Surgery: Ready for Prime Time and Outcome Prediction? Front. Cardiovasc. Med. 2016, 2, 39. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Caulfield, T.R.; Fiesel, F.C.; Moussaud-Lamodière, E.L.; Dourado, D.F.; Flores, S.C.; Springer, W. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput. Biol. 2014, 10, e1003935. [Google Scholar] [CrossRef] [PubMed]
- Celardo, I.; Lehmann, S.; Costa, A.C.; Loh, S.H.; Miguel Martins, L. dATF4 regulation of mitochondrial folate-mediated one-carbon metabolism is neuroprotective. Cell Death Differ. 2017, 24, 638–648, Correction in Cell Death Differ. 2019, 26, 1861. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Um, J.; Yoon, J.; Kim, H.; Lee, D.; Lee, Y.J.; Jee, H.J.; Jang, J.S.; Jang, Y.; Chung, J.; et al. Assessment of mitophagy in mt-Keima Drosophila revealed an essential role of the PINK1-Parkin pathway in mitophagy induction in vivo. FASEB J. 2019, 33, 9742–9751. [Google Scholar] [CrossRef]
- Masaldan, S.; Callegari, S.; Dewson, G. Therapeutic targeting of mitophagy in Parkinson’s disease. Biochem. Soc. Trans. 2022, 50, 783–797. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Protein | Mutation or Modification | Pathological Mechanism |
|---|---|---|
| Pink1 | I368N | Impossibility of binding to the outer mitochondrial membrane |
| Pink1 | p.Q456X | Decreasing of mRNA level |
| Pink1 | p.G411S | Partial loss of kinase activity |
| Parkin | UBL domain (1) | Decrease in Parkin phosphorylation |
| Parkin | UBL domain (2) | Parkin degradation |
| Pink1 | S-nitrosylation | Loss of kinase activity |
| Pink1 | G309D | Accumulation of α-synuclein |
| Therapeutic Compound | Therapeutic Strategy | Therapeutic Mechanism |
|---|---|---|
| Kinetin | PINK1 activator | Direct binding to the active site |
| Celastrol | PINK1 activator | Enhancing expression |
| Salidroside | PINK1 activator | Enhancing expression |
| No name | Parkin activator | Direct binding to the active site |
| USP30i | USP30 inhibitor | Direct binding to the active site |
| Factor | Advantages | Disadvantages |
|---|---|---|
| Investment attractiveness | Approximately 5–10% of PD patients have monogenic forms of the disease. Mutations encoding genes in Pink1 and/or Parkin account for 1–9% of all genetic PD. Therefore, considering the low percentage of subjects bearing these mutations, it does not seem very attractive to invest money and time in the development of novel activators of mitophagy. | |
| State of Development | Studies have shown the ability of potential drugs to reduce neuronal degeneration, which is a prerequisite for efficacy in the treatment of PD. |
|
| Choice of active compound |
|
|
| Target selection |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagov, A.V.; Goncharov, A.G.; Babich, O.O.; Larina, V.V.; Orekhov, A.N.; Melnichenko, A.A. Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease. Pharmaceutics 2022, 14, 2514. https://doi.org/10.3390/pharmaceutics14112514
Blagov AV, Goncharov AG, Babich OO, Larina VV, Orekhov AN, Melnichenko AA. Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease. Pharmaceutics. 2022; 14(11):2514. https://doi.org/10.3390/pharmaceutics14112514
Chicago/Turabian StyleBlagov, Alexander V., Andrey G. Goncharov, Olga O. Babich, Viktoriya V. Larina, Alexander N. Orekhov, and Alexandra A. Melnichenko. 2022. "Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease" Pharmaceutics 14, no. 11: 2514. https://doi.org/10.3390/pharmaceutics14112514
APA StyleBlagov, A. V., Goncharov, A. G., Babich, O. O., Larina, V. V., Orekhov, A. N., & Melnichenko, A. A. (2022). Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease. Pharmaceutics, 14(11), 2514. https://doi.org/10.3390/pharmaceutics14112514