
From Serendipity to Rational Identification of the 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one Core as a New Chemotype of AKT1 Inhibitors for Acute Myeloid Leukemia

 , , , , ,
, , , , , 
 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Computational Studies
2.1.1. Ligand Preparation
2.1.2. Similarity Search
2.1.3. Protein Preparation
2.1.4. Molecular Docking
2.1.5. Molecular Dynamics Simulations
2.2. Chemistry
2.2.1. Synthetic Procedures
2.2.2. Experimental Section
2.3. Enzymatic Assays
2.4. Cell-Based Assays
2.4.1. Cells and Compound Treatments
2.4.2. Colony Forming Unit (CFU) Assay
2.4.3. Cell Lysates Preparation and Western Blot Analysis
2.4.4. Cell Growth and Apoptosis Evaluation
2.4.5. Antibodies
2.4.6. Statistical Analysis
2.5. Validation Assays to Confirm T126 as a True Hit
2.5.1. Intrinsic Fluorescence Assay
2.5.2. Solubility Screen Assay to Determine Chemical Aggregation
2.5.3. UV-VIS Titration Assay with an Increasing Amount of MgCl2
3. Results and Discussion
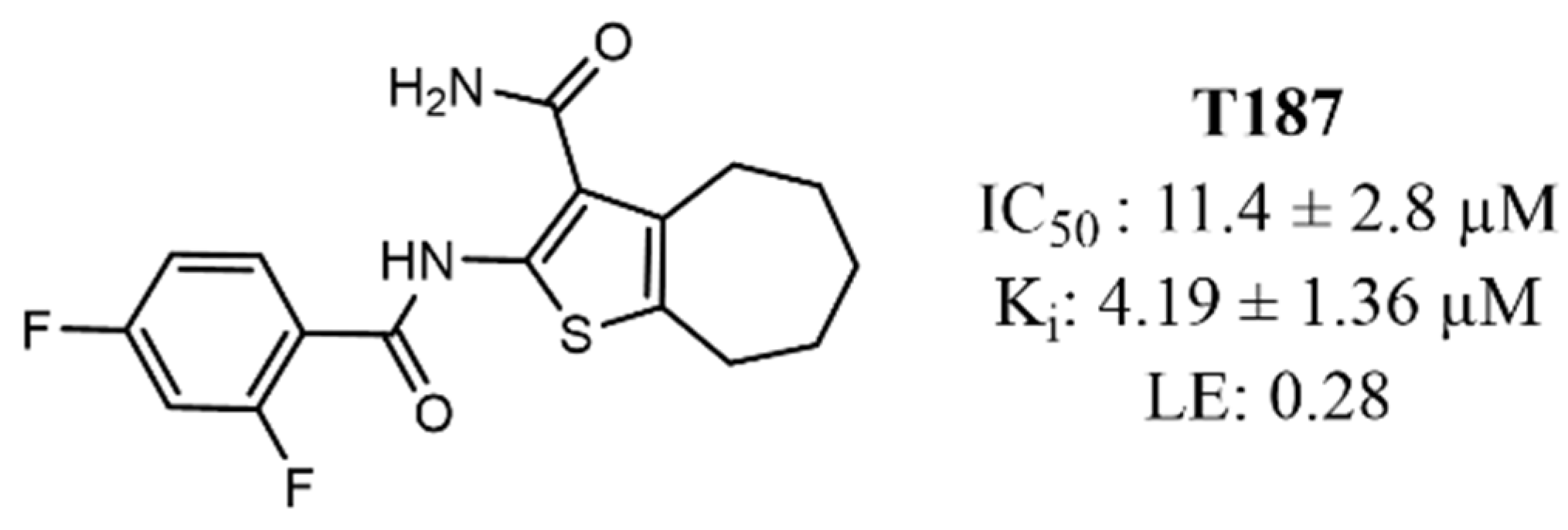
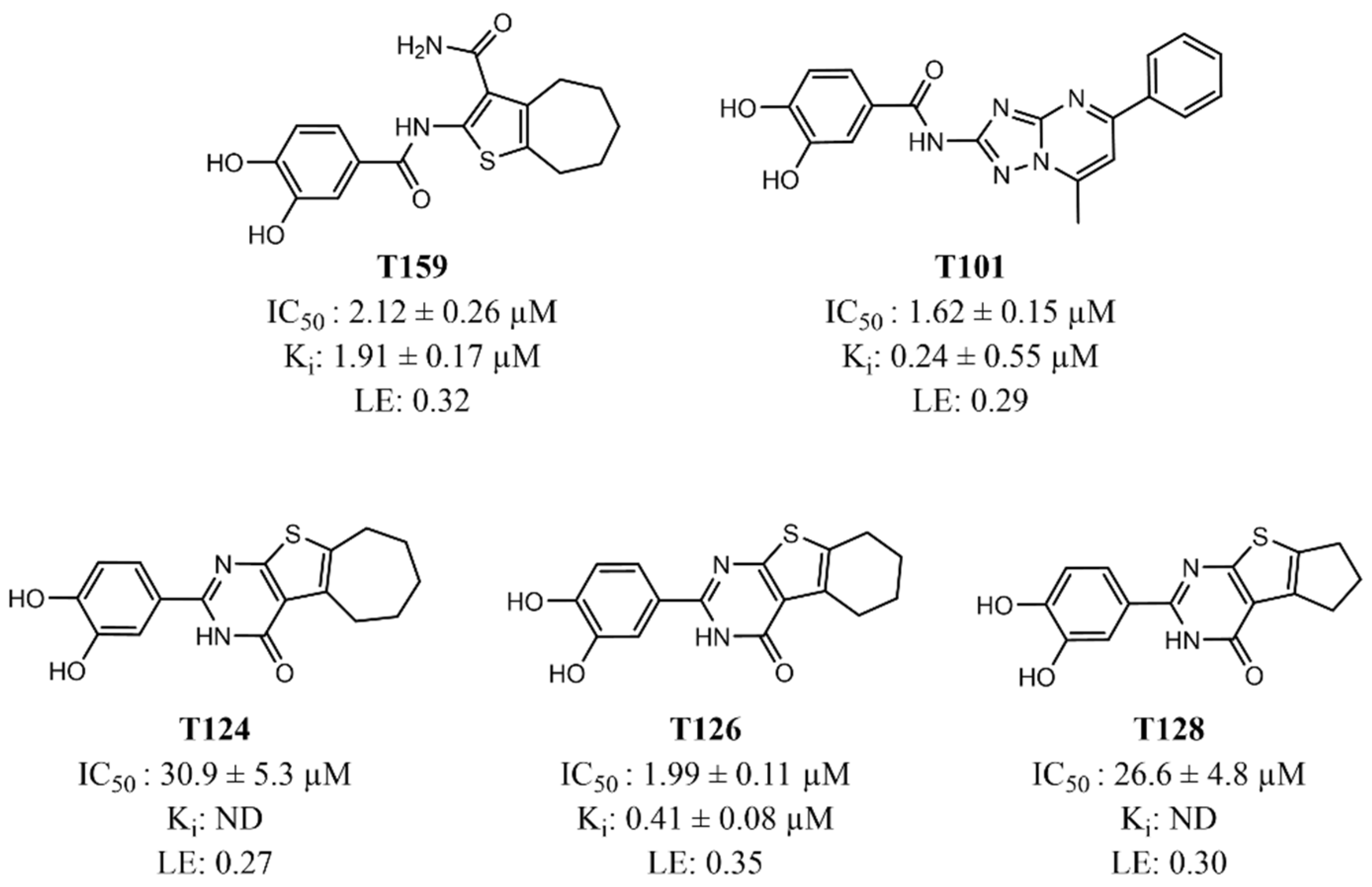
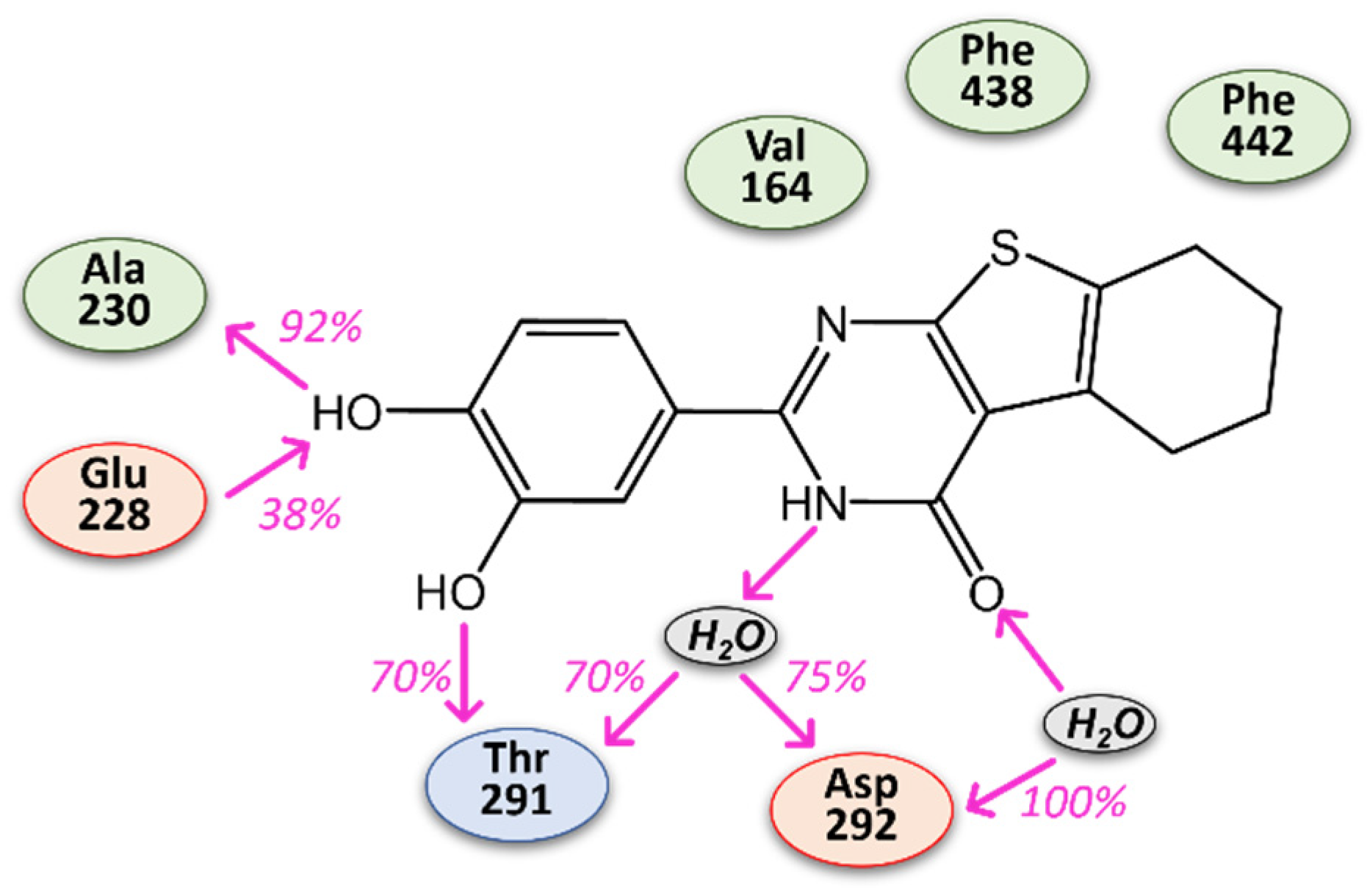
3.1. Computer-Aided Identification of Novel AKT1 Inhibitors
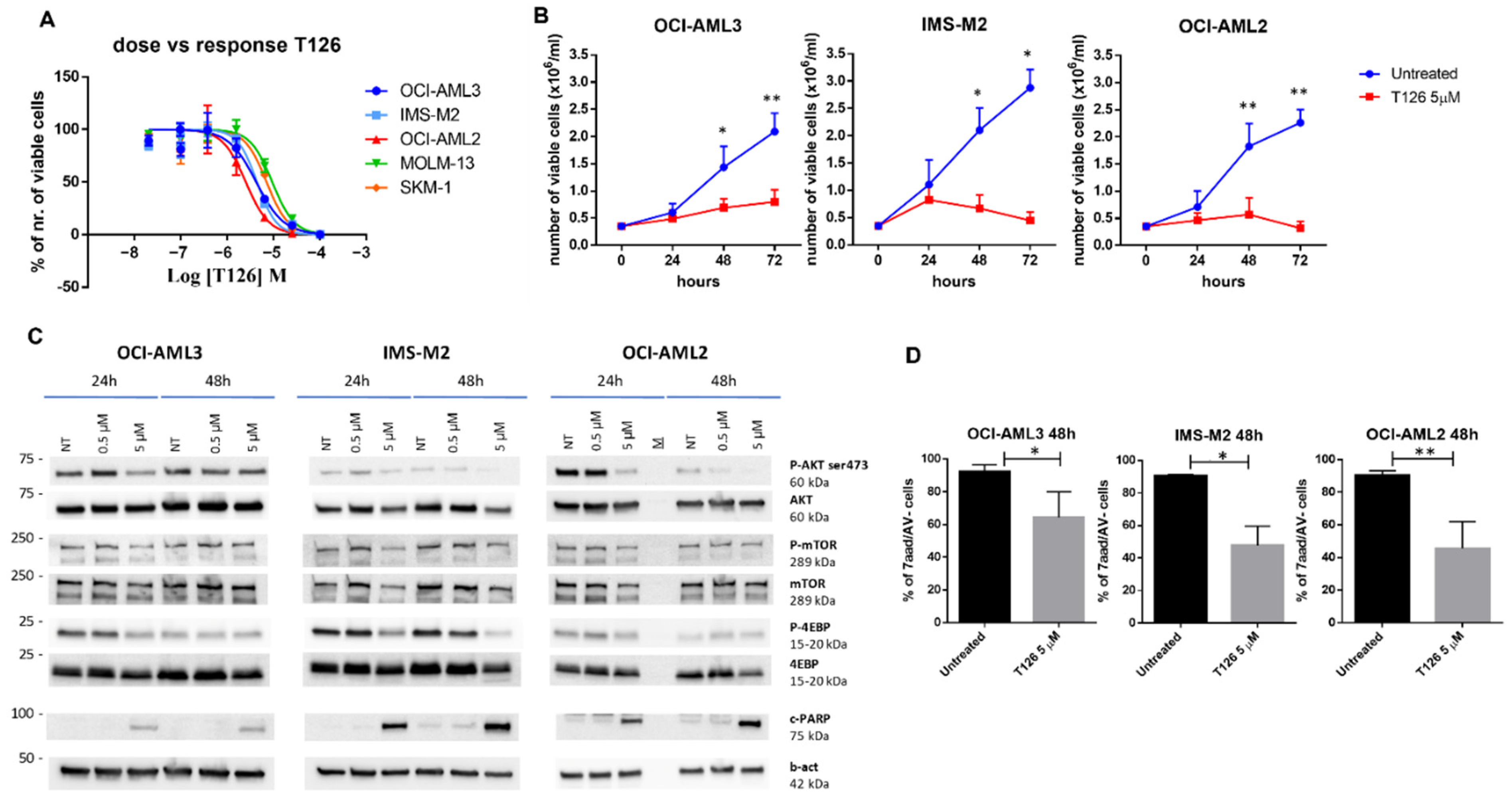
3.2. Evaluation of the Identified AKT1 Inhibitors on AML Cell Lines
3.3. Validation of T126 as a True Hit
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001, 2, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Nepstad, I.; Hatfield, K.J.; Gronningsaeter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, H.; Bradley, H.L.; Bunting, S.T.; Cooper, T.M.; Bunting, K.D. Capillary nano-immunoassay for Akt 1/2/3 and 4EBP1 phosphorylation in acute myeloid leukemia. J. Transl. Med. 2014, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Eng. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Tamburini, J.; Elie, C.; Bardet, V.; Chapuis, N.; Park, S.; Broet, P.; Cornillet-Lefebvre, P.; Lioure, B.; Ugo, V.; Blanchet, O.; et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 2007, 110, 1025–1028. [Google Scholar] [CrossRef]
- Recher, C.; Dos Santos, C.; Demur, C.; Payrastre, B. mTOR, a new therapeutic target in acute myeloid leukemia. Cell Cycle 2005, 4, 1540–1549. [Google Scholar] [CrossRef]
- Chow, S.; Minden, M.D.; Hedley, D.W. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp. Hematol. 2006, 34, 1183–1191. [Google Scholar] [CrossRef]
- Fransecky, L.; Mochmann, L.H.; Baldus, C.D. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell 2015, 3, 2. [Google Scholar] [CrossRef]
- Gallay, N.; Dos Santos, C.; Cuzin, L.; Bousquet, M.; Simmonet Gouy, V.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Recher, C. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009, 23, 1029–1038. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Cappellini, A.; Grafone, T.; Mantovani, I.; Ricci, F.; Billi, A.M.; Ottaviani, E.; Conte, R.; Martinelli, G.; Martelli, A.M. Detection of serine 473 phosphorylated Akt in acute myeloid leukaemia blasts by flow cytometry. Br. J. Haematol. 2004, 126, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.H.; Eom, J.I.; Cheong, J.W.; Maeng, H.O.; Kim, J.Y.; Jeung, H.K.; Lee, S.T.; Lee, M.H.; Hahn, J.S.; Ko, Y.W. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its significance as a prognostic variable. Leukemia 2003, 17, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, J.; Sheikh, A.; Niazi, A.K. Akt inhibitors: Mechanism of action and implications for anticancer therapeutics. Infect. Agent. Cancer 2013, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Hung, M.C. Induction of Akt activity by chemotherapy confers acquired resistance. J. Med. Assoc. 2009, 108, 180–194. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chappell, W.; Abrams, S.L.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; Libra, M.; et al. Targeting the translational apparatus to improve leukemia therapy: Roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia 2011, 25, 1064–1079. [Google Scholar] [CrossRef]
- Lin, K.H.; Rutter, J.C.; Xie, A.; Killarney, S.T.; Vaganay, C.; Benaksas, C.; Ling, F.; Sodaro, G.; Meslin, P.A.; Bassil, C.F.; et al. P2RY2-AKT activation is a therapeutically actionable consequence of XPO1 inhibition in acute myeloid leukemia. Nat. Cancer 2022, 3, 837–851. [Google Scholar] [CrossRef]
- Coleman, N.; Moyers, J.T.; Harbery, A.; Vivanco, I.; Yap, T.A. Clinical Development of AKT Inhibitors and Associated Predictive Biomarkers to Guide Patient Treatment in Cancer Medicine. Pharmgenomics. Pers. Med. 2021, 14, 1517–1535. [Google Scholar] [CrossRef]
- Lazaro, G.; Kostaras, E.; Vivanco, I. Inhibitors in AKTion: ATP-competitive vs allosteric. Biochem Soc. Trans. 2020, 48, 933–943. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2019.
- Rarey, M.; Dixon, J.S. Feature trees: A new molecular similarity measure based on tree matching. J. Comput. Aided Mol. Des. 1998, 12, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Cryst. D Biol. Cryst. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Addie, M.; Ballard, P.; Buttar, D.; Crafter, C.; Currie, G.; Davies, B.R.; Debreczeni, J.; Dry, H.; Dudley, P.; Greenwood, R.; et al. Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J. Med. Chem. 2013, 56, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Lin, J.; Wu, W.I.; Ballard, J.; Lee, B.B.; Gloor, S.L.; Vigers, G.P.; Morales, T.H.; Friedman, L.S.; Skelton, N.; et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal 2012, 5, ra37. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Massari, S.; Corona, A.; Distinto, S.; Desantis, J.; Caredda, A.; Sabatini, S.; Manfroni, G.; Felicetti, T.; Cecchetti, V.; Pannecouque, C.; et al. From cycloheptathiophene-3-carboxamide to oxazinone-based derivatives as allosteric HIV-1 ribonuclease H inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 55–74. [Google Scholar] [CrossRef]
- Desantis, J.; Nannetti, G.; Massari, S.; Barreca, M.L.; Manfroni, G.; Cecchetti, V.; Palu, G.; Goracci, L.; Loregian, A.; Tabarrini, O. Exploring the cycloheptathiophene-3-carboxamide scaffold to disrupt the interactions of the influenza polymerase subunits and obtain potent anti-influenza activity. Eur. J. Med. Chem. 2017, 138, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Masaoka, T.; Chung, S.; Caboni, P.; Rausch, J.W.; Wilson, J.A.; Taskent-Sezgin, H.; Beutler, J.A.; Tocco, G.; Le Grice, S.F. Exploiting drug-resistant enzymes as tools to identify thienopyrimidinone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 2013, 56, 5436–5445. [Google Scholar] [CrossRef] [PubMed]
- Desantis, J.; Massari, S.; Corona, A.; Astolfi, A.; Sabatini, S.; Manfroni, G.; Palazzotti, D.; Cecchetti, V.; Pannecouque, C.; Tramontano, E.; et al. 1,2,4-Triazolo[1,5-a]pyrimidines as a Novel Class of Inhibitors of the HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Molecules 2020, 25, 1183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Johnson, S.; Powell, D.; McGinnis, J.P.; Miranda, M.; Rabindran, S.K. Inhibition of tumor cell proliferation by thieno[2,3-d]pyrimidin-4(1H)-one-based analogs. Bioorg. Med. Chem. Lett. 2005, 15, 3763–3766. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; Martelli, M.P.; Dirks, W.G.; Bolli, N.; Liso, A.; Macleod, R.A.; Nicoletti, I.; Mannucci, R.; Pucciarini, A.; Bigerna, B.; et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 2005, 19, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.T.; Vu, H.A.; Iwasaki, R.; Nagamura, F.; Tojo, A.; Watanabe, T.; Sato, Y. Detection of exon 12 type A mutation of NPM1 gene in IMS-M2 cell line. Leuk Res. 2010, 34, 261–262. [Google Scholar] [CrossRef]
- Balusu, R.; Fiskus, W.; Rao, R.; Chong, D.G.; Nalluri, S.; Mudunuru, U.; Ma, H.; Chen, L.; Venkannagari, S.; Ha, K.; et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood 2011, 118, 3096–3106. [Google Scholar] [CrossRef]
- Sancineto, L.; Iraci, N.; Massari, S.; Attanasio, V.; Corazza, G.; Barreca, M.L.; Sabatini, S.; Manfroni, G.; Avanzi, N.R.; Cecchetti, V.; et al. Computer-aided design, synthesis and validation of 2-phenylquinazolinone fragments as CDK9 inhibitors with anti-HIV-1 Tat-mediated transcription activity. ChemMedChem 2013, 8, 1941–1953. [Google Scholar] [CrossRef]
- Astolfi, A.; Kudolo, M.; Brea, J.; Manni, G.; Manfroni, G.; Palazzotti, D.; Sabatini, S.; Cecchetti, F.; Felicetti, T.; Cannalire, R.; et al. Discovery of potent p38alpha MAPK inhibitors through a funnel like workflow combining in silico screening and in vitro validation. Eur. J. Med. Chem. 2019, 182, 111624. [Google Scholar] [CrossRef]
- Available online: https://scifinder.cas.org (accessed on 15 February 2022).
- Sabatini, S.; Manfroni, G.; Barreca, M.L.; Bauer, S.M.; Gargaro, M.; Cannalire, R.; Astolfi, A.; Brea, J.; Vacca, C.; Pirro, M.; et al. The Pyrazolobenzothiazine Core as a New Chemotype of p38 Alpha Mitogen-Activated Protein Kinase Inhibitors. Chem. Biol. Drug Des. 2015, 86, 531–545. [Google Scholar] [CrossRef]
- Astolfi, A.; Iraci, N.; Sabatini, S.; Barreca, M.L.; Cecchetti, V. p38alpha MAPK and Type I Inhibitors: Binding Site Analysis and Use of Target Ensembles in Virtual Screening. Molecules 2015, 20, 15842–15861. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Iraci, N.; Manfroni, G.; Barreca, M.L.; Cecchetti, V. A Comprehensive Structural Overview of p38alpha MAPK in Complex with Type I Inhibitors. ChemMedChem 2015, 10, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Rossi, R.; Venanzi, A.; Meggendorfer, M.; Perriello, V.M.; Martino, G.; Spinelli, O.; Ciurnelli, R.; Varasano, E.; Brunetti, L.; et al. Novel NPM1 exon 5 mutations and gene fusions leading to aberrant cytoplasmic nucleophosmin in AML. Blood 2021, 138, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Gionfriddo, I.; Brunetti, L.; Mezzasoma, F.; Milano, F.; Cardinali, V.; Ranieri, R.; Venanzi, A.; Pierangeli, S.; Vetro, C.; Spinozzi, G.; et al. Dactinomycin induces complete remission associated with nucleolar stress response in relapsed/refractory NPM1-mutated AML. Leukemia 2021, 35, 2552–2562. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef]
- Grandage, V.L.; Gale, R.E.; Linch, D.C.; Khwaja, A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia 2005, 19, 586–594. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling specificity in the Akt pathway in biology and disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharm. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astolfi, A.; Milano, F.; Palazzotti, D.; Brea, J.; Pismataro, M.C.; Morlando, M.; Tabarrini, O.; Loza, M.I.; Massari, S.; Martelli, M.P.; et al. From Serendipity to Rational Identification of the 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one Core as a New Chemotype of AKT1 Inhibitors for Acute Myeloid Leukemia. Pharmaceutics 2022, 14, 2295. https://doi.org/10.3390/pharmaceutics14112295
Astolfi A, Milano F, Palazzotti D, Brea J, Pismataro MC, Morlando M, Tabarrini O, Loza MI, Massari S, Martelli MP, et al. From Serendipity to Rational Identification of the 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one Core as a New Chemotype of AKT1 Inhibitors for Acute Myeloid Leukemia. Pharmaceutics. 2022; 14(11):2295. https://doi.org/10.3390/pharmaceutics14112295
Chicago/Turabian StyleAstolfi, Andrea, Francesca Milano, Deborah Palazzotti, Jose Brea, Maria Chiara Pismataro, Mariangela Morlando, Oriana Tabarrini, Maria Isabel Loza, Serena Massari, Maria Paola Martelli, and et al. 2022. "From Serendipity to Rational Identification of the 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one Core as a New Chemotype of AKT1 Inhibitors for Acute Myeloid Leukemia" Pharmaceutics 14, no. 11: 2295. https://doi.org/10.3390/pharmaceutics14112295
APA StyleAstolfi, A., Milano, F., Palazzotti, D., Brea, J., Pismataro, M. C., Morlando, M., Tabarrini, O., Loza, M. I., Massari, S., Martelli, M. P., & Barreca, M. L. (2022). From Serendipity to Rational Identification of the 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one Core as a New Chemotype of AKT1 Inhibitors for Acute Myeloid Leukemia. Pharmaceutics, 14(11), 2295. https://doi.org/10.3390/pharmaceutics14112295