
Development of WRAP5 Peptide Complexes for Targeted Drug/Gene Co-Delivery toward Glioblastoma Therapy


 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of TMZ-Loaded Peptides
2.2.2. Formation of Peptide/pDNA Complexes
2.2.3. Agarose Gel Electrophoresis
2.2.4. Characterization of Complexes
2.2.5. pDNA Protection Assay
2.2.6. Cell Culture
2.2.7. Live-Cell Imaging Assay
FITC Plasmid Labeling
Cellular Uptake/Internalization
2.2.8. Conventional Polymerase Chain Reaction (PCR)
Cells Transfection
cDNA Synthesis and PCR
2.2.9. p53 Protein Quantification
2.2.10. Cytotoxicity Study
2.2.11. Statistical Analysis
3. Results and Discussion
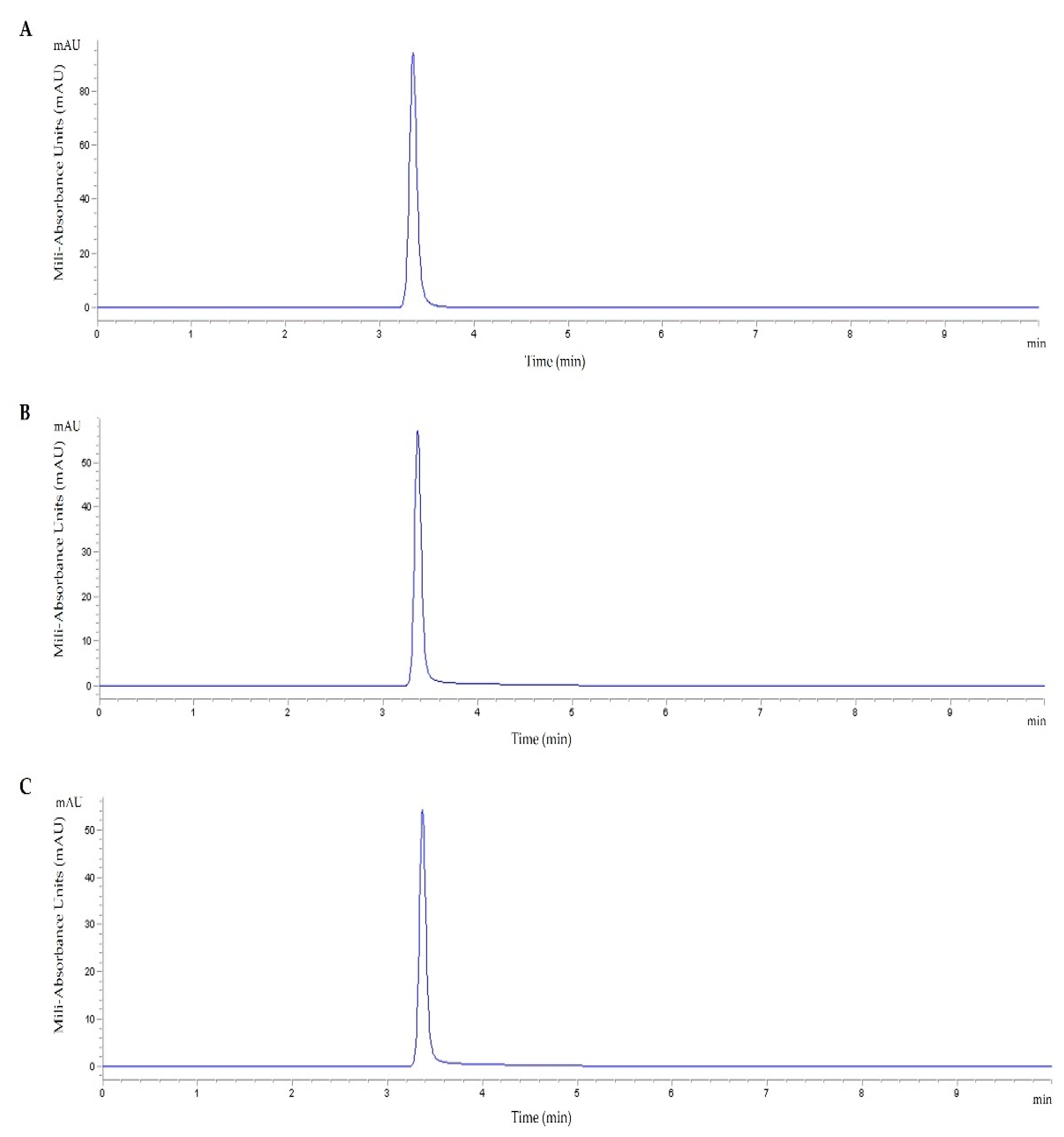
3.1. TMZ Loading Efficiency
3.2. pDNA Complexation
3.3. Physicochemical Properties of the Complexes
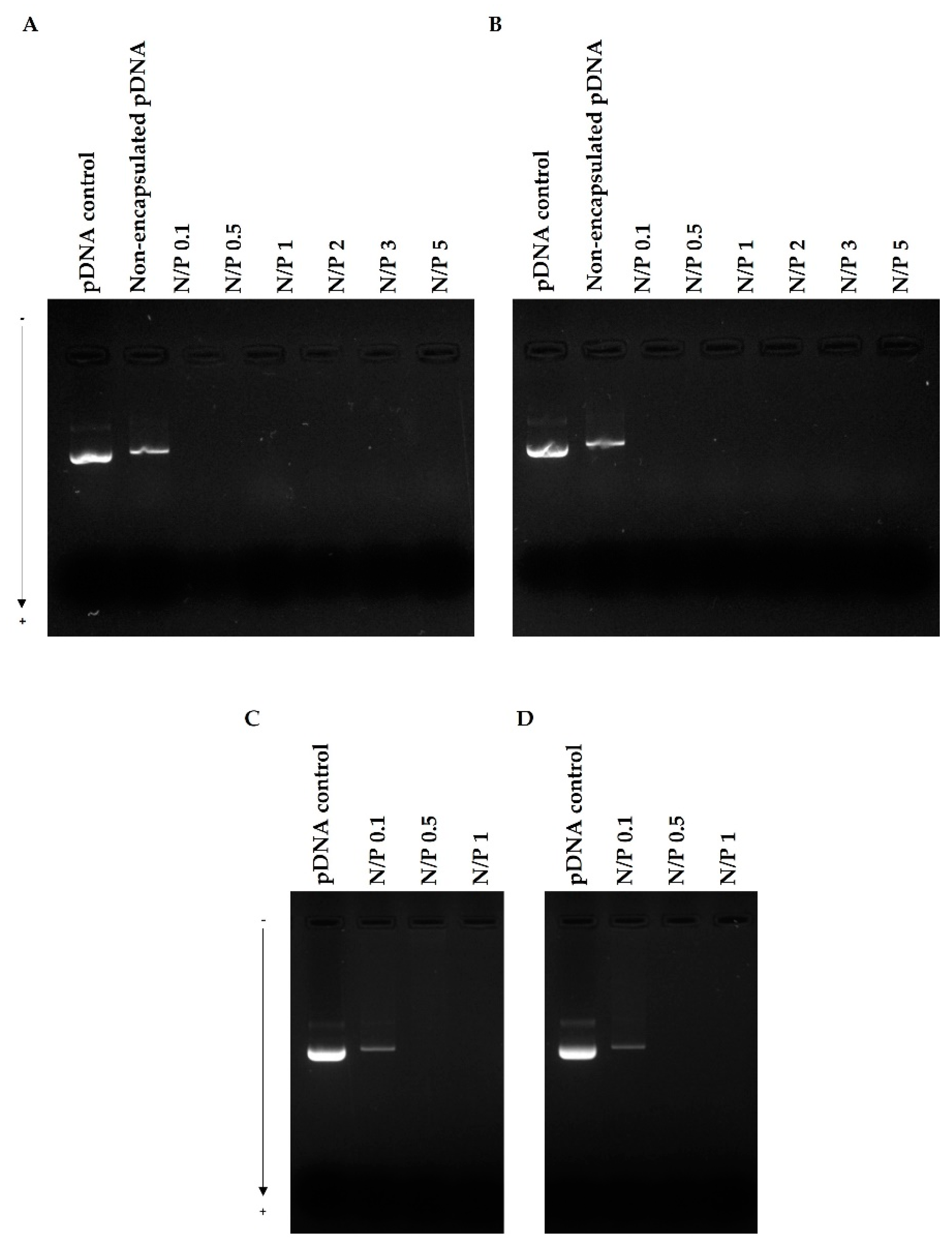
3.4. pDNA Protection Assay
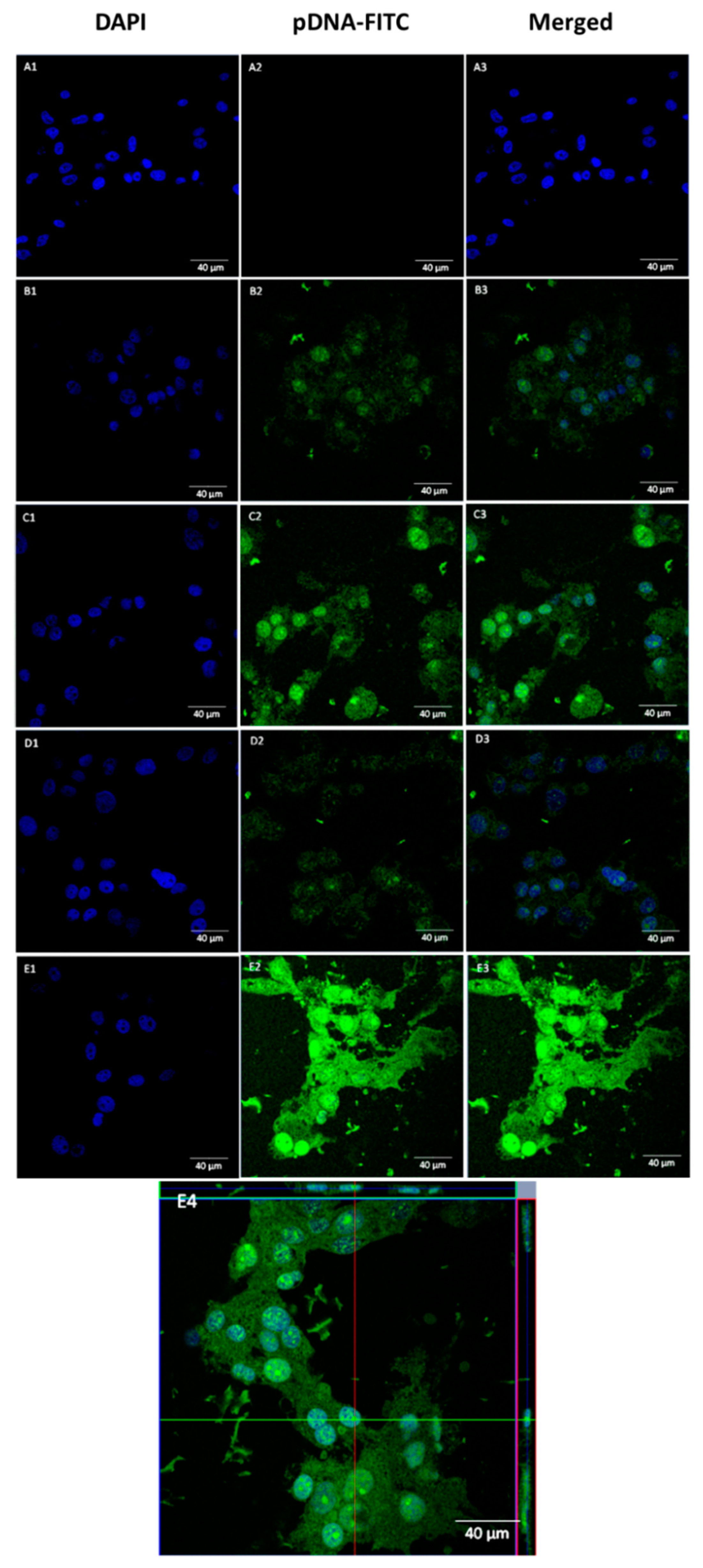
3.5. pDNA Cellular Internalization
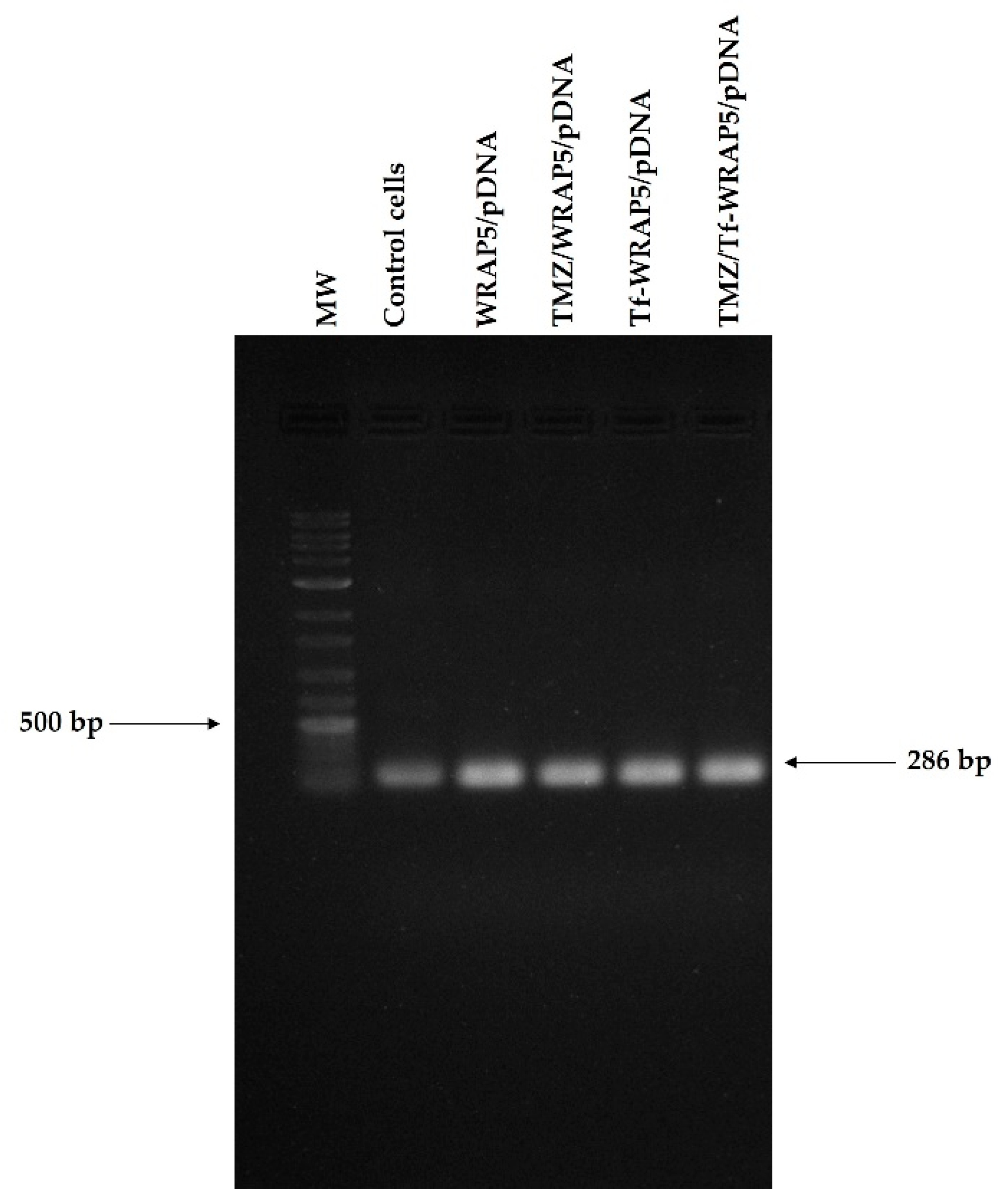
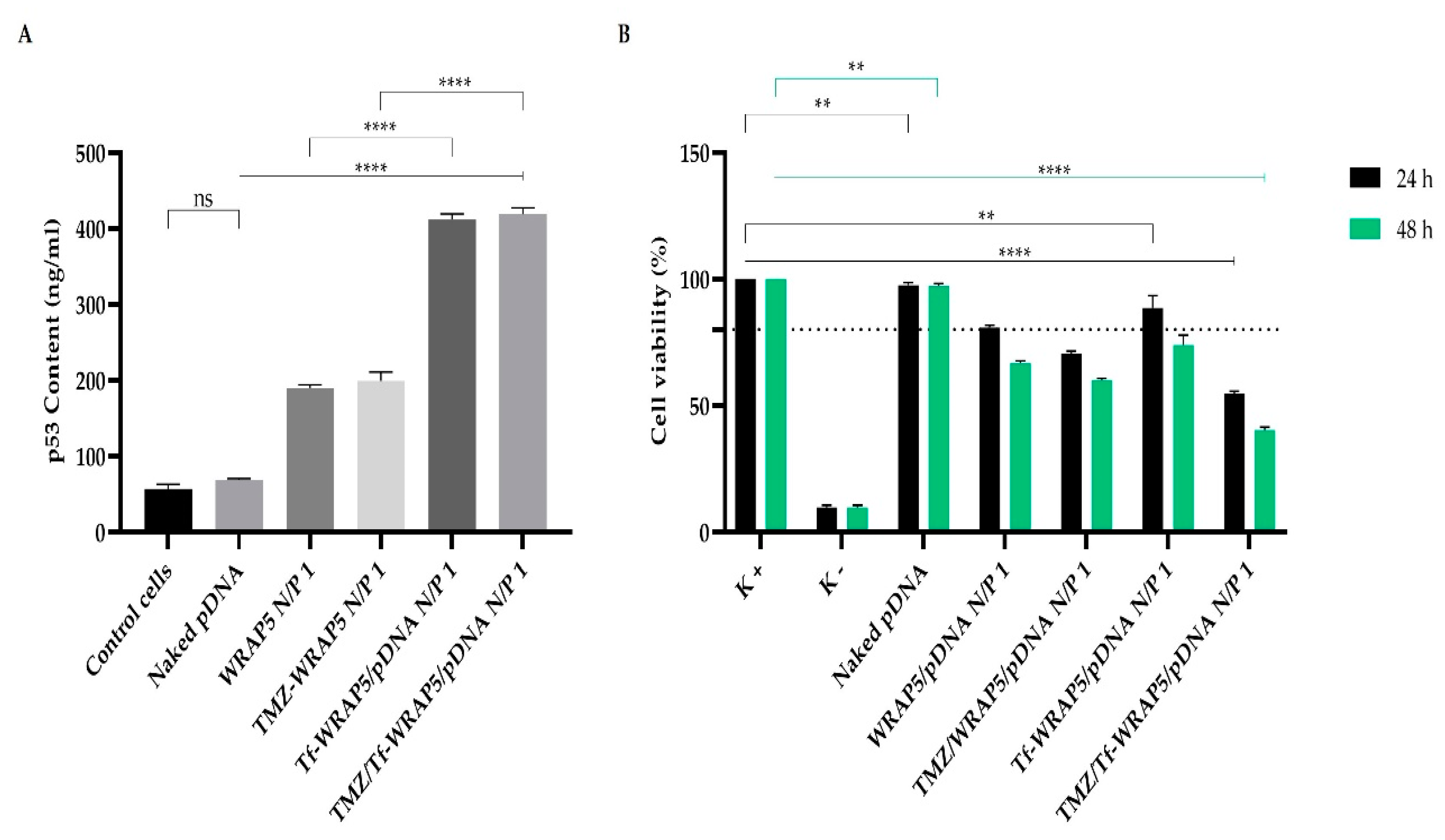
3.6. p53 Gene and Protein Expression
3.7. Cytotoxic Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodríguez Aguilar, L.; Vilchez, M.L.; Milla Sanabria, L.N. Targeting glioblastoma stem cells: The first step of photodynamic therapy. Photodiagn. Photodyn. Ther. 2021, 36, 102585. [Google Scholar] [CrossRef] [PubMed]
- Luís, Â.; Marcelino, H.; Rosa, C.; Domingues, F.C.; Pereira, L.; Cascalheira, J.F. The effects of cannabinoids on glioblastoma growth: A systematic review with meta-analysis of animal model studies. Eur. J. Pharmacol. 2020, 876, 173055. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Pillai, P.P. Current insights on extracellular vesicle-mediated glioblastoma progression: Implications in drug resistance and epithelial-mesenchymal transition. Biochim. Biophys. Acta (BBA) Gen. Subj. 2022, 1866, 130065. [Google Scholar] [CrossRef]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.; Carlisle, T.; Brown, H.; Hollenbeck, N.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef]
- Wenger, K.J.; Wagner, M.; You, S.-J.; Franz, K.; Harter, P.N.; Burger, M.C.; Voss, M.; Ronellenfitsch, M.W.; Fokas, E.; Steinbach, J.P.; et al. Bevacizumab as a last-line treatment for glioblastoma following failure of radiotherapy, temozolomide and lomustine. Oncol. Lett. 2017, 14, 1141–1146. [Google Scholar] [CrossRef]
- Aguilera-Márquez, J.d.R.; de Dios-Figueroa, G.T.; Reza-Saldivar, E.E.; Camacho-Villegas, T.A.; Canales-Aguirre, A.A.; Lugo-Fabres, P.H. Biomaterials: Emerging systems for study and treatment of glioblastoma. Neurol. Perspect. 2022, 2, S31–S42. [Google Scholar] [CrossRef]
- Tagde, P.; Tagde, P.; Tagde, S.; Bhattacharya, T.; Garg, V.; Akter, R.; Rahman, M.H.; Najda, A.; Albadrani, G.M.; Sayed, A.A.; et al. Natural bioactive molecules: An alternative approach to the treatment and control of glioblastoma multiforme. Biomed. Pharmacother. 2021, 141, 111928. [Google Scholar] [CrossRef]
- Liu, S.; Shi, W.; Zhao, Q.; Zheng, Z.; Liu, Z.; Meng, L.; Dong, L.; Jiang, X. Progress and prospect in tumor treating fields treatment of glioblastoma. Biomed. Pharmacother. 2021, 141, 111810. [Google Scholar] [CrossRef]
- Sousa, Â.; Faria, R.; Albuquerque, T.; Bhatt, H.; Biswas, S.; Queiroz, J.A.; Costa, D. Design of experiments to select triphenylphosphonium-polyplexes with suitable physicochemical properties for mitochondrial gene therapy. J. Mol. Liq. 2020, 302, 112488. [Google Scholar] [CrossRef]
- Zhang, P.; Li, X.; Xu, Q.; Wang, Y.; Ji, J. Polydopamine nanoparticles with different sizes for NIR-promoted gene delivery and synergistic photothermal therapy. Colloids Surf. B Biointerfaces 2021, 208, 112125. [Google Scholar] [CrossRef]
- Zheng, J.; Li, T.; Qi, S.; Qin, B.; Yu, J.; Chen, G. Neuroregenerative gene therapy to treat temporal lobe epilepsy in a rat model. Prog. Neurobiol. 2022, 208, 102198. [Google Scholar] [CrossRef] [PubMed]
- Saeb, S.; Assche, J.V.; Loustau, T.; Rohr, O.; Wallet, C.; Schwartz, C. Suicide gene therapy in cancer and HIV-1 infection: An alternative to conventional treatments. Biochem. Pharmacol. 2022, 197, 114893. [Google Scholar] [CrossRef]
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [CrossRef]
- Neves, A.R.; Sousa, A.; Faria, R.; Albuquerque, T.; Queiroz, J.A.; Costa, D. Cancer gene therapy mediated by RALA/plasmid DNA vectors: Nitrogen to phosphate groups ratio (N/P) as a tool for tunable transfection efficiency and apoptosis. Colloids Surf. B Biointerfaces 2020, 185, 110610. [Google Scholar] [CrossRef] [PubMed]
- Faria, R.; Sousa, Â.; Neves, A.R.; Queiroz, J.A.; Costa, D. Methotrexate-plasmid DNA polyplexes for cancer therapy: Characterization, cancer cell targeting ability and tuned in vitro transfection. J. Mol. Liq. 2019, 292, 111391. [Google Scholar] [CrossRef]
- Sousa, Â.; Almeida, A.M.; Faria, R.; Konate, K.; Boisguerin, P.; Queiroz, J.A.; Costa, D. Optimization of peptide-plasmid DNA vectors formulation for gene delivery in cancer therapy exploring design of experiments. Colloids Surf. B Biointerfaces 2019, 183, 110417. [Google Scholar] [CrossRef]
- Albuquerque, T.; Faria, R.; Sousa, Â.; Neves, A.R.; Queiroz, J.A.; Costa, D. Polymer-peptide ternary systems as a tool to improve the properties of plasmid DNA vectors in gene delivery. J. Mol. Liq. 2020, 309, 113157. [Google Scholar] [CrossRef]
- Kavakli, I.H.; Gul, S.; Turkay, M. Identification of novel small molecules targeting core clock proteins to regulate circadian rhythm. Curr. Opin. Chem. Eng. 2022, 35, 100730. [Google Scholar] [CrossRef]
- Harakandi, C.; Nininahazwe, L.; Xu, H.; Liu, B.; He, C.; Zheng, Y.-C.; Zhang, H. Recent advances on the intervention sites targeting USP7-MDM2-p53 in cancer therapy. Bioorg. Chem. 2021, 116, 105273. [Google Scholar] [CrossRef]
- Mei, Y.; Liang, D.; Wang, T.; Yu, D. Gaining insights into relevance across cancers based on mutation features of TP53 gene. Biochem. Biophys. Rep. 2021, 28, 101165. [Google Scholar] [CrossRef]
- Konate, K.; Dussot, M.; Aldrian, G.; Vaissière, A.; Viguier, V.; Neira, I.F.; Couillaud, F.; Vivès, E.; Boisguerin, P.; Deshayes, S. Peptide-Based Nanoparticles to Rapidly and Efficiently “Wrap ’n Roll” siRNA into Cells. Bioconjug. Chem. 2019, 30, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, I.; Heidari, R.; Sadeghian, S.; Raee, M.J.; Negahdaripour, M. Potential of cell-penetrating peptides (CPPs) in delivery of antiviral therapeutics and vaccines. Eur. J. Pharm. Sci. 2022, 169, 106094. [Google Scholar] [CrossRef] [PubMed]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef]
- Boisguérin, P.; Konate, K.; Josse, E.; Vivès, E.; Deshayes, S. Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery. Biomedicines 2021, 9, 583. [Google Scholar] [CrossRef]
- Deshayes, S.; Konate, K.; Dussot, M.; Chavey, B.; Vaissière, A.; Van, T.N.N.; Aldrian, G.; Padari, K.; Pooga, M.; Vivès, E.; et al. Deciphering the internalization mechanism of WRAP:siRNA nanoparticles. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183252. [Google Scholar] [CrossRef]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. BioMed. Res. Int. 2015, 2015, 320941. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, M.J.; Jung, H.H.; Chang, W.S.; Choi, H.S.; Rachmilevitch, I.; Zadicario, E.; Chang, J.W. One-Year Outcome of Multiple Blood-Brain Barrier Disruptions with Temozolomide for the Treatment of Glioblastoma. Front. Oncol. 2020, 10, 1663. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Alahmari, A. Blood-Brain Barrier Overview: Structural and Functional Correlation. Neural Plast. 2021, 2021, 6564585. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Fang, C.; Chang, J. Blood-Brain Barrier Breakdown: An Emerging Biomarker of Cognitive Impairment in Normal Aging and Dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Sun, Y.; Wei, J.; Qiu, X.; Meng, F.; Storm, G.; Zhong, Z. Selective transferrin coating as a facile strategy to fabricate BBB-permeable and targeted vesicles for potent RNAi therapy of brain metastatic breast cancer in vivo. J. Control. Release 2021, 337, 521–529. [Google Scholar] [CrossRef]
- Ramalho, M.J.; Loureiro, J.A.; Coelho, M.A.N.; Pereira, M.C. Transferrin Receptor-Targeted Nanocarriers: Overcoming Barriers to Treat Glioblastoma. Pharmaceutics 2022, 14, 279. [Google Scholar] [CrossRef]
- Koneru, T.; McCord, E.; Pawar, S.; Tatiparti, K.; Sau, S.; Iyer, A.K. Transferrin: Biology and Use in Receptor-Targeted Nanotherapy of Gliomas. ACS Omega 2021, 6, 8727–8733. [Google Scholar] [CrossRef]
- Tran, K.; Brice, R.; Yao, L. Bioscaffold-based study of glioblastoma cell behavior and drug delivery for tumor therapy. Neurochem. Int. 2021, 147, 105049. [Google Scholar] [CrossRef]
- Sheykhzadeh, S.; Luo, M.; Peng, B.; White, J.; Abdalla, Y.; Tang, T.; Mäkilä, E.; Voelcker, N.H.; Tong, W.Y. Transferrin-targeted porous silicon nanoparticles reduce glioblastoma cell migration across tight extracellular space. Sci. Rep. 2020, 10, 2320. [Google Scholar] [CrossRef]
- Luo, M.; Lewik, G.; Ratcliffe, J.C.; Choi, C.H.J.; Mäkilä, E.; Tong, W.Y.; Voelcker, N.H. Systematic Evaluation of Transferrin-Modified Porous Silicon Nanoparticles for Targeted Delivery of Doxorubicin to Glioblastoma. ACS Appl. Mater. Interfaces 2019, 11, 33637–33649. [Google Scholar] [CrossRef]
- Zhou, W.; Tan, W.; Huang, X.; Yu, H.G. Doxorubicin combined with Notch1-targeting siRNA for the treatment of gastric cancer. Oncol. Lett. 2018, 16, 2805–2812. [Google Scholar] [CrossRef]
- Xu, C.; Liu, W.; Hu, Y.; Li, W.; Di, W. Bioinspired tumor-homing nanoplatform for co-delivery of paclitaxel and siRNA-E7 to HPV-related cervical malignancies for synergistic therapy. Theranostics 2020, 10, 3325–3339. [Google Scholar] [CrossRef]
- Chen, Z.; Lai, X.; Song, S.; Zhu, X.; Zhu, J. Nanostructured lipid carriers based temozolomide and gene co-encapsulated nanomedicine for gliomatosis cerebri combination therapy. Drug Deliv. 2016, 23, 1369–1373. [Google Scholar] [CrossRef]
- Peng, Y.; Huang, J.; Xiao, H.; Wu, T.; Shuai, X. Codelivery of temozolomide and siRNA with polymeric nanocarrier for effective glioma treatment. Int. J. Nanomed. 2018, 13, 3467–3480. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Sun, S.; Xu, H.; Zhao, Z.; Han, Z.; Jia, J.; Wu, D.; Lu, J.; Liu, H.; Yu, R. Combined Delivery of Temozolomide and siPLK1 Using Targeted Nanoparticles to Enhance Temozolomide Sensitivity in Glioma. Int. J. Nanomed. 2020, 15, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef] [PubMed]
- Mitha, A.T.; Rekha, M.R. Multifunctional polymeric nanoplexes for anticancer co-delivery of p53 and mitoxantrone. J. Mater. Chem. B 2014, 2, 8005–8016. [Google Scholar] [CrossRef]
- Misra, S.K.; Naz, S.; Kondaiah, P.; Bhattacharya, S. A cationic cholesterol based nanocarrier for the delivery of p53-EGFP-C3 plasmid to cancer cells. Biomaterials 2014, 35, 1334–1346. [Google Scholar] [CrossRef]
- Shchors, K.; Persson, A.I.; Rostker, F.; Tihan, T.; Lyubynska, N.; Li, N.; Swigart, L.B.; Berger, M.S.; Hanahan, D.; Weiss, W.A.; et al. Using a preclinical mouse model of high-grade astrocytoma to optimize p53 restoration therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E1480–E1489. [Google Scholar] [CrossRef]
- Gazaille, C.; Sicot, M.; Saulnier, P.; Eyer, J.; Bastiat, G. Local Delivery and Glioblastoma: Why Not Combining Sustained Release and Targeting? Front. Med. Technol. 2021, 3, 791596. [Google Scholar] [CrossRef]
- Peng, H.; Guo, X.; He, J.; Duan, C.; Yang, M.; Zhang, X.; Zhang, L.; Fu, R.; Wang, B.; Wang, D.; et al. Intracranial delivery of synthetic mRNA to suppress glioblastoma. Mol. Ther. Oncolytics 2022, 24, 160–170. [Google Scholar] [CrossRef]
- Faria, R.; Vivés, E.; Boisguerin, P.; Sousa, A.; Costa, D. Development of Peptide-Based Nanoparticles for Mitochondrial Plasmid DNA Delivery. Polymers 2021, 13, 1836. [Google Scholar] [CrossRef]
- Choi, E.; Han, J.; Tan, X.; Oh, J.; Lee, D.; Rhim, T.; Lee, M. Combined delivery of temozolomide and the thymidine kinase gene for treatment of glioblastoma. J. Drug Target. 2017, 25, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Ak, G.; Ünal, A.; Karakayalı, T.; Özel, B.; Selvi Günel, N.; Hamarat Şanlıer, Ş. Brain-targeted, drug-loaded solid lipid nanoparticles against glioblastoma cells in culture. Colloids Surf. B Biointerfaces 2021, 206, 111946. [Google Scholar] [CrossRef]
- Wang, T.; Pickard, A.J.; Gallo, J.M. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016, 36, 3289–3299. [Google Scholar]
- Rodriguez de Anda, D.A.; Ohannesian, N.; Martirosyan, K.S.; Chew, S.A. Effects of solvent used for fabrication on drug loading and release kinetics of electrosprayed temozolomide-loaded PLGA microparticles for the treatment of glioblastoma. J. Biomed. Mater. Res. B Appl. Biomater. 2019, 107, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, S.; Tomeh, M.A.; Wilkinson, R.N.; Hill, C.; Brown, S.; Zhao, X. Designed Antitumor Peptide for Targeted siRNA Delivery into Cancer Spheroids. ACS Appl. Mater. Interfaces 2021, 13, 49713–49728. [Google Scholar] [CrossRef]
- Hu, Y.; Zhu, Y.; Sutherland, N.D.; Wilson, D.R.; Pang, M.; Liu, E.; Staub, J.R.; Berlinicke, C.A.; Zack, D.J.; Green, J.J.; et al. Size-Controlled and Shelf-Stable DNA Particles for Production of Lentiviral Vectors. Nano Lett. 2021, 21, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Albuquerque, T.; Faria, R.; Paul, M.; Biswas, S.; Sousa, Â.; Costa, D. Development of Tailor-Made Dendrimer Ternary Complexes for Drug/Gene Co-Delivery in Cancer. Pharmaceutics 2021, 13, 1256. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Mohanty, R.P.; Liu, X.; Ghosh, D. Electrostatic driven transport enhances penetration of positively charged peptide surfaces through tumor extracellular matrix. Acta Biomater. 2020, 113, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Pedersen, J.N.; Marie, R. Size and surface charge characterization of nanoparticles with a salt gradient. Nat. Commun. 2020, 11, 2337. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Hasan, A.; Primavera, R.; Wilson, R.J.; Thakor, A.S.; Kevadiya, B.D. Cellular uptake and retention of nanoparticles: Insights on particle properties and interaction with cellular components. Mater. Today Commun. 2020, 25, 101692. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]
- Manzanares, D.; Ceña, V. Endocytosis: The Nanoparticle and Submicron Nanocompounds Gateway into the Cell. Pharmaceutics 2020, 12, 371. [Google Scholar] [CrossRef]
- Chong, Z.X.; Yeap, S.K.; Ho, W.Y. Transfection types, methods and strategies: A technical review. PeerJ 2021, 9, e11165. [Google Scholar] [CrossRef]
- Santi, M.; Maccari, G.; Mereghetti, P.; Voliani, V.; Rocchiccioli, S.; Ucciferri, N.; Luin, S.; Signore, G. Rational Design of a Transferrin-Binding Peptide Sequence Tailored to Targeted Nanoparticle Internalization. Bioconjug. Chem. 2017, 28, 471–480. [Google Scholar] [CrossRef]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Liu, D.-z.; Cheng, Y.; Cai, R.-q.; Wang, B.D.W.-w.; Cui, H.; Liu, M.; Zhang, B.-l.; Mei, Q.-b.; Zhou, S.-y. The enhancement of siPLK1 penetration across BBB and its anti glioblastoma activity in vivo by magnet and transferrin co-modified nanoparticle. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 991–1003. [Google Scholar] [CrossRef]
- Slobodin, B.; Dikstein, R. So close, no matter how far: Multiple paths connecting transcription to mRNA translation in eukaryotes. EMBO Rep. 2020, 21, e50799. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.S.; Eusébio, D.; Neves, A.R.; Albuquerque, T.; Bhatt, H.; Biswas, S.; Costa, D.; Sousa, Â. Synthesis and Characterization of Mannosylated Formulations to Deliver a Minicircle DNA Vaccine. Pharmaceutics 2021, 13, 673. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | TMZ Loading Efficiency (%) |
|---|---|
| TMZ/WRAP5 | 60.1 ± 4.8 |
| TMZ/Tf-WRAP5 | 66.4 ± 8.3 |
| System | N/P | Size (nm) | PdI | Zeta Potential (mV) | CC (%) |
|---|---|---|---|---|---|
| WRAP5/pDNA | N/P 0.1 | 398 ± 4 | 0.447 ± 0.06 | +1.49 ± 0.60 | 51.33 ± 3.06 |
| N/P 0.5 | 387 ± 3 | 0.387 ± 0.03 | +3.03 ± 0.29 | 81.67 ± 2.08 | |
| N/P 1 | 272 ± 5 | 0.300 ± 0.03 | +4.03 ± 0.11 | 89.67 ± 4.04 | |
| Tf-WRAP5/pDNA | N/P 0.1 | 349 ± 9 | 0.543 ± 0.03 | +1.78 ± 0.51 | 50.26 ± 1.53 |
| N/P 0.5 | 323 ± 5 | 0.387 ± 0.04 | +3.60 ± 0.30 | 89.33 ± 3.22 | |
| N/P 1 | 233± 6 | 0.327 ± 0.03 | +4.43 ± 0.05 | 94.33 ± 0.58 | |
| TMZ/WRAP5/pDNA | N/P 0.1 | 382 ± 5 | 0.284 ± 0.05 | +2.84 ± 0.19 | 54.56 ± 2.88 |
| N/P 0.5 | 318 ± 8 | 0.276 ± 0.04 | +7.85 ± 0.23 | 74.89 ± 1.90 | |
| N/P 1 | 179 ± 4 | 0.410 ± 0.04 | +12.55 ± 0.42 | 92.56 ± 1.81 | |
| TMZ/Tf-WRAP5/pDNA | N/P 0.1 | 392 ± 5 | 0.399 ± 0.03 | +3.93 ± 0.29 | 52.89 ± 2.15 |
| N/P 0.5 | 252 ± 6 | 0.379 ± 0.04 | +7.90 ± 0.22 | 71.44 ± 2.46 | |
| N/P 1 | 182.9 ± 4 | 0.396 ± 0.04 | +11.94 ± 0.29 | 89.56 ± 2.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves, A.R.; Albuquerque, T.; Faria, R.; Gonçalves, A.M.; Santos, C.; Vivès, E.; Boisguérin, P.; Passarinha, L.A.; Sousa, Â.; Costa, D. Development of WRAP5 Peptide Complexes for Targeted Drug/Gene Co-Delivery toward Glioblastoma Therapy. Pharmaceutics 2022, 14, 2213. https://doi.org/10.3390/pharmaceutics14102213
Neves AR, Albuquerque T, Faria R, Gonçalves AM, Santos C, Vivès E, Boisguérin P, Passarinha LA, Sousa Â, Costa D. Development of WRAP5 Peptide Complexes for Targeted Drug/Gene Co-Delivery toward Glioblastoma Therapy. Pharmaceutics. 2022; 14(10):2213. https://doi.org/10.3390/pharmaceutics14102213
Chicago/Turabian StyleNeves, Ana Raquel, Tânia Albuquerque, Rúben Faria, Ana M. Gonçalves, Cecília Santos, Eric Vivès, Prisca Boisguérin, Luís A. Passarinha, Ângela Sousa, and Diana Costa. 2022. "Development of WRAP5 Peptide Complexes for Targeted Drug/Gene Co-Delivery toward Glioblastoma Therapy" Pharmaceutics 14, no. 10: 2213. https://doi.org/10.3390/pharmaceutics14102213
APA StyleNeves, A. R., Albuquerque, T., Faria, R., Gonçalves, A. M., Santos, C., Vivès, E., Boisguérin, P., Passarinha, L. A., Sousa, Â., & Costa, D. (2022). Development of WRAP5 Peptide Complexes for Targeted Drug/Gene Co-Delivery toward Glioblastoma Therapy. Pharmaceutics, 14(10), 2213. https://doi.org/10.3390/pharmaceutics14102213