
Nano-Dry-Melting: A Novel Technology for Manufacturing of Pharmaceutical Amorphous Solid Dispersions

Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process Technology | Advantages | Disadvantages | Requirements for Processability |
|---|---|---|---|
| Solvent Based Method | |||
| Solvent-based in general | Moderate temperature, Prevention of degradation [7], Molecular level mixing [10], | Organic solvent: HSEQ * risk, [7] Economic burden, secondary drying step required [7], Expensive [10], Phase separation possible [10] | Solvent with sufficient drug solubility, low toxicity, high volatility and low explosion risk [7] |
| Spray Drying (SD) | Easy scale up [7], Single step, Control of powder shape/form, Continuous batch manufacturing [10] | - | - |
| Fluidized bed granulation/ layering | Products exhibit good flowability and tableting properties [11], Reduction in additional downstream processing steps (Coating, Granulation), Can prevent stability problems [3] | Limited drug load [11] | Core required |
| Co-precipitation | Easy scale up [5], Higher PSD* than SD particles, Superior compaction profile, High drug load possible [3] | Post-processing required (filtering, drying) [5], Risk of recrystallization during supersaturated state in antisolvent [3] | Antisolvent for all components required [12], Antisolvent and solvent must be miscible [3] |
| Freeze Drying Spray-Freeze Drying | High robustness and reliability, Minimal thermal stress, Minimal risk of phase separation [7], Highly porous particles enabling fast dissolution [3] | Expensive, Large equipment, Cryoprotective may be required [7] | Sufficient drug solubility in water or inorganic solvent miscible with water [7] |
| Processes that include supercritical fluids (SCF) | Can generate very small particles, Low temperatures, Not dependent on organic solvent, Health and environmentally friendly [10], Low production costs [3] | Difficult to scale up, Cost-intensive equipment [7], Low solubility of most pharmaceutic compounds in CO2 [10] | - |
| Fusion-Based Method | |||
| Fusion-based in general | No solvent required, No drying required [7] | High thermal load, Possible degradation [7] | Drug polymer miscibility, Thermostable substances [7] |
| Hot Melt Extrusion (HME) | High throughput, Low costs [13], Continuous manufacturing, Furrow mixing of components, Easy scale up, Drug release can be tailored [7], Shaping of product form [10] | High shear stress, Varying residence time, Miscibility determines process temperature [13] | Polymer of processible viscosity (low Tg *) Often: plasticizer(s) required |
| Melt Agglomeration | Use of standard granulation equipment, High variability in batch size, Useful for water sensitive drugs [7] | - | Carrier required [7] |
| Milling | Less thermal load (than HME), Included PSD * reduction | High mechanical stress, Degree and robustness of amorphization low/limited [7], Drying necessary [5], Typically only employed in lab scale [3] | - |
| Kinetisol® | Highly reduced processing times compared to HME (20s), Lower temperatures (than HME) [3,11], High drug load possible [3] | High mechanical load, Less stable product, High risk of drug degradation [3] | - |
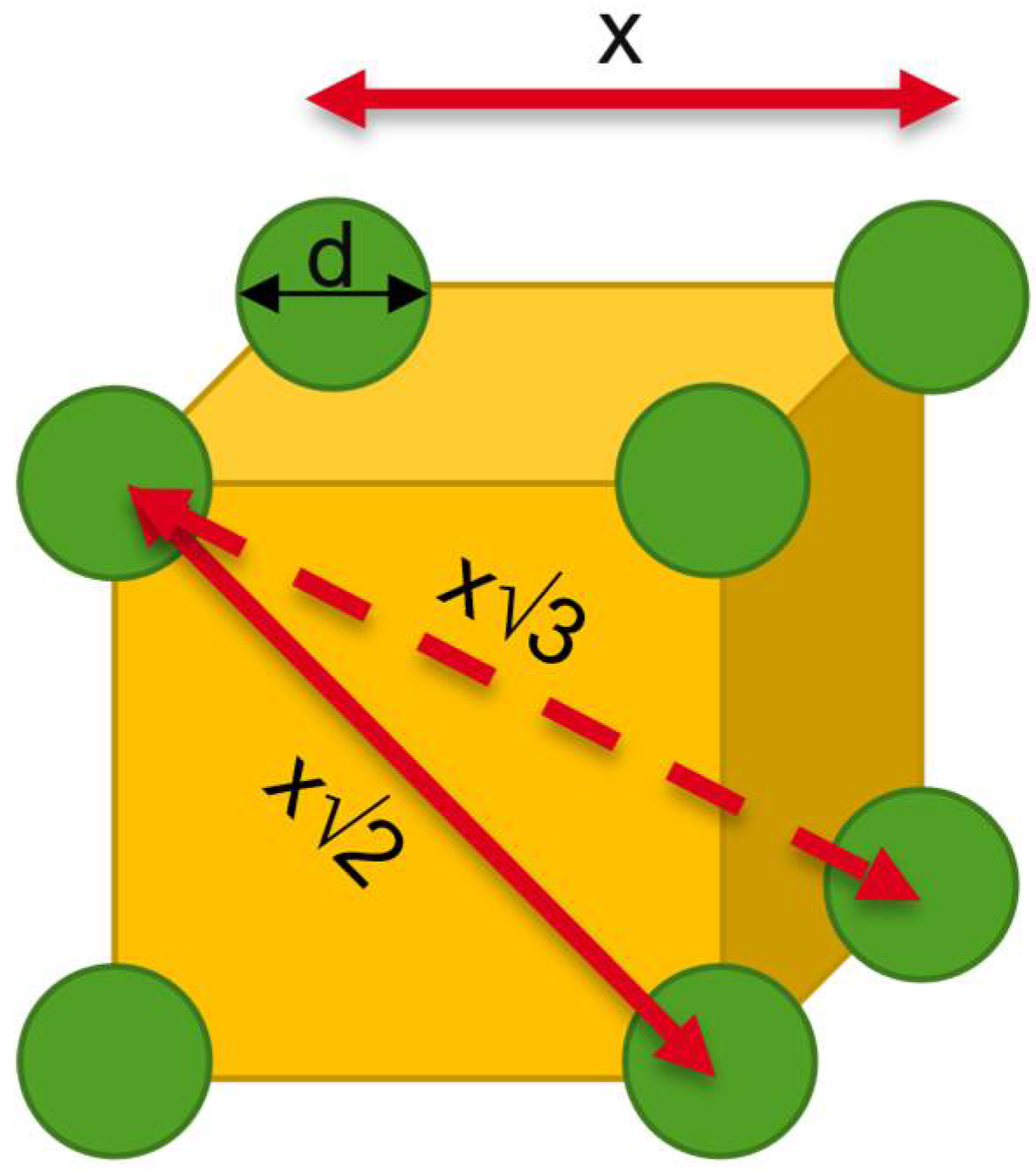
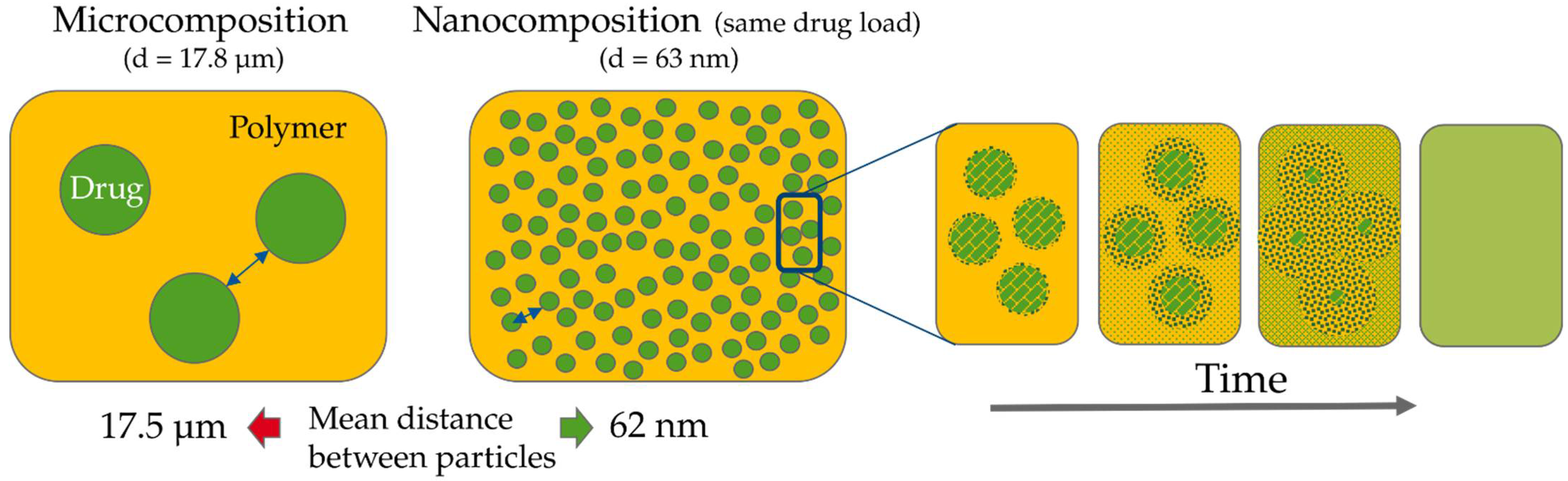
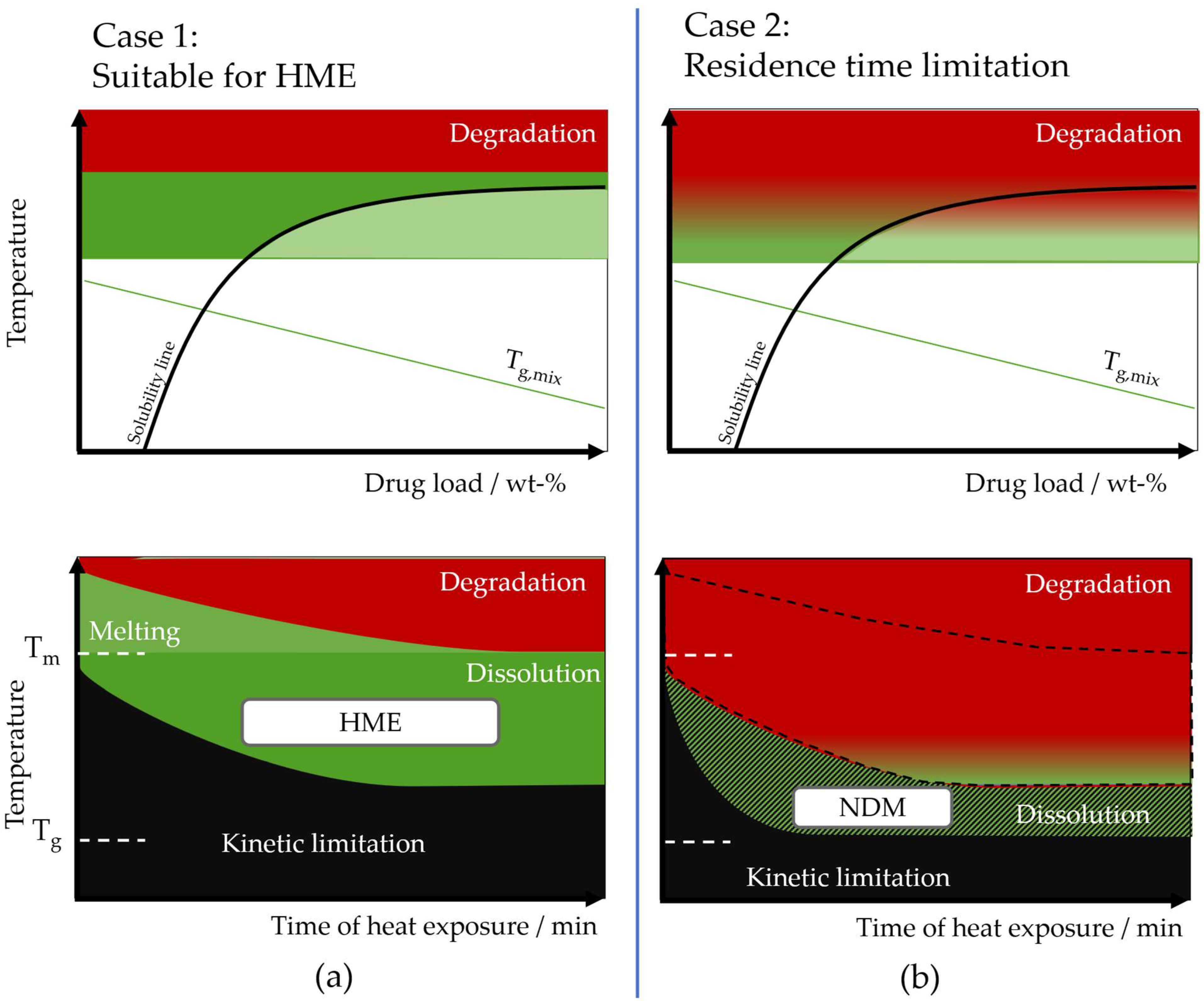
Theoretical Background of Nano-Dry-Melting
2. Materials and Methods
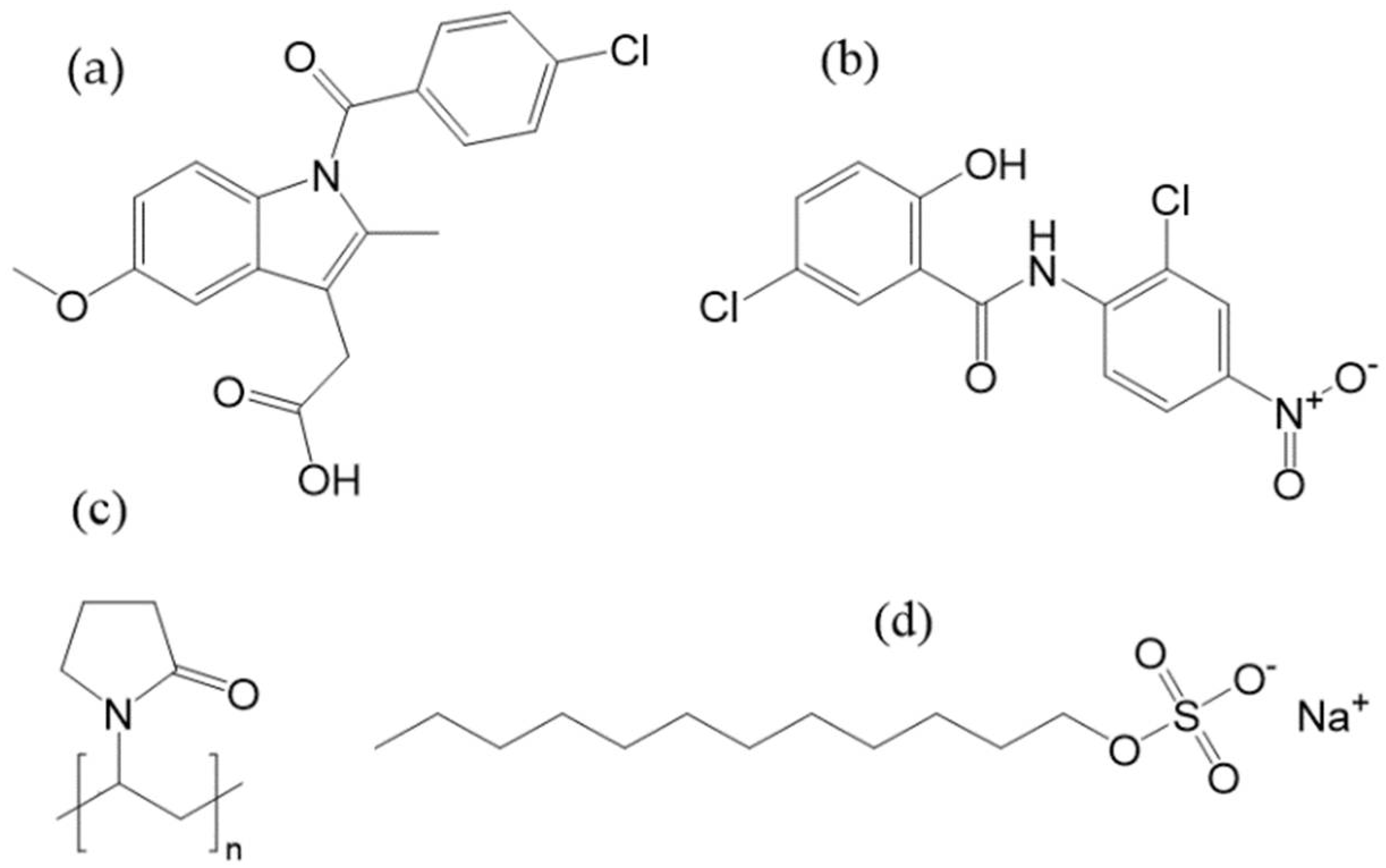
2.1. Materials
2.2. Methods
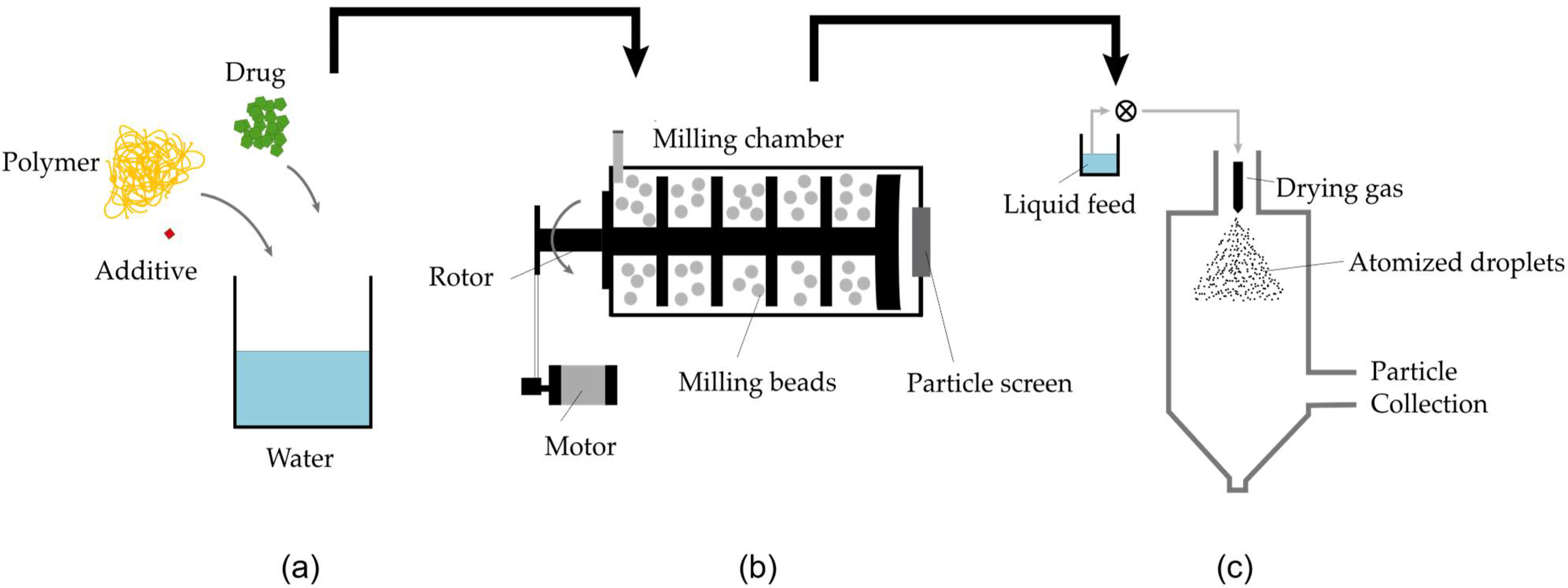
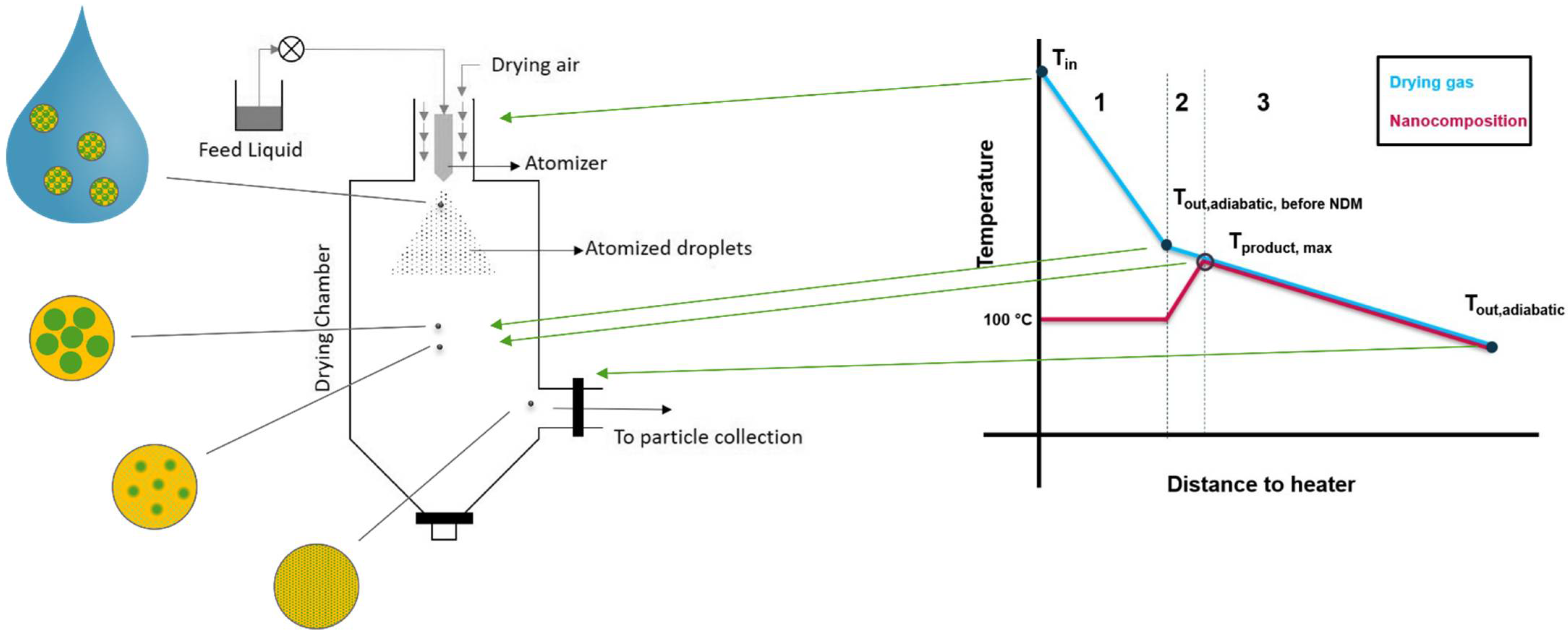
2.2.1. Technical Implementation of NDM Process
2.2.2. Milling
2.2.3. Spray Drying
2.2.4. Preparation of Physical Mixture
2.2.5. Experiments with the Kofler-Bench
2.2.6. Freeze-Drying
2.2.7. Determination of Process Conditions
- The heat transferred to the suspension will be consumed for the evaporation of water first, which is an endothermic process. It can therefore be assumed that the solid product will only heat up beyond 100 °C after most of the water is removed, and the polymer and nanoparticles are in solid state. Subsequently, the solids will heat up until the dissolution of the nanoparticle into the polymer starts.
- Since typical spray dryers in industry are not well isolated systems, the process can be assumed as non-adiabatic, meaning that the temperature will drop proportionally to the distance to the heater [26]. This heat loss is due to convection with the surroundings and heat radiation. The former one can be expressed as a function of the temperature gradient between inside the drying system and outside the system using Equation (11). The heat loss Qloss is proportional to the contact surface between dryer and surrounding A, the specific thermal conductivity κ and the temperature gradient ΔT between the dryer and the surroundings [27]. Since the temperature gradient itself changes with the distance of the heater, its course is hard to predict:
- 3.
- The energy balance of the spray dryer can be simplified as seen in Equation (12). The energy that is entering the spray dryer with incoming drying gas, Ein, is reduced to the outgoing energy, Eout, by the energy needed for evaporation, Evap., for DisP, EDisP., and the heat loss due to convection with the surroundings, Qloss. All three are summarized to Eloss (Equation (13)):
- 4.
- The maximum temperature that the dried product can reach (Tmax, product) in this process, is the temperature of the drying gas after evaporation is completed, but before the DisP starts. This temperature becomes accessible by calculating an adiabatic process with . The resulting heat loss is presented in Equation (14). It is only dependent on the energy needed for the evaporation of water. Accordingly, Tproduct,max is the outlet temperature in case of an adiabatic process, without any heat loss due to DisP: (Equation (15)):
- 5.
- Calculating this adiabatic outlet-temperature can be done graphically with the Mollier-diagram or calculated using the underlying Equation (16). The later one was done in this work. Cp are the specific isobar heat capacities, are the mass flows and is the molar evaporation enthalpy of water [28]. The used parameters are listed in the Table 3.
2.2.8. Characterization Techniques
3. Results
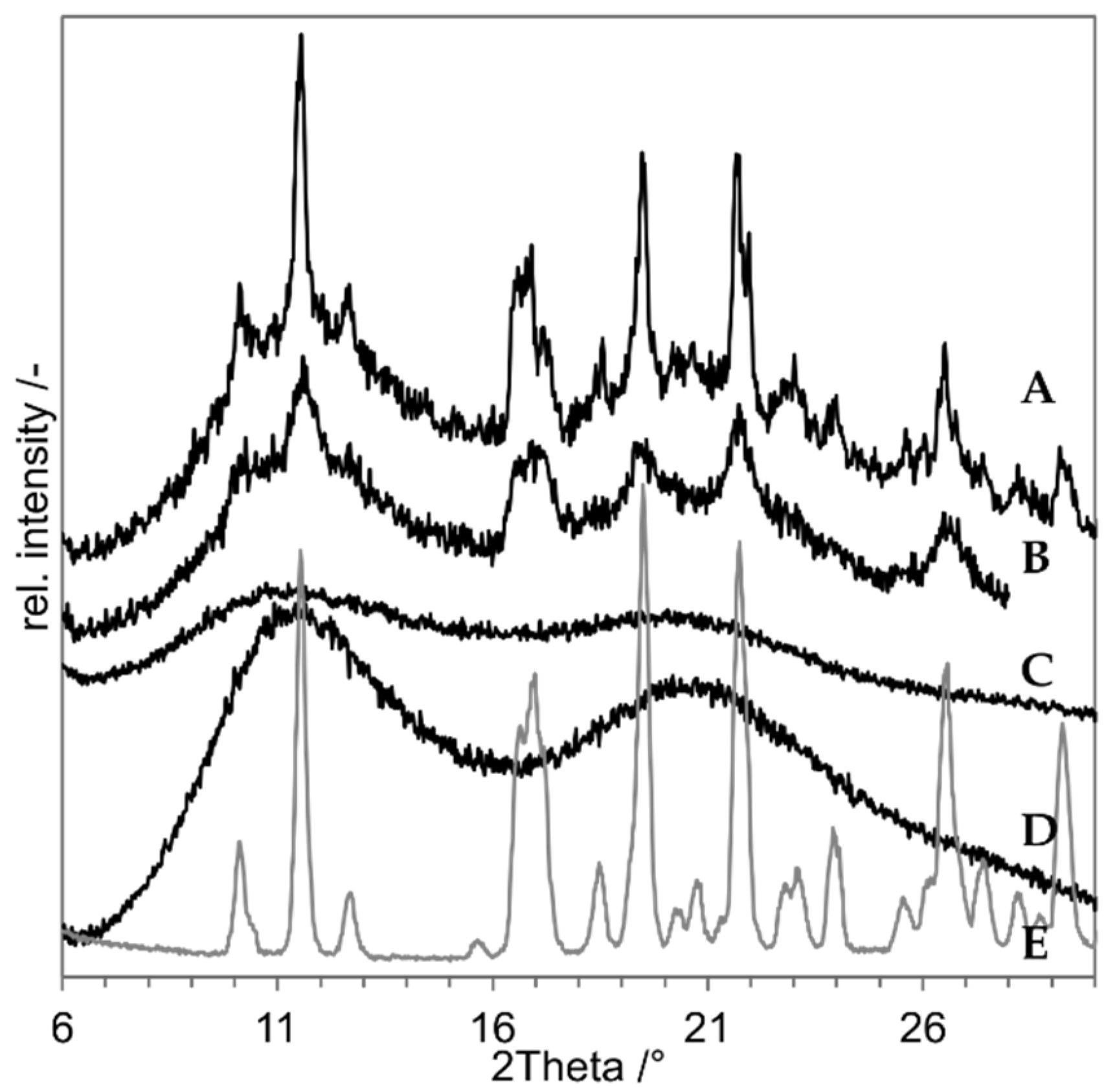
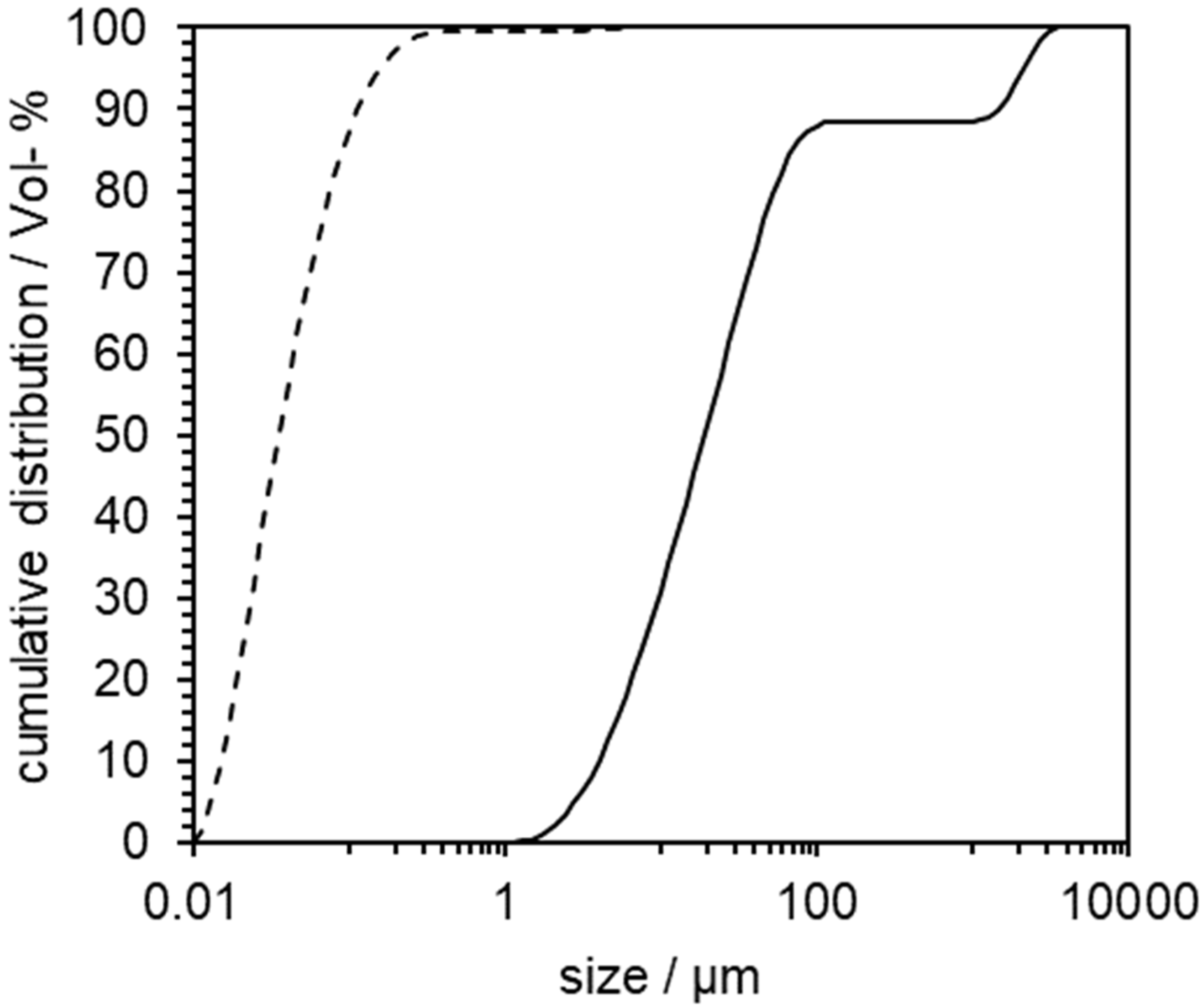
3.1. Preliminary Experiments with Indomethacin
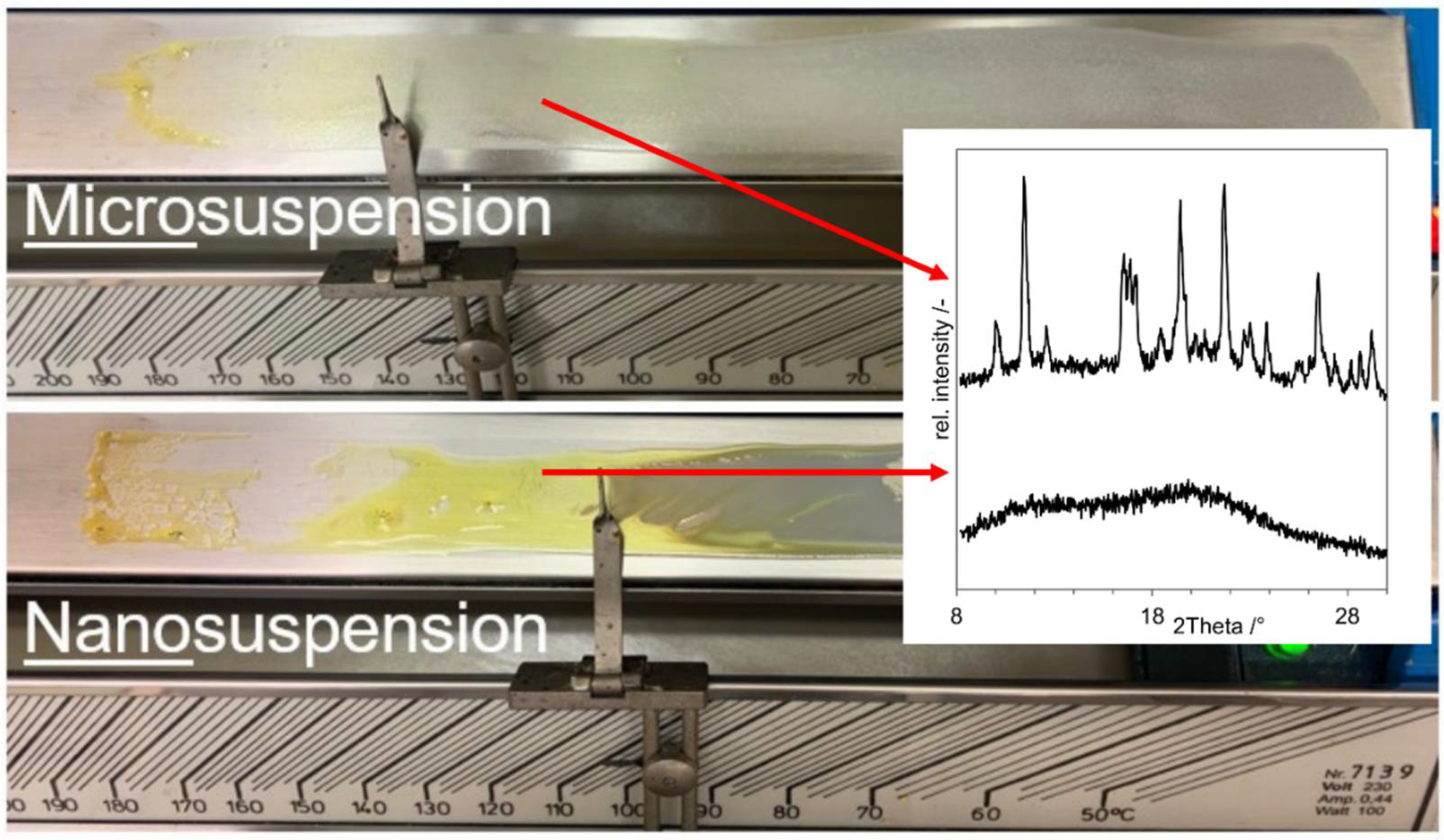
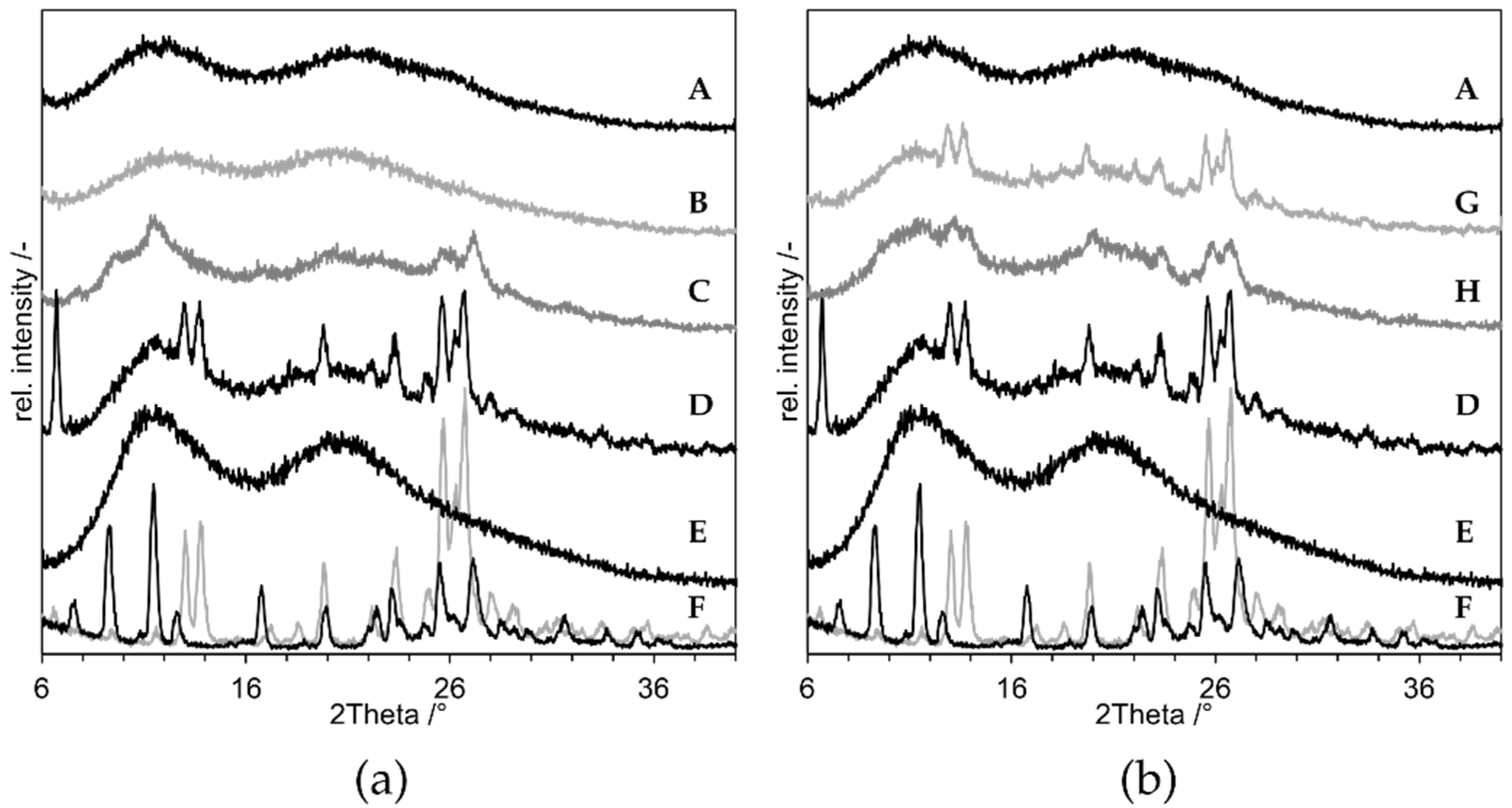
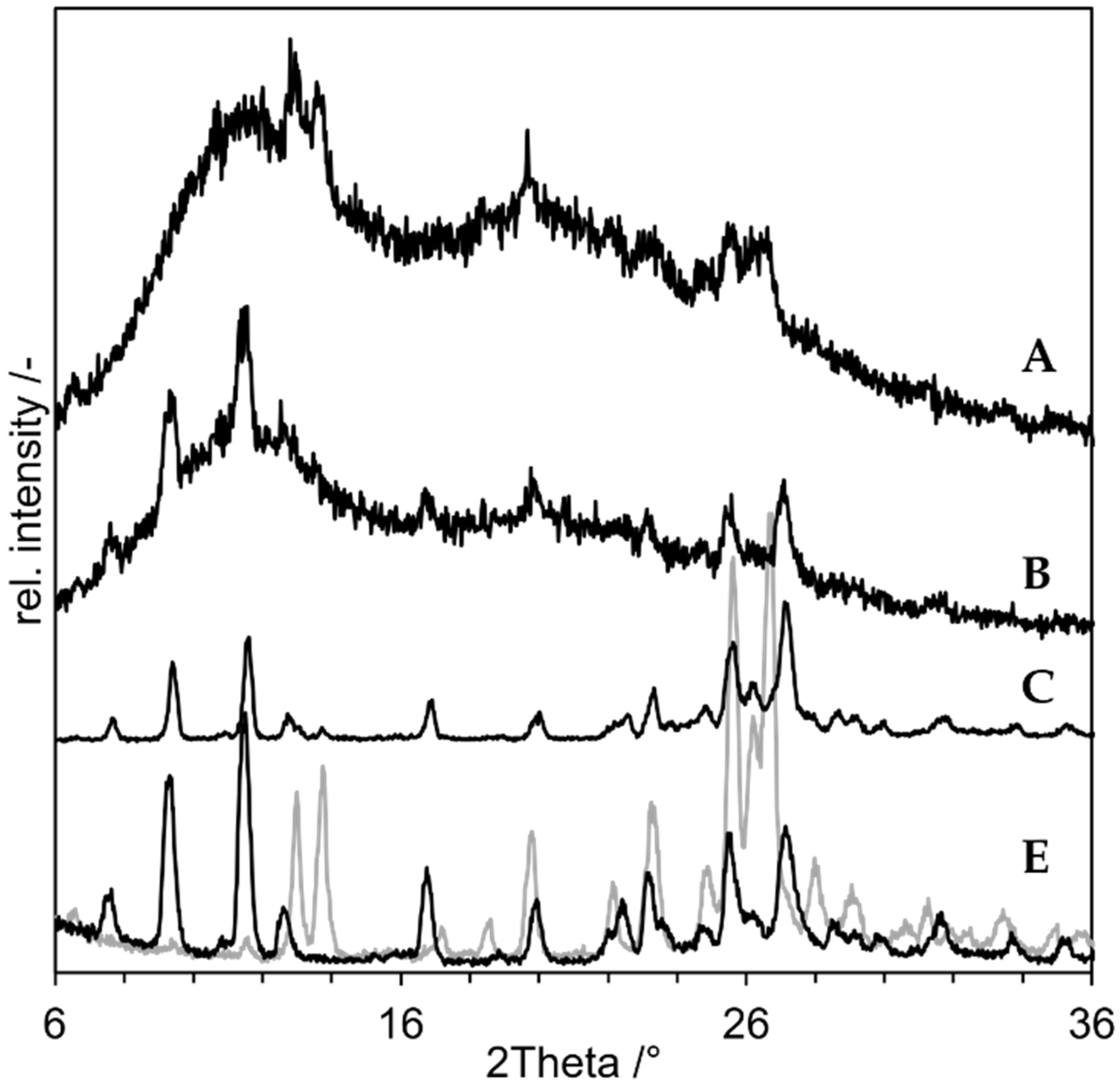
3.2. Proof of Concept
- As negative control, a physical mixture of the components (“PM”, D in Figure 8);
- As positive control, an ASD prepared by solvent evaporation from organic solution (”ASD-SB”, A in Figure 8);
- Microcompositions which were spray dried with Tin = 220 °C and Tin = 120 °C (“Micro-DM”, G and H in Figure 8);
- Nanocompositions which were spray dried with Tin = 220 °C and Tin = 120 °C (“NDM”, B and C in Figure 8).
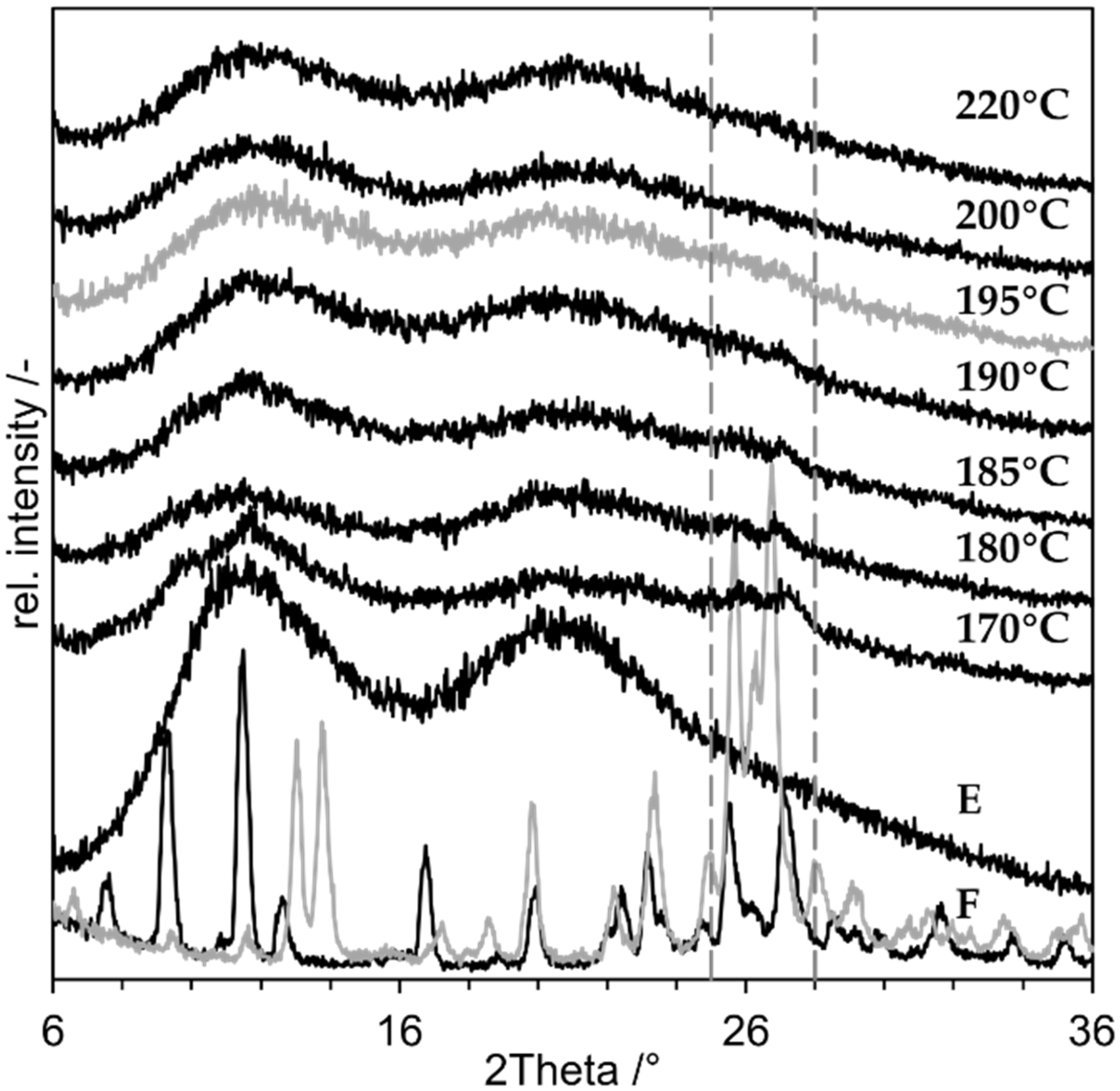
3.3. Minimum Process Temperature
3.4. Impact of Monohydrate Formation
4. Discussion
4.1. Feasibility of Nano-Dry-Melting
4.2. Nano-Dry-Melting as Novel, Alternative Process Technology
- It only requires water and no organic solvents (in contrast to solvent-based methods), which is advantageous for economic as well as for ecological reasons;
- It exposes the sample to lower temperatures for a shorter time (in contrast to fusion-based methods) which minimizes thermal stress to the product.
4.3. Analytical Considerations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bryant, M.J.; Black, S.N.; Blade, H.; Docherty, R.; Maloney, A.G.P.; Taylor, S.C. The CSD Drug Subset: The Changing Chemistry and Crystallography of Small Molecule Pharmaceuticals. J. Pharm. Sci. 2019, 108, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.Y.; Boersen, N.A.; Hui, H.W.; Chow, S.F.; Wan, K.Y.; Chow, A.H.L. Delivery of poorly soluble compounds by amorphous solid dispersions. Curr. Pharm. Des. 2014, 20, 303–324. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Tony Zhou, Q. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Shah, N. (Ed.) Amorphous Solid Dispersions: Theory and Practice; Springer Science and Business Media: Dordrecht, The Netherlands, 2014; ISBN 978-1-4939-1597-2. [Google Scholar]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Dedroog, S.; Huygens, C.; van den Mooter, G. Chemically identical but physically different: A comparison of spray drying, hot melt extrusion and cryo-milling for the formulation of high drug loaded amorphous solid dispersions of naproxen. Eur. J. Pharm. Biopharm. 2019, 135, 1–12. [Google Scholar] [CrossRef]
- Sihorkar, V.; Dürig, T. Chapter 5—The role of polymers and excipients in developing amorphous solid dispersions: An industrial perspective. In Drug Delivery Aspects: Volume 4: Expectations and Realities of Multifunctional Drug Delivery Systems; Shegokar, R., Ed.; Elsevier: San Diego, CA, USA, 2020; pp. 79–113. ISBN 978-0-12-821222-6. [Google Scholar]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Iyer, R.; Petrovska Jovanovska, V.; Berginc, K.; Jaklič, M.; Fabiani, F.; Harlacher, C.; Huzjak, T.; Sanchez-Felix, M.V. Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development. Pharmaceutics 2021, 13, 1682. [Google Scholar] [CrossRef]
- Hou, H.H.; Rajesh, A.; Pandya, K.M.; Lubach, J.W.; Muliadi, A.; Yost, E.; Jia, W.; Nagapudi, K. Impact of Method of Preparation of Amorphous Solid Dispersions on Mechanical Properties: Comparison of Coprecipitation and Spray Drying. J. Pharm. Sci. 2019, 108, 870–879. [Google Scholar] [CrossRef]
- Seiler, V.K.; Theil, F.; Nagel, N.; van Lishaut, H. Adding a New Dimension to the Amorphous Solid Dispersion Phase Diagram: Studying Dissolution Kinetics of Crystalline Drugs in a Polymer Matrix Using Temperature Dependent XRPD and DSC. J. Pharm. Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.G. Nano-Dry Melting. WO-2021084139-A1, 23 March 2020. [Google Scholar]
- Li, M.; Azad, M.; Davé, R.; Bilgili, E. Nanomilling of Drugs for Bioavailability Enhancement: A Holistic Formulation-Process Perspective. Pharmaceutics 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Moseson, D.E.; Taylor, L.S. The application of temperature-composition phase diagrams for hot melt extrusion processing of amorphous solid dispersions to prevent residual crystallinity. Int. J. Pharm. 2018, 553, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Anhalt, K.; Geissler, S.; Harms, M.; Weigandt, M.; Fricker, G. Development of a new method to assess nanocrystal dissolution based on light scattering. Pharm. Res. 2012, 29, 2887–2901. [Google Scholar] [CrossRef]
- Bisrat, M.; Anderberg, E.K.; Barnett, M.I.; Nyström, C. Physicochemical aspects of drug release. XV. Investigation of diffusional transport in dissolution of suspended, sparingly soluble drugs. Int. J. Pharm. 1992, 80, 191–201. [Google Scholar] [CrossRef]
- Hintz, R.; Johnson, K. The effect of particle size distribution on dissolution rate and oral absorption. Int. J. Pharm. 1989, 51, 9–17. [Google Scholar] [CrossRef]
- Galli, C. Experimental determination of the diffusion boundary layer width of micron and submicron particles. Int. J. Pharm. 2006, 313, 114–122. [Google Scholar] [CrossRef]
- Hao, T.; Riman, R.E. Calculation of interparticle spacing in colloidal systems. J. Colloid Interface Sci. 2006, 297, 374–377. [Google Scholar] [CrossRef]
- Baird, J.A.; van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Van Tonder, E.C.; Maleka, T.S.P.; Liebenberg, W.; Song, M.; Wurster, D.E.; de Villiers, M.M. Preparation and physicochemical properties of niclosamide anhydrate and two monohydrates. Int. J. Pharm. 2004, 269, 417–432. [Google Scholar] [CrossRef]
- Ousset, A.; Meeus, J.; Robin, F.; Schubert, M.; Somville, P.; Dodou, K. Comparison of a Novel Miniaturized Screening Device with Büchi B290 Mini Spray-Dryer for the Development of Spray-Dried Solid Dispersions (SDSDs). Processes 2018, 6, 129. [Google Scholar] [CrossRef]
- Mini Spray Dryer B-290: Technical Data Sheet. Available online: https://assets.buchi.com/image/upload/v1605791039/pdf/Technical-Datasheet/TDS_11594126_B-290.pdf (accessed on 30 August 2022).
- Baker, C.G.J.; McKenzie, K.A. Energy Consumption of Industrial Spray Dryers. Dry. Technol. 2005, 23, 365–386. [Google Scholar] [CrossRef]
- VDI-Wärmeatlas: Mit 320 Tabellen; VDI-Gesellschaft Verfahrenstechnik und Chemieingenieurwesen, 11., bearb. und erw. Aufl.; Springer Vieweg: Berlin, Germany, 2013; ISBN 978-3-642-19981-3.
- Atkins, P.W.; Paula, J.d.; Bär, M. Physikalische Chemie, 5th ed.; Wiley-VCH Verl.: Weinheim, Germany, 2013; ISBN 3527682899. [Google Scholar]
- Stephan, K.; Mayinger, F. Thermodynamik: Band 1: Einstoffsysteme. Grundlagen und technische Anwendungen, 15th ed.; Springer: Berlin, Heidelberg, Germany, 1998; ISBN 978-3-662-13213-5. [Google Scholar]
- ChemicalBook. Indometacin|53-86-1. Available online: https://www.chemicalbook.com/ChemicalProductProperty_EN_CB1750267.htm (accessed on 19 September 2022).
- chemspider. Niclosamide|C13H8Cl2N2O4. Available online: http://www.chemspider.com/Chemical-Structure.4322.html (accessed on 27 January 2021).
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Von Der Ges. Der Wiss. Zu Göttingen Math. Phys. Kl. 1918, 1918, 98–100. [Google Scholar]
- Knopp, M.M.; Olesen, N.E.; Holm, P.; Langguth, P.; Holm, R.; Rades, T. Influence of Polymer Molecular Weight on Drug-Polymer Solubility: A Comparison between Experimentally Determined Solubility in PVP and Prediction Derived from Solubility in Monomer. J. Pharm. Sci. 2015, 104, 2905–2912. [Google Scholar] [CrossRef]
- Brady, J.; Dürig, T.; Lee, P.I.; Li, J.-X. Polymer Properties and Characterization. In Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice, 2nd ed.; Qiu, Y., Chen, Y., Zhang, G.G., Yu, L., Mantri, R.V., Eds.; Elsevier Science: Saint Louis, MO, USA, 2016; pp. 181–223. ISBN 9780128024478. [Google Scholar]
- Ma, X.; Müller, F.; Huang, S.; Lowinger, M.; Liu, X.; Schooler, R.; Williams Iii, R.O. Influence of Carbamazepine Dihydrate on the Preparation of Amorphous Solid Dispersions by Hot Melt Extrusion. Pharmaceutics 2020, 12, 379. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R. Particle engineering in pharmaceutical solids processing: Surface energy considerations. Curr. Pharm. Des. 2015, 21, 2677–2694. [Google Scholar] [CrossRef]
- Jain, M.; Ganesh, L.; Manoj, B.; Randhir, C.; Shashikant, B.; Chirag, S. Spray Drying in Pharmaceutical Industry: A Review. Res. J. Pharm. Dos. Technol. 2012, 4, 74–79. [Google Scholar]
- Bhonsale, S.; Muñoz López, C.A.; van Impe, J. Global Sensitivity Analysis of a Spray Drying Process. Processes 2019, 7, 562. [Google Scholar] [CrossRef]
- Schmitz-Schug, I.; Foerst, P.; Kulozik, U. Impact of the spray drying conditions and residence time distribution on lysine loss in spray dried infant formula. Dairy Sci. Technol. 2013, 93, 443–462. [Google Scholar] [CrossRef]
- Jeantet, R.; Ducept, F.; Dolivet, A.; Méjean, S.; Schuck, P. Residence time distribution: A tool to improve spray-drying control. Dairy Sci. Technol. 2008, 88, 31–43. [Google Scholar] [CrossRef]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-Melt Extrusion: From Theory to Application in Pharmaceutical Formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef] [PubMed]
- Kyeremateng, S.O.; Voges, K.; Dohrn, S.; Sobich, E.; Lander, U.; Weber, S.; Gessner, D.; Evans, R.C.; Degenhardt, M. A Hot-Melt Extrusion Risk Assessment Classification System for Amorphous Solid Dispersion Formulation Development. Pharmaceutics 2022, 14, 1044. [Google Scholar] [CrossRef]
- Mujumdar, A.S. Handbook of Industrial Drying; CRC Press: Boca Raton, FL, USA, 2006; ISBN 9780429136092. [Google Scholar]
- Lee, S.L.; O’Connor, T.F.; Yang, X.; Cruz, C.N.; Chatterjee, S.; Madurawe, R.D.; Moore, C.M.V.; Yu, L.X.; Woodcock, J. Modernizing Pharmaceutical Manufacturing: From Batch to Continuous Production. J. Pharm Innov. 2015, 10, 191–199. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Theoretical and experimental investigation of drug-polymer interaction and miscibility and its impact on drug supersaturation in aqueous medium. Eur. J. Pharm. Biopharm. 2016, 107, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Lauer, M.E.; Grassmann, O.; Siam, M.; Tardio, J.; Jacob, L.; Page, S.; Kindt, J.H.; Engel, A.; Alsenz, J. Atomic force microscopy-based screening of drug-excipient miscibility and stability of solid dispersions. Pharm. Res. 2011, 28, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Purohit, H.S.; Taylor, L.S. Phase separation kinetics in amorphous solid dispersions upon exposure to water. Mol. Pharm. 2015, 12, 1623–1635. [Google Scholar] [CrossRef]













| Ingredient | Function | Indomethacin Suspension | Niclosamide Suspension | ||
|---|---|---|---|---|---|
| Concentration in Suspension/wt.-% | Concentration in Solid/wt.-% | Concentration in Suspension/wt.-% | Concentration in Solid/wt.-% | ||
| Indomethacin | Drug | 6.6 | 33 | ||
| Niclosamide | Drug | 1.89 | 19.7 | ||
| PVP K25 | Stabilizer | 7.56 | 78.8 | ||
| PVP K12 | Stabilizer | 13.2 | 66 | ||
| SDS | Surfactant | 0.02 | 1 | 0.15 | 1.5 |
| Water | Fluid | 80 | - | 90.40 | - |
| Term | Description | Value | Reference |
|---|---|---|---|
| Specific heat capacity of air | [29] | ||
| specific enthalpy of vaporization of water | 225,700 | [29] | |
| Mass flow of drying gas | Depending on experimental setup | ||
| Mass flow of water in feed | Depending on experimental setup | ||
| Inlet temperature of drying gas | Depending on experimental setup | Direct setpoint on spray dryer |
| Property | Conventional SD | SD for DM |
|---|---|---|
| Temperature | As low as possible T ~ Tvap.H20 * | As high as necessary T ~ Tg > Tvap.H20 * |
| Residence time | Shortest residence time possible | Sufficiently long residence time for Evaporation + DisP |
| Droplet formation | Gentle nozzle pressure to generate big droplets resulting in big particles with good flowability properties | Very high nozzle pressure to create small droplets and enable fast evaporation |
| Residual moisture | Generally higher due to slow evaporation rate, Residual moisture can be removed in additional drying process | Generally low since evaporation rate should be high |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermeling, M.; Nueboldt, C.; Heumann, R.; Hoheisel, W.; Breitkreutz, J. Nano-Dry-Melting: A Novel Technology for Manufacturing of Pharmaceutical Amorphous Solid Dispersions. Pharmaceutics 2022, 14, 2145. https://doi.org/10.3390/pharmaceutics14102145
Hermeling M, Nueboldt C, Heumann R, Hoheisel W, Breitkreutz J. Nano-Dry-Melting: A Novel Technology for Manufacturing of Pharmaceutical Amorphous Solid Dispersions. Pharmaceutics. 2022; 14(10):2145. https://doi.org/10.3390/pharmaceutics14102145
Chicago/Turabian StyleHermeling, Malin, Christoph Nueboldt, Roman Heumann, Werner Hoheisel, and Joerg Breitkreutz. 2022. "Nano-Dry-Melting: A Novel Technology for Manufacturing of Pharmaceutical Amorphous Solid Dispersions" Pharmaceutics 14, no. 10: 2145. https://doi.org/10.3390/pharmaceutics14102145
APA StyleHermeling, M., Nueboldt, C., Heumann, R., Hoheisel, W., & Breitkreutz, J. (2022). Nano-Dry-Melting: A Novel Technology for Manufacturing of Pharmaceutical Amorphous Solid Dispersions. Pharmaceutics, 14(10), 2145. https://doi.org/10.3390/pharmaceutics14102145