
Two Novel Palbociclib-Resorcinol and Palbociclib-Orcinol Cocrystals with Enhanced Solubility and Dissolution Rate


 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cocrystals Preparation
2.3. Single Crystal X-ray Diffraction (SCXRD)
2.4. Hirshfeld Surface Analysis
2.5. Molecular Electrostatic Potential Analysis
2.6. Powder X-ray Diffraction (PXRD)
2.7. Thermal Analyses
2.8. Attenuated Total Reflection Fourier’s Transform Infrared Spectroscopy (ATR-FTIR)
2.9. Scanning Electron Microscopy (SEM)
2.10. Intrinsic Dissolution Rate (IDR) Experiments
2.11. In Vitro Dissolution Studies
2.12. Stability Studies
2.13. In Vivo Pharmacokinetic (PK) Experiments
2.14. In Vitro Cytotoxicity Assay
3. Results
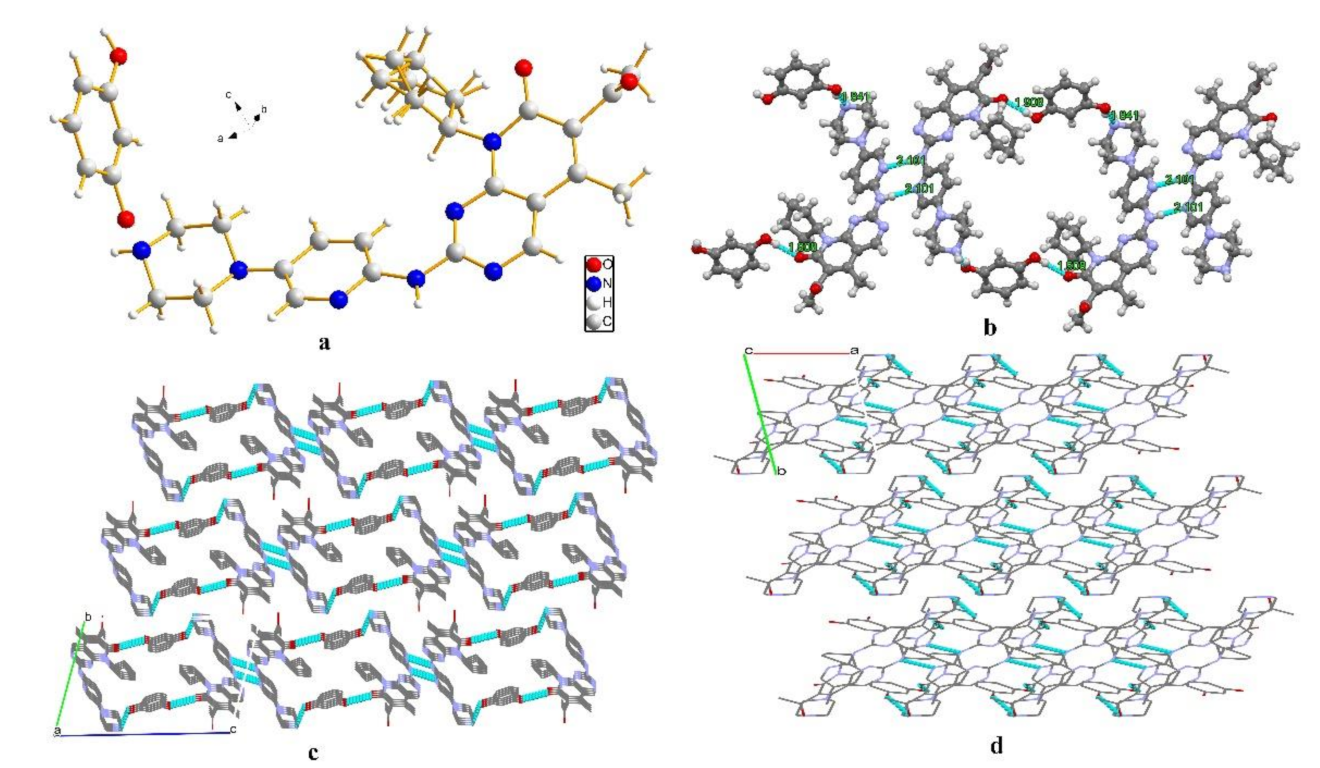
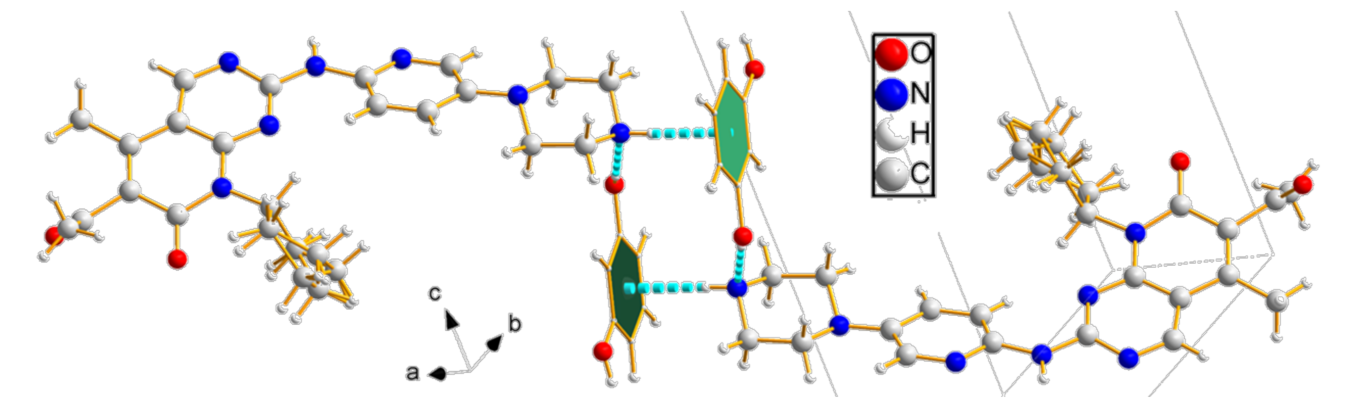
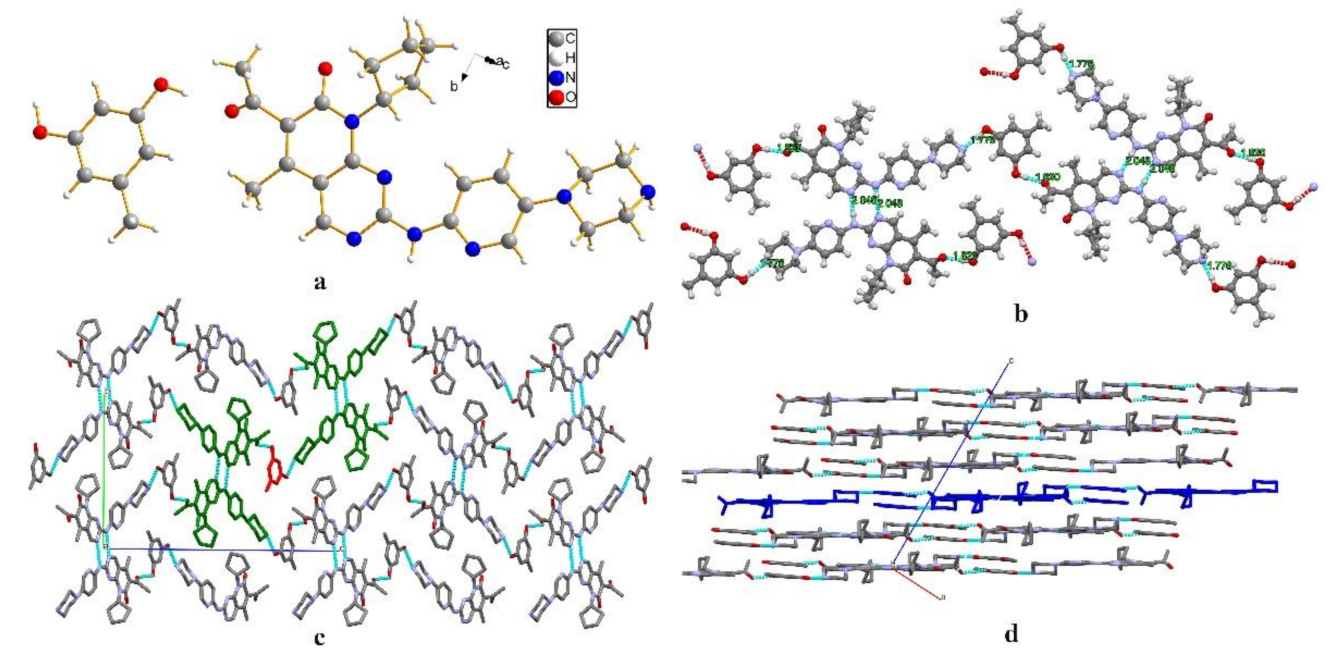
3.1. Single-Crystal X-ray Diffraction (SCXRD)
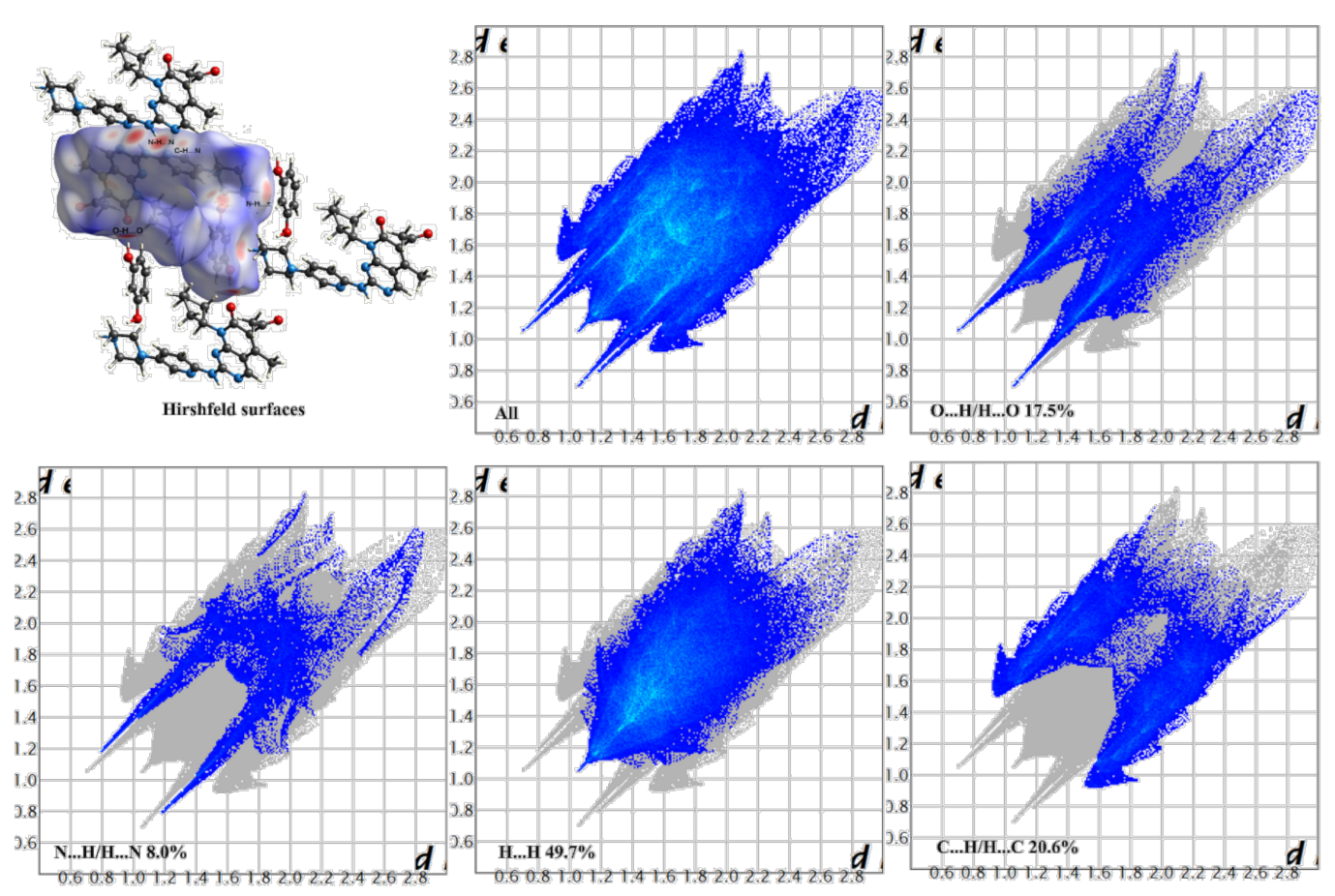
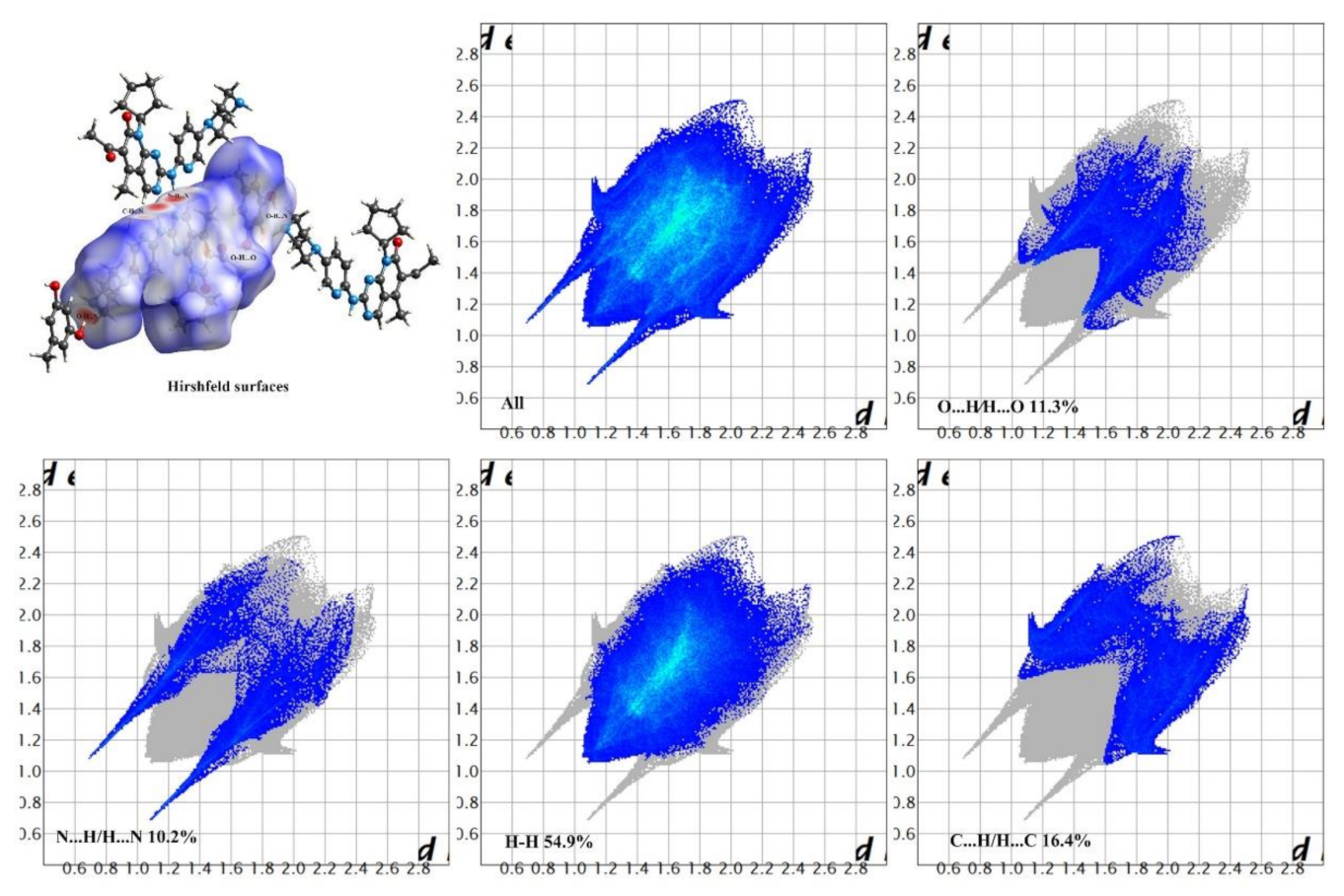
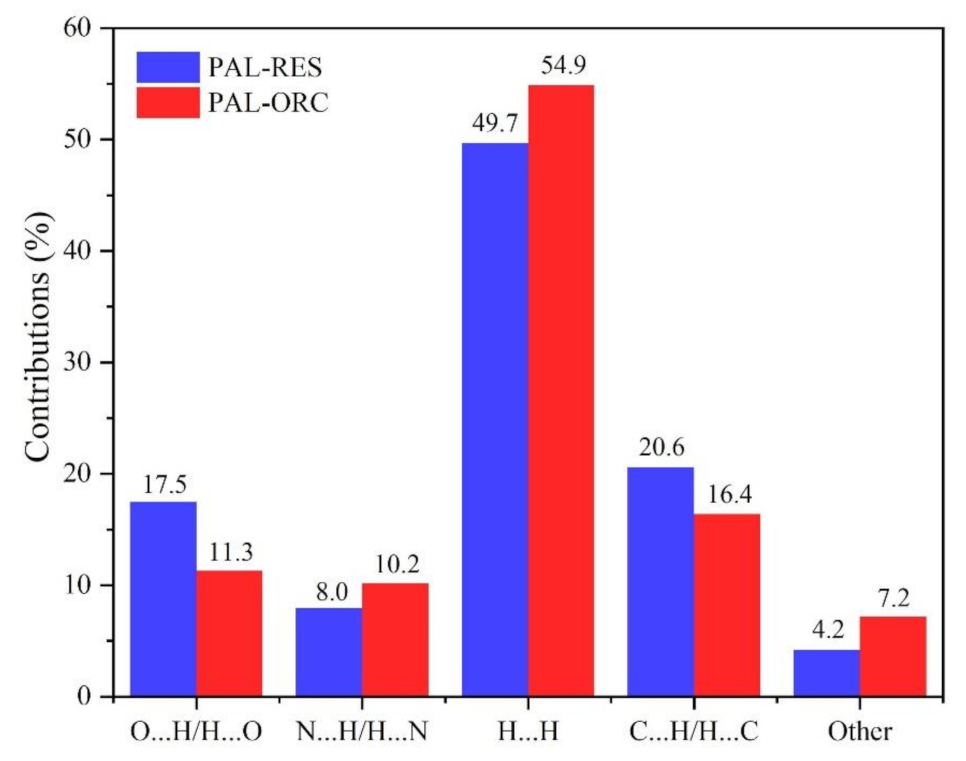
3.2. Hirshfeld Surfaces and 2D Fingerprint Plots
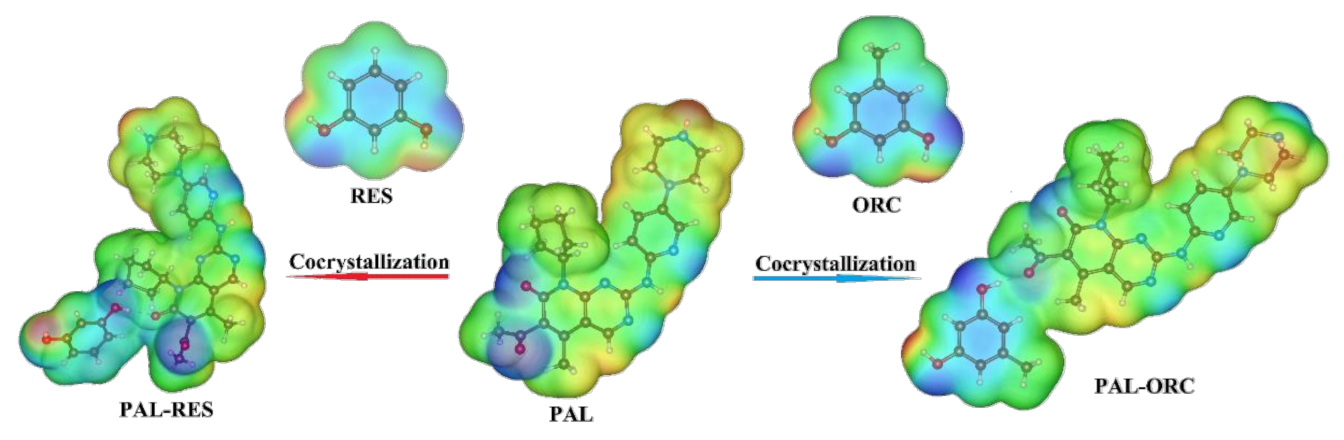
3.3. Molecular Electrostatic Potential Analysis
3.4. Powder X-ray Diffraction Analysis (PXRD)
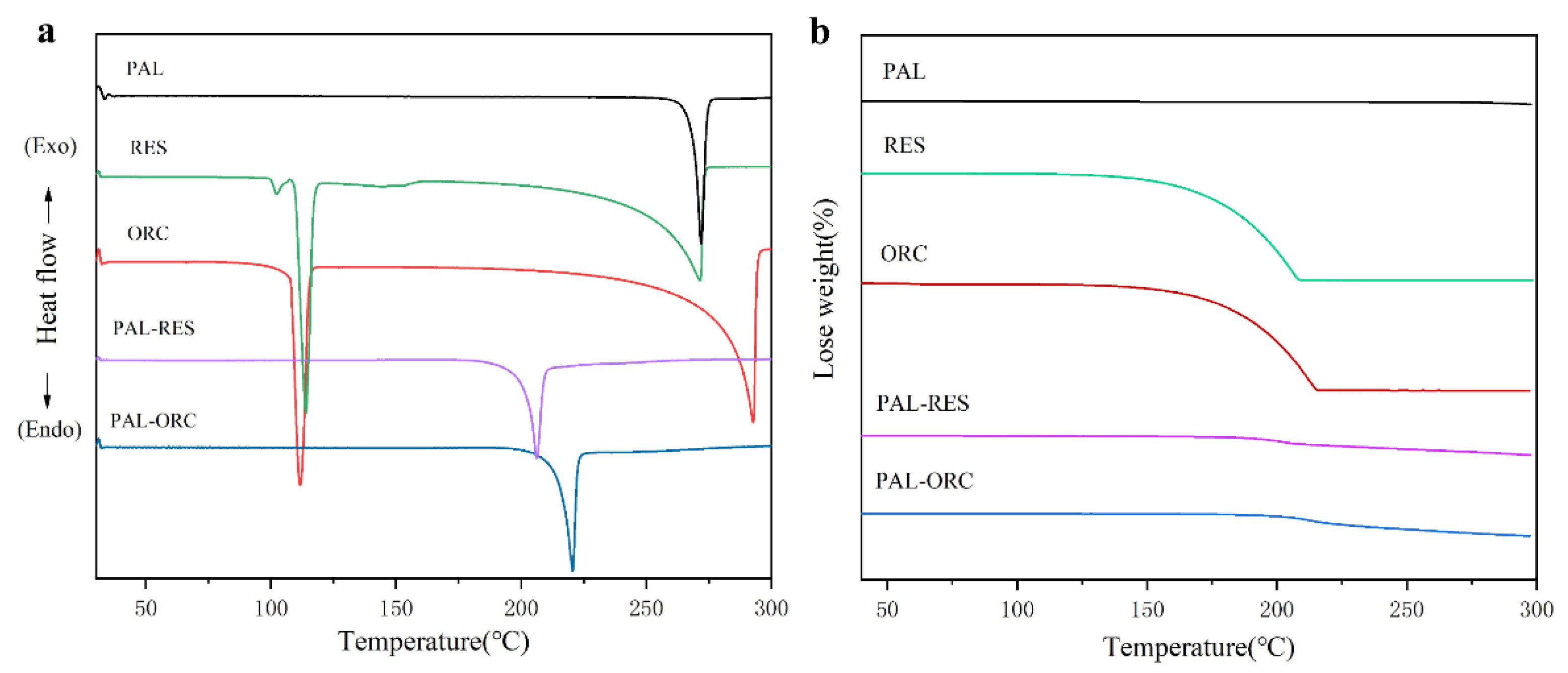
3.5. Thermal Analysis
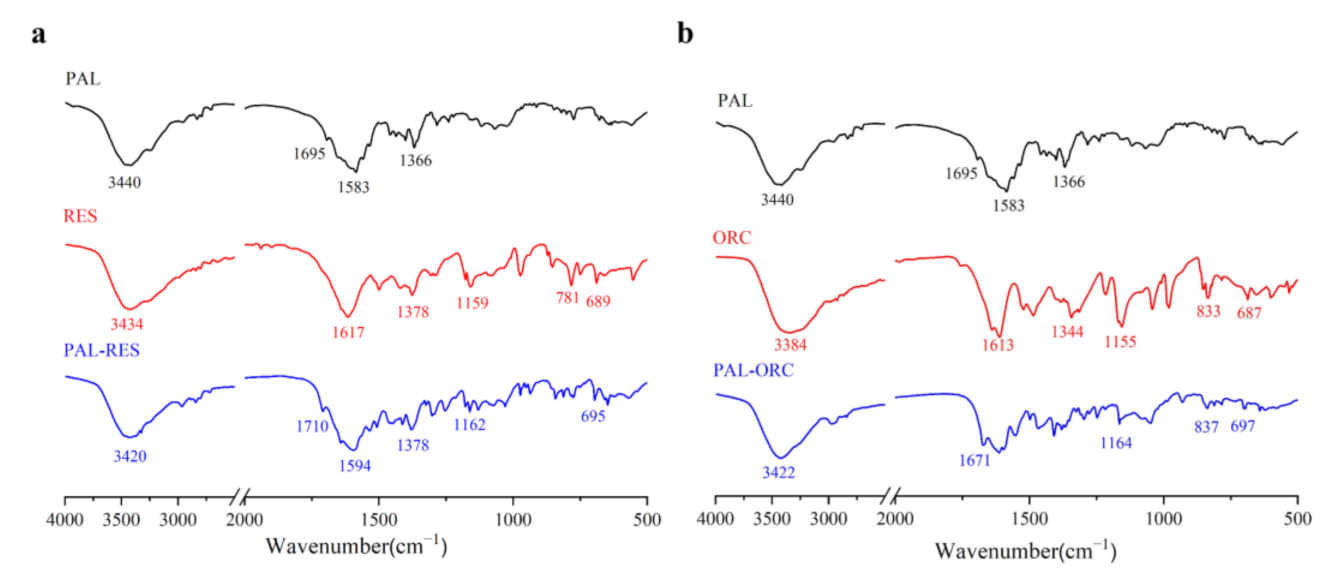
3.6. Attenuated Total Reflection Fourier’s Transform Infrared Spectroscopy (ATR-FTIR)
3.7. Scanning Electron Microscopy (SEM) Analysis
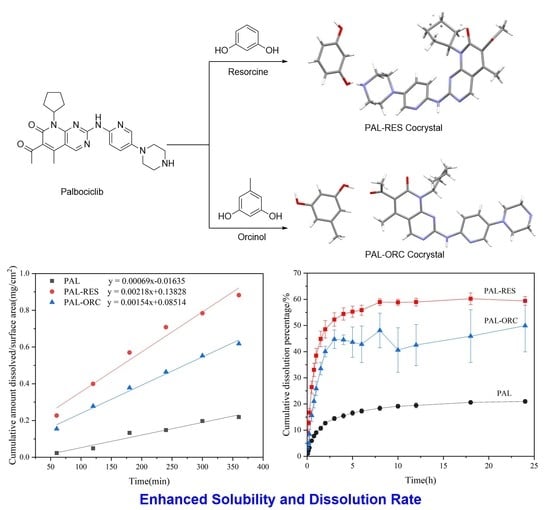
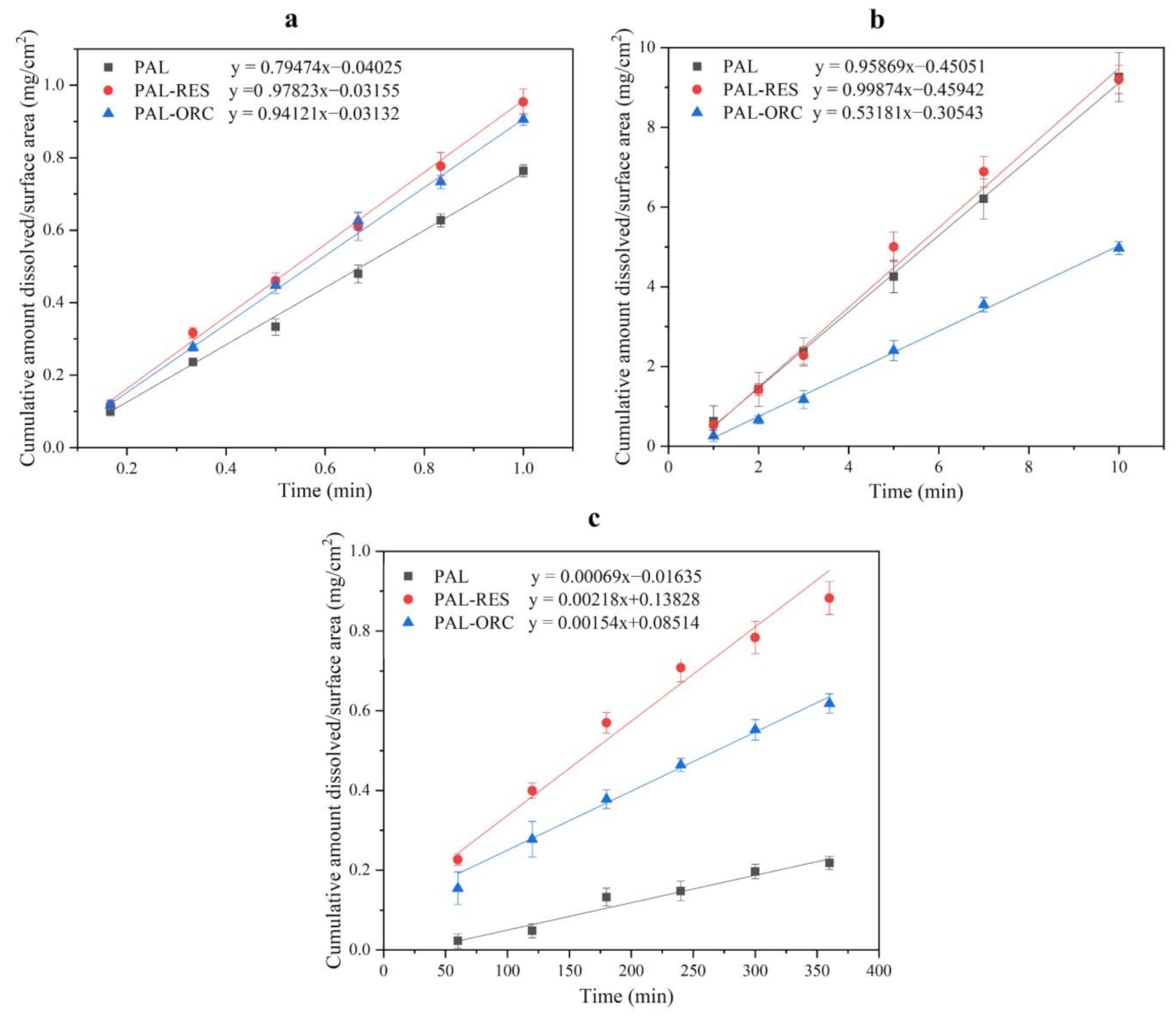
3.8. Intrinsic DissolutionRate (IDR) Experiments
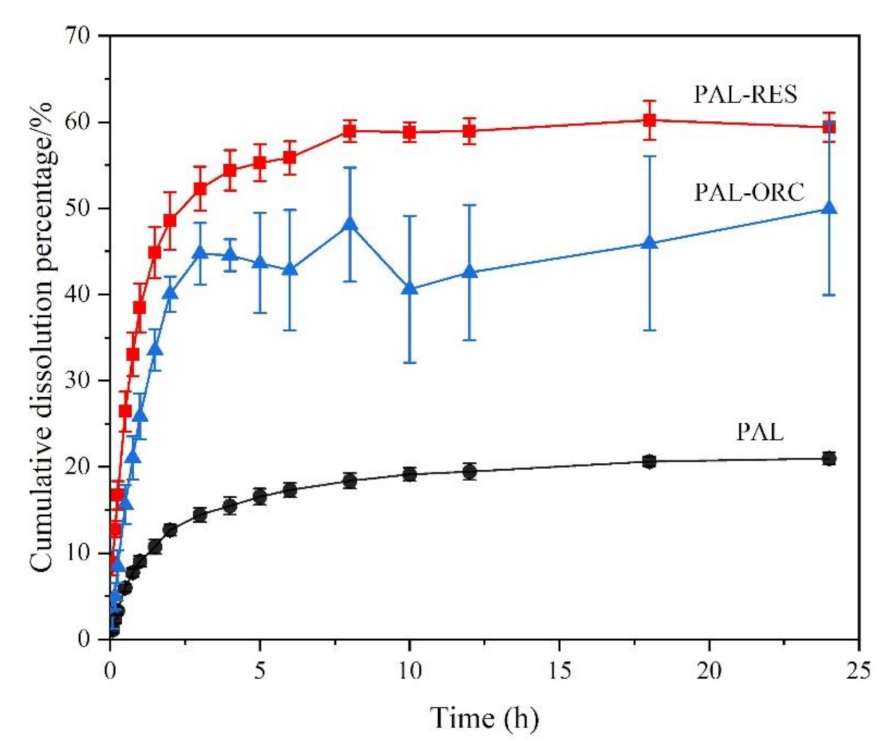
3.9. Powder Dissolution Analysis
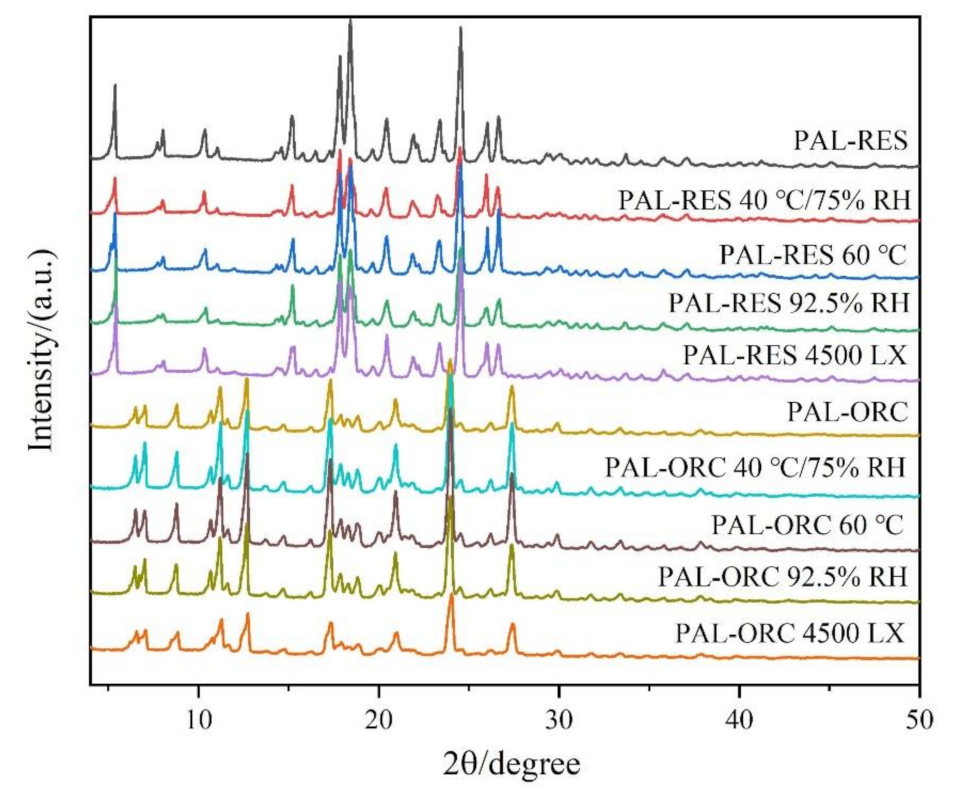
3.10. Stability
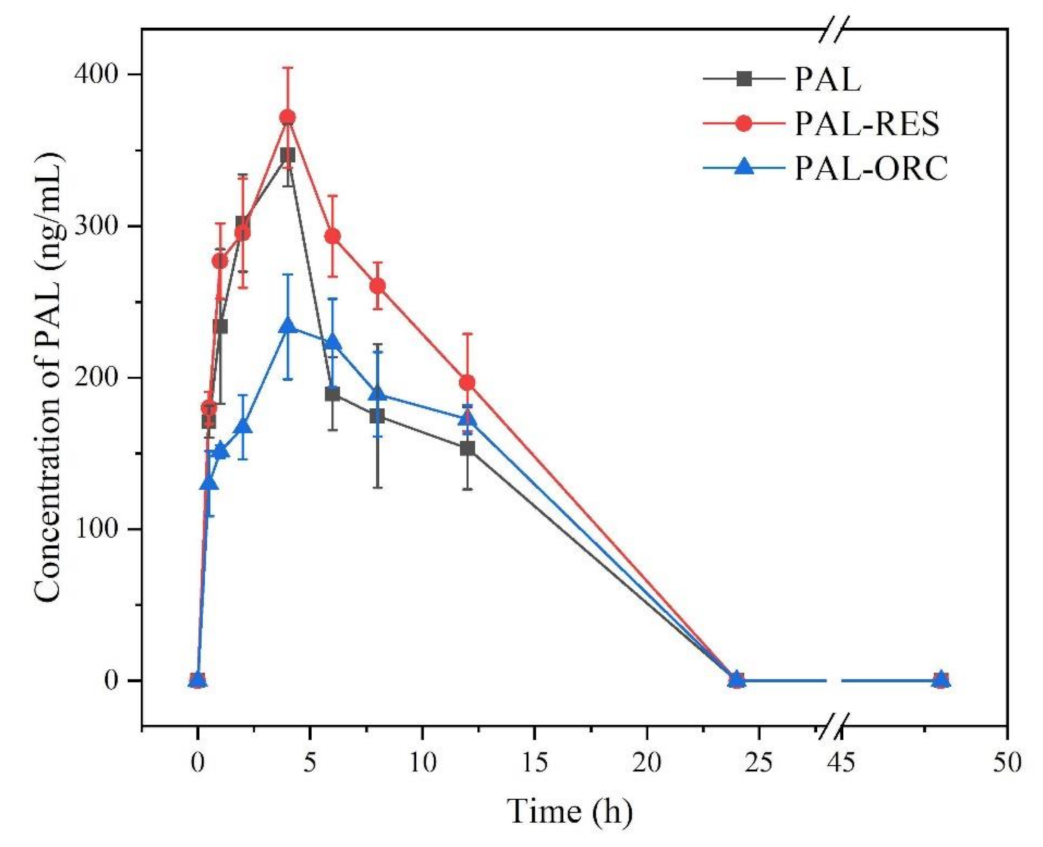
3.11. In Vivo Pharmacokinetic (PK) Experiments
3.12. In Vitro Cytotoxicity Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jirat, J.; Ondo, D.; Babor, M.; Ridvan, L.; Soos, M. Complex methodology for rational design of Apremilast-benzoic acid co-crystallization process. Int. J. Pharm. 2019, 570, 118639. [Google Scholar] [CrossRef]
- Xm, A.; Sh, B.; Mbla, C.; Xu, L.A.; Xl, C.; Ysa, C. Influence of mechanical and thermal energy on nifedipine amorphous solid dispersions prepared by hot melt extrusion: Preparation and physical stability—Sciencedirect. Int. J. Pharm. 2019, 561, 324–334. [Google Scholar]
- Guthrie, S.M.; Smilgies, D.M.; Giri, G. Controlling polymorphism in pharmaceutical compounds using solution shearing. Cryst. Growth Des. 2018, 2, 602–606. [Google Scholar] [CrossRef]
- Fu, Q.; Lu, H.D.; Xie, Y.F.; Liu, J.Y.; Han, Y.; Gong, N.B. Salt formation of two bcs ii drugs (indomethacin and naproxen) with (1r, 2r)-1,2-diphenylethylenediamine: Crystal structures, solubility and thermodynamics analysis. J. Mol. Struct. 2019, 1185, 281–289. [Google Scholar] [CrossRef]
- RaoKhandavilli, U.B.; Gangavaram, S.; Rajesh, G. High solubility crystalline hydrates of na and k furosemide salts. CrystEngComm 2014, 16, 4842–4852. [Google Scholar] [CrossRef]
- Boothroyd, S.; Kerridge, A.; Broo, A.; Buttar, D.; Anwar, J. Why do some molecules form hydrates or solvates? Cryst. Growth Des. 2018, 18, 1903–1908. [Google Scholar] [CrossRef]
- Patel, D.J.; Puranik, P.K. Pharmaceutical Co-crystal: An Emerging Technique to enhance Physicochemical properties of drugs. Int. J. Chemtech Res. 2020, 13, 283–290. [Google Scholar] [CrossRef]
- Bhatt, J.A.; Bahl, D.; Morris, K.; Stevens, L.L.; Haware, R.V. Structure-mechanics and improved tableting performance of the drug-drug cocrystal metformin:salicylic acid. Eur.J. Pharm. Biopharm. 2020, 153, 23–35. [Google Scholar] [CrossRef]
- Tomar, S.; Chakraborti, S.; Jindal, A.; Grewal, M.K.; Chadha, R. Cocrystals of diacerein: Towards the development of improved biopharmaceutical parameters. Int. J. Pharm. 2020, 574, 118942. [Google Scholar] [CrossRef]
- Lee, C.; Cho, A.Y.; Yoon, W.; Yun, H.; Kang, J.W.; Lee, J. Cocrystal Formation via Resorcinol–Urea Interactions: Naringenin and Carbamazepine. Cryst. Growth Des. 2019, 19, 3807–3814. [Google Scholar] [CrossRef]
- Budziak-Wieczorek, I.; Macioek, U. Synthesis and Characterization of a (-)-Epicatechin and Barbituric Acid Cocrystal: Single-Crystal X-ray Diffraction and Vibrational Spectroscopic Studies. ACS Omega 2021, 6, 8199–8209. [Google Scholar] [CrossRef]
- Abbas, N.; Latif, S.; Fatima, K.; Hussain, A.; Shamim, R. Amelioration of physicochemical, pharmaceutical, and pharmacokinetic properties of lornoxicam by cocrystallization with a novel coformer. Drug Dev. Ind. Pharm. 2021, 47, 498–508. [Google Scholar]
- Bose, P.; Chandra, S.; Das, A.; Roy, T.; Mukherjee, L. Enhancement of solubility of an oral hypoglycaemic drug, glimeperide by the technique cocrystallisation. J. Pharm. Sci. 2021, 12, 1092. [Google Scholar]
- Vasilev, N.A.; Surov, A.O.; Voronin, A.P.; Drozd, K.V.; Perlovich, G.L. Novel cocrystals of itraconazole: Insights from phase diagrams, formation thermodynamics and solubility. Int. J. Pharm. 2021, 599, 120441. [Google Scholar] [CrossRef]
- Yang, D.; Cao, J.; Jiao, L.; Yang, S.; Zhang, L.; Lu, Y.; Du, G. Solubility and Stability Advantages of a New Cocrystal of Berberine Chloride with Fumaric Acid. ACS Omega 2020, 5, 8283–8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Is, A.; Svd, B. Cocrystallization of carbamazepine with amides: Cocrystal and eutectic phases with improved dissolution. J. Mol. Struct. 2019, 1193, 398–415. [Google Scholar]
- Kavanagh, O.N.; Wang, C.; Walker, G.M.; Sun, C.C. Modulation of the powder properties of lamotrigine by crystal forms. Int. J. Pharm. 2021, 595, 120274. [Google Scholar] [CrossRef]
- Rai, S.K.; Gunnam, A.; Mannava, M.; Nangia, A.K. Improving the Dissolution Rate of Anticancer Drug Dabrafenib. Cryst. Growth Des. 2020, 20, 1035–1046. [Google Scholar] [CrossRef]
- Bommaka, M.K.; Mannava, M.K.C.; Suresh, K.; Gunnam, A.; Nangia, A. Entacapone: Improving Aqueous Solubility, Diffusion Permeability, and Cocrystal Stability with Theophylline. Cryst. Growth Des. 2018, 18, 6061–6069. [Google Scholar] [CrossRef]
- Almansa, C.; Mercè, R.; Tesson, N.; Farran, J.; Tomàs, J.; Plata-Salamán, C. Co-crystal of Tramadol Hydrochloride–Celecoxib (ctc): A Novel API–API Co-crystal for the Treatment of Pain. Cryst. Growth Des. 2017, 17, 1884–1892. [Google Scholar] [CrossRef]
- Gascon, N.; Almansa, C.; Merlos, M.; Vela, J.M.; Encina, G.; Morte, A.; Smith, K.; Plata-Salamán, C. Co-crystal of tramadol-celecoxib: Preclinical and clinical evaluation of a novel analgesic. Expert Opin. Investig. Drugs 2019, 28, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Guerain, M.; Guinet, Y.; Correia, N.T.; Paccou, L.; Hédoux, A. Polymorphism and stability of ibuprofen/nicotinamide cocrystal: The effect of the crystalline synthesis method. Int. J. Pharm. 2020, 584, 119454. [Google Scholar] [CrossRef]
- Hiendrawan, S.; Veriansyah, B.; Widjojokusumo, E.; Soewandhi, S.N.; Wikarsa, S.; Tjandrawinata, R.R. Physicochemical and mechanical properties of paracetamol cocrystal with 5-nitroisophthalic acid. Int. J. Pharm. 2016, 497, 106–113. [Google Scholar] [CrossRef]
- Yin, H.-M.; Wu, N.; Zhou, B.-J.; Hong, M.-H.; Zhu, B.; Qi, M.-H.; Ren, G.-B. Slow-Release Drug–Drug Cocrystals of Oxaliplatin with Flavonoids: Delaying Hydrolysis and Reducing Toxicity. Cryst. Growth Des. 2021, 21, 75–85. [Google Scholar] [CrossRef]
- Wang, J.; Dai, X.-L.; Lu, T.-B.; Chen, J.-M. Temozolomide–Hesperetin Drug–Drug Cocrystal with Optimized Performance in Stability, Dissolution, and Tabletability. Cryst. Growth Des. 2021, 21, 838–846. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Zhao, M.-Y.; Bu, F.-Z.; Niu, Y.-Y.; Yu, Y.-M.; Li, Y.-T.; Yan, C.-W.; Wu, Z.-Y. Cocrystallization of Amantadine Hydrochloride with Resveratrol: The First Drug–Nutraceutical Cocrystal Displaying Synergistic Antiviral Activity. Cryst. Growth Des. 2021, 21, 2763–2776. [Google Scholar] [CrossRef]
- Thipparaboina, R.; Kumar, D.; Chavan, R.B.; Shastri, N.R. Multidrug co-crystals: Towards the development of effective therapeutic hybrids. Drug Discov. Today 2016, 481–490. [Google Scholar] [CrossRef]
- Zhou, J.; Li, L.; Zhang, H.; Xu, J.; Huang, D.; Gong, N.; Han, W.; Yang, X.; Zhou, Z. Crystal structures, dissolution and pharmacokinetic study on a novel phosphodiesterase-4 inhibitor chlorbipram cocrystals. Int. J. Pharm. 2020, 576, 118984. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Palbociclib: First global approval. Drugs 2015, 75, 543–551. [Google Scholar] [CrossRef]
- Gopalan, P.K.; Pinder, M.C.; Chiappori, A.; Ivey, A.M.; Villegas, A.G.; Kaye, F.J. A phase II clinical trial of the CDK 4/6 inhibitor palbociclib (PD 0332991) in previously treated, advanced non-small cell lung cancer (NSCLC) patients with inactivated CDKN2A. J. Clin. Oncol. 2014, 32, 8077. [Google Scholar] [CrossRef]
- Jin, H.P.; Park, H.; Kim, K.H.; Kim, J.S.; Choi, I.S.; Roh, E.Y.; Ji, E.K.; Chang, M. Acute Lymphoblastic Leukemia in a Patient Treated with Letrozole and Palbociclib. J. Breast Cancer 2020, 23, 100–106. [Google Scholar]
- Xu, Y.; Yao, Y.; Park, W.D.; Derebail, S.; Munshi, N.C. Enhancing the Immune Surveillance in Multiple Myeloma Via CDK4/6 Inhibition. Blood 2020, 136, 33–34. [Google Scholar] [CrossRef]
- Tamura, K. Differences of cyclin-dependent kinase 4/6 inhibitor, palbociclib and abemaciclib, in breast cancer. Jpn. J. Clin. Oncol. 2019, 49, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Kwapisz, D. Cyclin-dependent kinase 4/6 inhibitors in breast cancer: Palbociclib, ribociclib, and abemaciclib. Breast Cancer Res. Treat. 2017, 166, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Klamerus, K.J.; Yuhas, L.M.; Pawlak, S.; Plotka, A.; O’Gorman, M. Impact of acid-reducing agents on the pharmacokinetics of palbociclib, a weak base with ph-dependent solubility, with different food intake conditions. Clin. Pharm. Drug Dev. 2017, 6, 614–626. [Google Scholar] [CrossRef]
- Xu, H.; Yu, S.; Liu, Q.; Yuan, X.; Mani, S.; Pestell, R.G.; Wu, K. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J. Hematol. Oncol. 2017, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Rocca, A.; Farolfi, A.; Bravaccini, S.; Schirone, A.; Amadori, D. Palbociclib (PD 0332991): Targeting the cell cycle machinery in breast cancer. Expert Opin. Pharm. 2014, 15, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Shi, X.-S.; Tian, F.; Anne, Z.; Chen, Q.; Huang, L.-N.; Hong, G. Palbociclib Amorphous. State. Patent No. CN108017629A, 5 May 2018. [Google Scholar]
- Aimei, S. Novel Palbociclib Crystal. Form. Patent CN108929321A, 4 December 2018. [Google Scholar]
- Lui, Z.-T.; Li, X.-F.; Geng, Q.; Xue, Y. Palbociclib Pharmaceutical Composition and Method for Preparing. Same. Patent CN108066312A, 20 March 2020. [Google Scholar]
- Zhang, M.; Xiong, X.; Suo, Z.; Hou, Q.; Gan, N.; Tang, P. Co-amorphous palbociclib–organic acid systems with increased dissolution rate, enhanced physical stability and equivalent biosafety. RSC Adv. 2019, 9, 3946–3955. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.-Y.; Kim, P.; Kim, G.-Y.; Lee, J.-Y.; Song, K.-H.; Lee, M.-J.; Yoon, W.; Yun, H.; Choi, G.J. Preparation and Characterization of Aripiprazole Cocrystals with Coformers of Multihydroxybenzene Compounds. Cryst. Growth Des. 2017, 17, 6641–6652. [Google Scholar] [CrossRef]
- Paul, M.; Desiraju, G.R. From a Binary to a Quaternary Cocrystal: An Unusual Supramolecular Synthon. Angew. Chem. Int. Ed. 2019, 58, 12027–12031. [Google Scholar] [CrossRef] [Green Version]
- Shimpi, M.R.; Alhayali, A.; Cavanagh, K.L.; Rodríguez-Hornedo, N.; Velaga, S.P. Tadalafil–Malonic Acid Cocrystal: Physicochemical Characterization, pH-Solubility, and Supersaturation Studies. Cryst. Growth Des. 2018, 18, 4378–4387. [Google Scholar] [CrossRef]
- Liu, W.; Ma, R.; Liang, F.; Duan, C.; Zhang, G.; Chen, Y.; Hao, C. New Cocrystals of Antipsychotic Drug Aripiprazole: Decreasing the Dissolution through Cocrystallization. Molecules 2021, 26, 2414. [Google Scholar] [CrossRef] [PubMed]
- Bofill, L.; Sande, D.D.; Barbas, R.; Prohens, R. A New and Highly Stable Cocrystal of Vitamin D3 for Use in Enhanced Food Supplements. Cryst. Growth Des. 2021, 21, 1418–1423. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of ShelX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The orca quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Abidi, S.; Azim, Y.; Gupta, A.K.; Pradeep, C.P. Mechanochemical synthesis and structural characterization of three novel cocrystals of dimethylglyoxime with n-heterocyclic aromatic compounds and acetamide. J. Mol. Struct. 2017, 1150, 103–111. [Google Scholar] [CrossRef]
- Hong, M.; Li, S.; Ji, W.; Qi, M.H.; Ren, G.B. Cocrystals of lenvatinib with sulfamerazine and salicylic acid: Crystal structure, equilibrium solubility, stability study, and anti-hepatoma activity. Cryst. Growth Des. 2021, 21, 3714–3727. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 101–108. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Wang, L.Y.; Dai, J.K.; Liu, F.; Li, Y.T.; Wu, Z.Y. The comparative study of cocrystal/salt in simultaneously improving solubility and permeability of acetazolamide. J. Mol. Struct. 2019, 1184, 225–232. [Google Scholar] [CrossRef]
- Dhumal, R.S.; Kelly, A.L.; York, P.; Coates, P.D.; Paradkar, A. Cocrystalization and simultaneous agglomeration using hot melt extrusion. Pharm. Res. 2010, 27, 2725–2733. [Google Scholar] [CrossRef] [PubMed]
- Amo, E.M.; Vellonen, K.S.; Kidron, H.; Urtti, A. Intravitreal clearance and volume of distribution of compounds in rabbits: In silico prediction and pharmacokinetic simulations for drug development. Eur. J. Pharm. Biopharm. 2015, 95, 215–226. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | PAL-RES | PAL-ORC |
|---|---|---|
| Empirical formula | C30H35N7O4 | C31H37N7O4 |
| Formula weight | 557.65 | 571.67 |
| Temperature/K | 100.00 (10) | 104.15 |
| Crystal system | triclinic | monoclinic |
| Space group | P-1 | P21/c |
| a/Å | 6.63890 (10) | 6.5309 (2) |
| b/Å | 12.4152 (2) | 16.8792 (4) |
| c/Å | 18.2788 (3) | 25.8097 (6) |
| α/° | 73.114 (2) | 90 |
| β/° | 81.7870 (10) | 92.819 (2) |
| γ/° | 78.1760 (10) | 90 |
| Volume/Å3 | 1405.46 (4) | 2841.72 (13) |
| Z | 2 | 4 |
| ρcalcg/cm3 | 1.318 | 1.336 |
| μ/mm−1 | 0.732 | 0.091 |
| F(000) | 592.0 | 1216.0 |
| Radiation | CuKα (λ = 0.154178) | MoKα (λ = 0.71073) |
| 2θ/° | 5.072 to 151.986 | 2.884 to 58.07 |
| Goodness-of-fit on F2 | 1.064 | 1.067 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0456, wR2 = 0.1190 | R1 = 0.0417, wR2 = 0.1005 |
| Final R indexes [all data] | R1 = 0.0537, wR2 = 0.1241 | R1 = 0.0553, wR2 = 0.1064 |
| Largest diff. peak/hole/e Å−3 | 0.30/−0.28 | 0.26/−0.24 |
| CCDC NO. | 2096399 | 2096409 |
| Compounds | D-H·A | d(D-H)/Å | d(H·A)/Å | d(D·A)/Å | ∠D-H·A/° | Symmetry Code |
|---|---|---|---|---|---|---|
| PAL-RES | O3-H3 N7 | 0.84 | 1.85 | 2.6455 (18) | 157.5 | |
| O4-H4C·O1 | 0.84 | 1.91 | 2.7477 (15) | 177.8 | 1 − X, 1 − Y, 1 − Z | |
| PAL-ORC | C7-H7·N6 | 0.95 | 2.32 | 2.9242 (16) | 120.6 | |
| C11-H11·N31 | 0.95 | 2.50 | 3.2444 (16) | 135.4 | 2 − X, 1 − Y, 1 − Z | |
| C20-H20A·O2 | 0.99 | 2.60 | 3.1118 (18) | 112.1 | ||
| C23-H23A·O2 | 0.99 | 2.33 | 2.9490 (17) | 119.3 | ||
| C25-H25·O1 | 0.95 | 2.65 | 3.3288 (17) | 128.7 | ||
| N2-H2C·O3 | 0.885 (17) | 2.642 (18) | 3.4934 (16) | 161.9 (15) | 2 + X, ½ − Y, ½ + Z | |
| N4-H4·N5 | 0.88 | 2.05 | 2.9238 (14) | 173.0 | 2 − X, 1 − Y, 1 − Z | |
| O3-H3·O1 | 0.91 | 1.82 | 2.7346 (14) | 178.2 | ||
| O4-H4AB·N23 | 0.99 | 1.78 | 2.7642 (15) | 174.1 | −3 + X, ½ − Y, −½ + Z |
| PAL | PAL in PAL-RES | PAL in PAL-ORC | |
|---|---|---|---|
| Cmax (ng/mL) | 346.69 | 371.57 | 233.46 |
| Tmax (h) | 4 | 4 | 4 |
| T1/2 (h) | 19.89 | 10.33 | 17.36 |
| Vd (L/kg) | 20.48 | 12.05 | 19.26 |
| MRT (h) | 5.36 | 5.73 | 6.15 |
| AUC0−t (ng∙h/mL) | 2616.48 | 3245.47 | 2254.08 |
| Frel | - | 1.24 | 0.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, C.; Liu, W.; Tao, Y.; Liang, F.; Chen, Y.; Xiao, X.; Zhang, G.; Chen, Y.; Hao, C. Two Novel Palbociclib-Resorcinol and Palbociclib-Orcinol Cocrystals with Enhanced Solubility and Dissolution Rate. Pharmaceutics 2022, 14, 23. https://doi.org/10.3390/pharmaceutics14010023
Duan C, Liu W, Tao Y, Liang F, Chen Y, Xiao X, Zhang G, Chen Y, Hao C. Two Novel Palbociclib-Resorcinol and Palbociclib-Orcinol Cocrystals with Enhanced Solubility and Dissolution Rate. Pharmaceutics. 2022; 14(1):23. https://doi.org/10.3390/pharmaceutics14010023
Chicago/Turabian StyleDuan, Chenxin, Wenwen Liu, Yunwen Tao, Feifei Liang, Yanming Chen, Xinyi Xiao, Guisen Zhang, Yin Chen, and Chao Hao. 2022. "Two Novel Palbociclib-Resorcinol and Palbociclib-Orcinol Cocrystals with Enhanced Solubility and Dissolution Rate" Pharmaceutics 14, no. 1: 23. https://doi.org/10.3390/pharmaceutics14010023
APA StyleDuan, C., Liu, W., Tao, Y., Liang, F., Chen, Y., Xiao, X., Zhang, G., Chen, Y., & Hao, C. (2022). Two Novel Palbociclib-Resorcinol and Palbociclib-Orcinol Cocrystals with Enhanced Solubility and Dissolution Rate. Pharmaceutics, 14(1), 23. https://doi.org/10.3390/pharmaceutics14010023