
Quantitative Analysis of the Phase Transition Mechanism Underpinning the Systemic Self-Assembly of a Mechanopharmaceutical Device

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
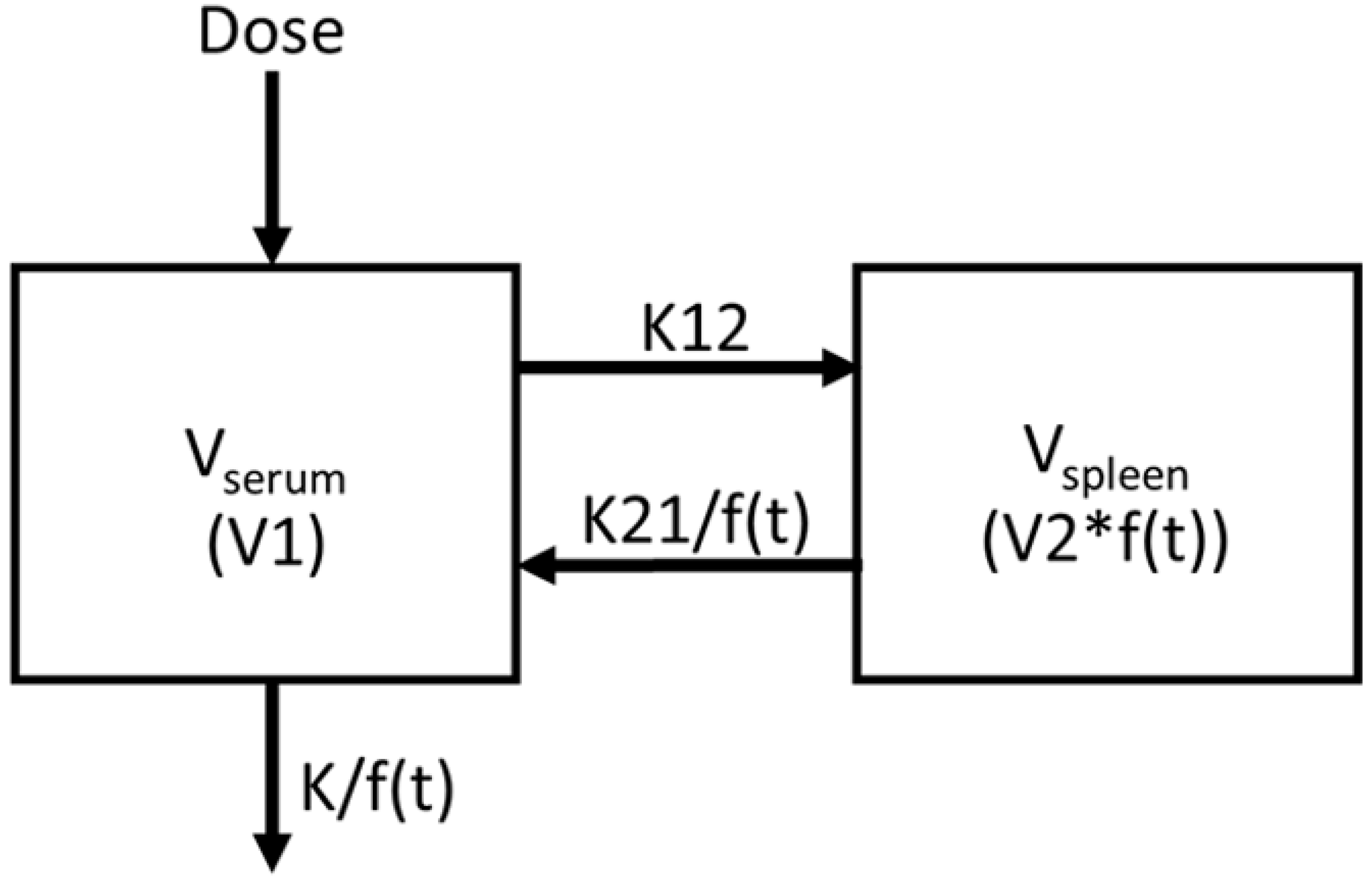
2.1. Introduction of Nonlinearity into 2-Compartment PK Model
2.2. Data Acquisition and Compartmental Pharmacokinetics Modeling
2.3. Parameter Sensitivity Analysis (PSA)
2.4. Total Volume of Distribution and Half-Life Calculations
2.5. Dose Dependent Mass Sequestration Analysis
3. Results
3.1. Comparative Analysis of Sigmoidal Functions and Utility of the RSR Equation to Describe the Mechanistic Underpinnings of CFZ Pharmacokinetics
3.2. Preliminary Analysis of Parameters on Growth Function f(t)
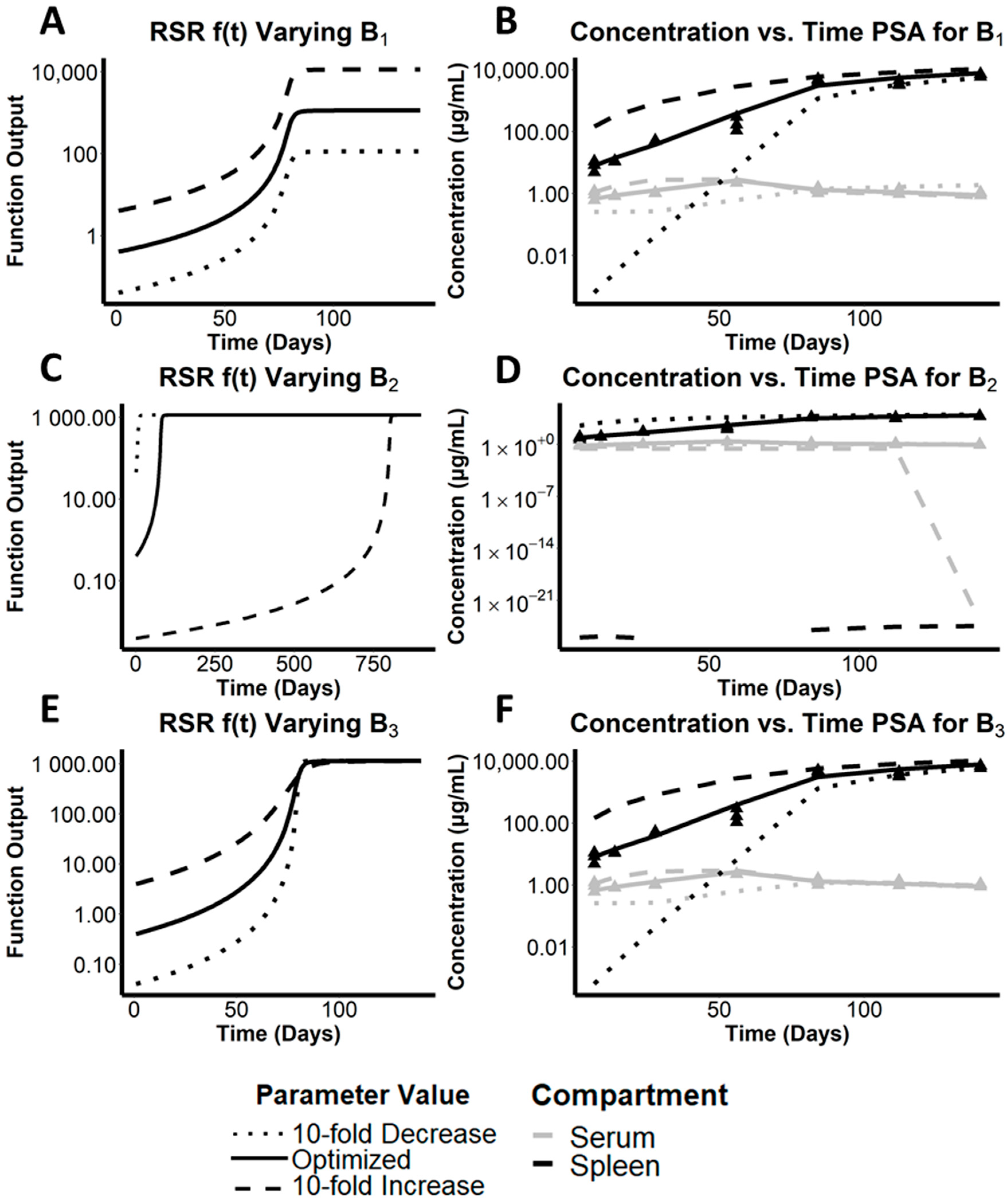
3.3. Assessing the Effect of the Maximal Cargo Capacity of the Spleen on the Systemic Pharmacokinetics of CFZ
3.4. Quantitative Analysis of the Relative Importance of Different Parameters to the Simulated Systemic Pharmacokinetics of CFZ
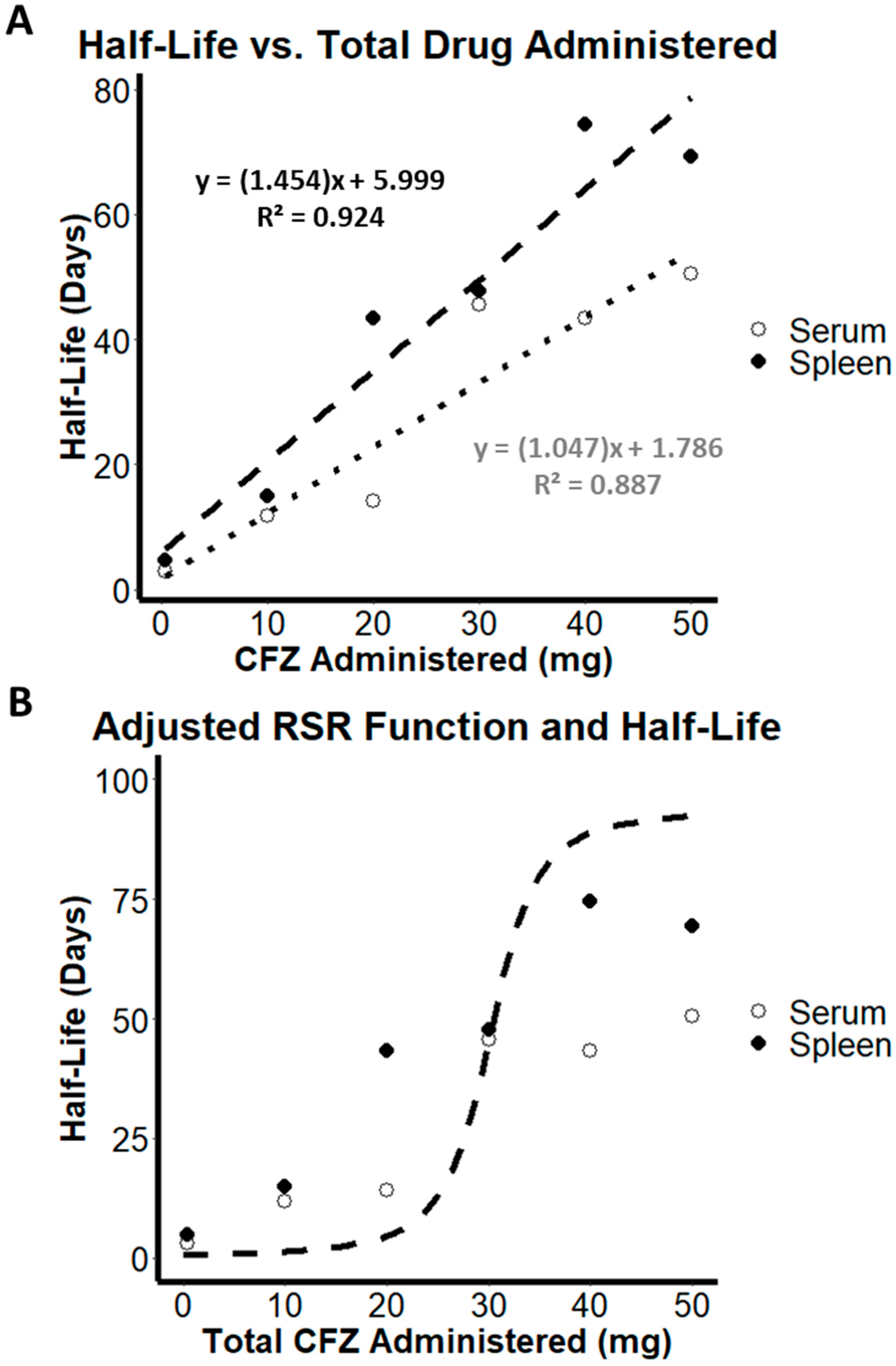
3.5. Estimation of Increasing Half-Life as a Function of Increasing Volume of Distribution
3.6. Patterns and Evidence of Load-Dependent Pharmacokinetics from Variable Dosing Data
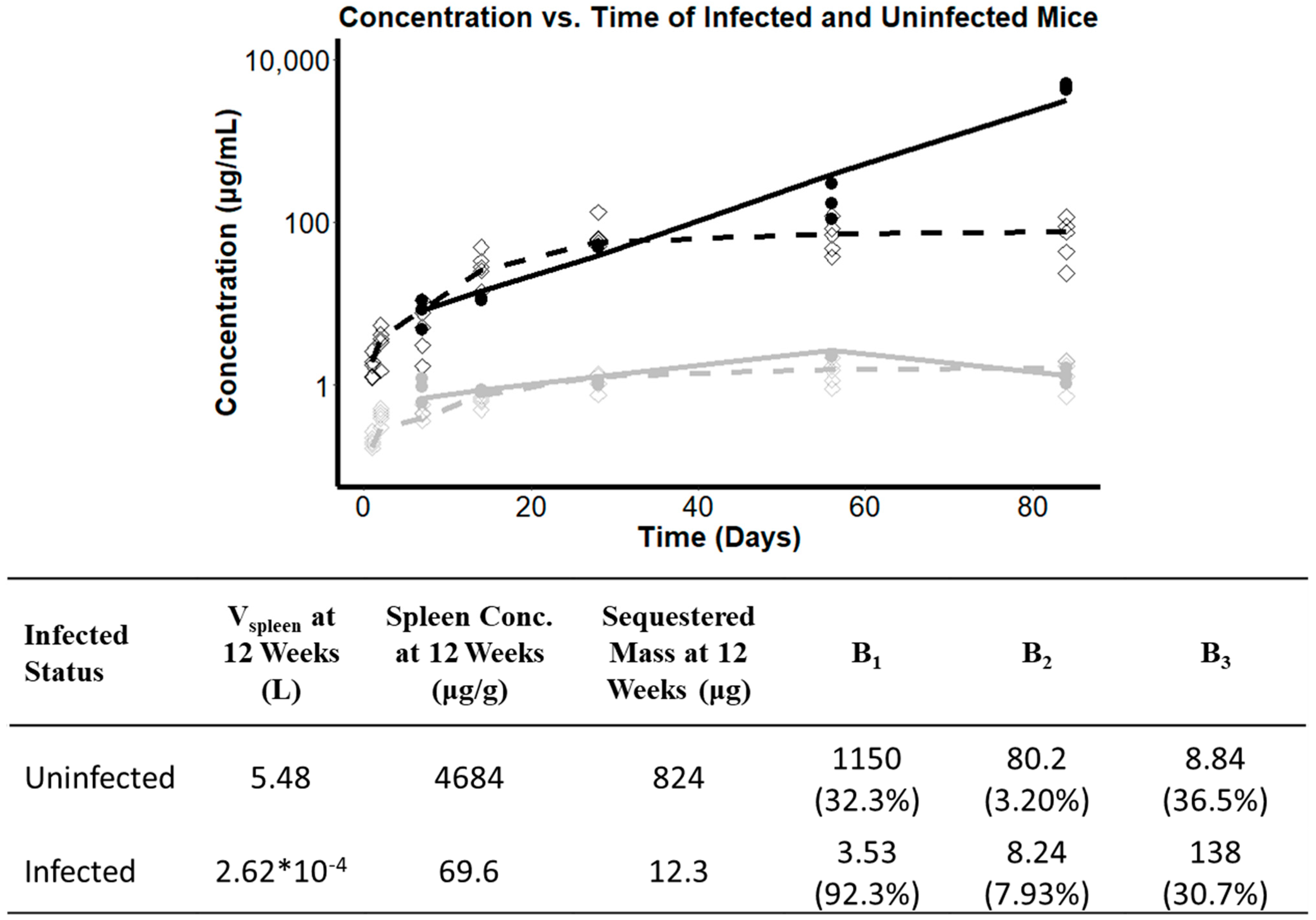
3.7. Investigating the Effect of Infection Status on the Pharmacokinetics of CFZ
4. Discussion
4.1. Exploring the RSR Function as a Tool to Describe Soluble-to-Insoluble Phase Transitions in a Population Pharmacokinetics Model
4.2. Infection Status as Covariate in CFZ Accumulation
4.3. Load-Dependent Drug Sequestration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Willmer, A.R.; Dunne, S.; Swanson, R.; Almeida, D.; Ammerman, N.C.; Stringer, K.A.; Capparelli, E.V.; Rosania, G.R. An Adaptive Biosystems Engineering Approach towards Modeling the Soluble-to-Insoluble Phase Transition of Clofazimine. Pharmaceutics 2021. accepted. [Google Scholar]
- Arbiser, J.; Moschella, S. Clofazimine: A review of its medical uses and mechanisms of action. J. Am. Acad. Dermatol. 1995, 32, 241–247. [Google Scholar] [CrossRef]
- Cholo, M.C.; Steel, H.C.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current status and future prospects. J. Antimicrob. Chemother. 2011, 67, 290–298. [Google Scholar] [CrossRef]
- Ammerman, N.C.; Swanson, R.V.; Tapley, A.; Moodley, C.; Ngcobo, B.; Adamson, J.; Dorasamy, A.; Moodley, S.; Mgaga, Z.; Bester, L.A.; et al. Clofazimine has delayed antimicrobial activity against Mycobacterium tuberculosis both in vitro and in vivo. J. Antimicrob. Chemother. 2017, 72, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Yin, X.; Meng, X.; Chan, J.F.W.; Ye, Z.W.; Riva, L.; Pache, L.; Chan, C.C.Y.; Lai, P.M.; Chan, C.C.S.; et al. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature 2021, 593, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Dual Therapy With Interferon Beta-1b and Clofazimine for COVID-19—Full Text View. Full Text View—ClinicalTrials.gov. Available online: clinicaltrials.gov/ct2/show/NCT04465695 (accessed on 14 September 2021).
- Anderson, R.; Smit, M.J. Clofazimine and B669 inhibit the proliferative responses and Na+, K(+)-adenosine triphosphatase activity of human lymphocytes by a lysophospholipid-dependent mechanism. Biochem. Pharmacol. 1993, 46, 2029–2038. [Google Scholar] [CrossRef]
- Wan, W.; Zhu, S.; Li, S.; Shang, W.; Zhang, R.; Li, H.; Liu, W.; Xiao, G.; Peng, K.; Zhang, L. High-Throughput screening of an FDA-approved drug library identifies inhibitors against arenaviruses and SARS-CoV-2. ACS Infect. Dis. 2020, 38. [Google Scholar] [CrossRef]
- Rzeczycki, P.; Woldemichael, T.; Willmer, A.; Murashov, M.D.; Baik, J.; Keswani, R.; Yoon, G.S.; Stringer, K.A.; Rodriguez-Hornedo, N.; Rosania, G.R. An Expandable Mechanopharmaceutical Device (1): Measuring the Cargo Capacity of Macrophages in a Living Organism. Pharm. Res. 2018, 36, 12. [Google Scholar] [CrossRef]
- Swanson, R.V.; Adamson, J.; Moodley, C.; Ngcobo, B.; Ammerman, N.C.; Dorasamy, A.; Moodley, S.; Mgaga, Z.; Tapley, A.; Bester, L.A.; et al. Pharmacokinetics and Pharmacodynamics of Clofazimine in a Mouse Model of Tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 3042–3051. [Google Scholar] [CrossRef] [Green Version]
- Baik, J.; Stringer, K.A.; Mane, G.; Rosania, G.R. Multiscale Distribution and Bioaccumulation Analysis of Clofazimine Reveals a Massive Immune System-Mediated Xenobiotic Sequestration Response. Antimicrob. Agents Chemother. 2012, 57, 1218–1230. [Google Scholar] [CrossRef] [Green Version]
- Rzeczycki, P.; Yoon, G.S.; Keswani, R.K.; Sud, S.; Baik, J.; Murashov, M.D.; Bergin, I.L.; Stringer, K.A.; Rosania, G.R. An Expandable Mechanopharmaceutical Device (2): Drug Induced Granulomas Maximize the Cargo Sequestering Capacity of Macrophages in the Liver. Pharm. Res. 2018, 36, 3. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.; Rosania, G.R. Macrophages Sequester Clofazimine in an Intracellular Liquid Crystal-Like Supramolecular Organization. PLoS ONE 2012, 7, e47494. [Google Scholar] [CrossRef] [PubMed]
- Keswani, R.K.; Baik, J.; Yeomans, L.; Hitzman, C.; Johnson, A.M.; Pawate, A.S.; Kenis, P.J.; Rodriguez-Hornedo, N.; Stringer, K.A.; Rosania, G.R. Chemical Analysis of Drug Biocrystals: A Role for Counterion Transport Pathways in Intracellular Drug Disposition. Mol. Pharm. 2015, 12, 2528–2536. [Google Scholar] [CrossRef] [Green Version]
- Woldemichael, T.; Keswani, R.K.; Rzeczycki, P.M.; Murashov, M.D.; LaLone, V.; Gregorka, B.; Swanson, J.A.; Stringer, K.A.; Rosania, G.R. Reverse Engineering the Intracellular Self-Assembly of a Functional Mechanopharmaceutical Device. Sci. Rep. 2018, 8, 2934. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.H.; Hartmann-Siantar, C.; Fisher, D.; Descalle, M.A.; Daly, T.; Lehmann, J.; Lewis, M.R.; Hoffman, T.; Smith, J.; Situ, P.D.; et al. Evaluation of Beta-Absorbed Fractions in a Mouse Model for 90Y, 188Re, 166Ho, 149Pm, 64Cu, and 177Lu Radionuclides. Cancer Biother. Radiopharm. 2005, 20, 436–449. [Google Scholar] [CrossRef]
- Chicurel, M.; Garcia, E.; Goodsaid, F. Modulation of macrophage lysosomal pH by Mycobacterium tuberculosis-derived proteins. Infect. Immun. 1988, 56, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queval, C.J.; Song, O.R.; Carralot, J.P.; Saliou, J.M.; Bongiovanni, A.; Deloison, G.; Deboosere, N.; Jouny, S.; Iantomasi, R.; Delorme, V.; et al. Mycobacterium tuberculosis Controls Phagosomal Acidification by Targeting CISH-Mediated Signaling. Cell Rep. 2017, 20, 3188–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDougall, A.C.; Horsfall, W.R.; Hede, J.E.; Chaplin, A.J. Splenic infarction and tissue accumulation of crystals associated with the use of clofazimine (Lamprene; B663) in the treatment of pyoderma gangrenosum. Br. J. Dermatol. 1980, 102, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Jopling, W.H. Complications of treatment with clofazimine (Lamprene: B663). Lepr. Rev. 1976, 47, 1–3. [Google Scholar] [CrossRef]
- Sandler, E.D.; Ng, V.L.; Hadley, W.K. Clofazimine crystals in alveolar macrophages from a patient with the acquired immunodeficiency syndrome. Arch. Pathol. Lab. Med. 1992, 116, 541–543. [Google Scholar] [PubMed]
- Harbeck, R.J.; Worthen, G.S.; Lebo, T.D.; Peloquin, C.A. Clofazimine crystals in the cytoplasm of pulmonary macrophages. Ann. Pharmacother. 1999, 33, 250. [Google Scholar] [CrossRef]
- Abdelwahab, M.T.; Wasserman, S.; Brust, J.C.; Gandhi, N.R.; Meintjes, G.; Everitt, D.; Diacon, A.; Dawson, R.; Wiesner, L.; Svensson, E.M.; et al. Clofazimine pharmacokinetics in patients with TB: Dosing implications. J. Antimicrob. Chemother. 2020, 75, 3269–3277. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | RMSLE for 10-Fold Increase | RMSLE for 10-Fold Decrease | OFV for 10-Fold Increase | OFV for 10-Fold Decrease | P* | CI | CV% |
|---|---|---|---|---|---|---|---|
| B1 | 0.239 | 0.321 | 316.419 | 209.998 | 1150 | [449, 1150] | 32.3% |
| B2 | 0.749 | 0.297 | 335.703 | 326.066 | 80.2 | [76.7, 82] | 3.20% |
| B3 | 0.237 | 0.314 | 317.759 | 211.809 | 8.84 | [4.92, 13.1] | 36.5% |
| Study | Dosing Regimen | Total Duration | Route of Administration | CFZ Load at 2 Weeks (mg) | CFZ Load at 8 Weeks (mg) | Total Drug Load (mg) |
|---|---|---|---|---|---|---|
| 1 | 25 mg/kg/d, 5 days per week | 20 weeks | Oral Gavage | 5 | 20 | 50 |
| 2 | 36 mg/kg/d, 7 days per week | 8 weeks | Mixed in Feed | 18.9 * | 50.4 | 50.4 |
| 3 † | 6.25 mg/kg/d, 5 days per week | 12 weeks | Oral Gavage | 1.25 | 5 | 7.5 |
| 4 † | 12.5 mg/kg/d, 5 days per week | 12 weeks | Oral Gavage | 2.5 | 10 | 15 |
| 5 † | 25 mg/kg/d, 5 days per week | 12 weeks | Oral Gavage | 5 | 20 | 30 |
| 6 † | 50 mg/kg for 1 day, 25 mg/kg for day 2 to week 2 (5 days per week), 25 mg/kg/d for weeks 2 through 12 (3 days per week) | 12 weeks | Oral Gavage | 5.5 | 14.5 | 20.5 |
| 7 † | 100 mg/kg for 1 day, 75 mg/kg for day 2, 50 mg/kg for day 3 to week 2 (5 days per week), 25 mg/kg/d for weeks 2 through 12 (3 days per week) | 12 weeks | Oral Gavage | 11.5 | 20.5 | 26.5 |
| 8 † | 200 mg/kg for 1 day, 100 mg/kg for day 2, 75 mg/kg for day 3 to week 2 (5 days per week), 50 mg/kg/d for weeks 2 through 12 (3 days per week) | 12 weeks | Oral Gavage | 18 | 27 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunne, S.; Willmer, A.R.; Swanson, R.; Almeida, D.; Ammerman, N.C.; Stringer, K.A.; Capparelli, E.V.; Rosania, G.R. Quantitative Analysis of the Phase Transition Mechanism Underpinning the Systemic Self-Assembly of a Mechanopharmaceutical Device. Pharmaceutics 2022, 14, 15. https://doi.org/10.3390/pharmaceutics14010015
Dunne S, Willmer AR, Swanson R, Almeida D, Ammerman NC, Stringer KA, Capparelli EV, Rosania GR. Quantitative Analysis of the Phase Transition Mechanism Underpinning the Systemic Self-Assembly of a Mechanopharmaceutical Device. Pharmaceutics. 2022; 14(1):15. https://doi.org/10.3390/pharmaceutics14010015
Chicago/Turabian StyleDunne, Steven, Andrew R. Willmer, Rosemary Swanson, Deepak Almeida, Nicole C. Ammerman, Kathleen A. Stringer, Edmund V. Capparelli, and Gus R. Rosania. 2022. "Quantitative Analysis of the Phase Transition Mechanism Underpinning the Systemic Self-Assembly of a Mechanopharmaceutical Device" Pharmaceutics 14, no. 1: 15. https://doi.org/10.3390/pharmaceutics14010015
APA StyleDunne, S., Willmer, A. R., Swanson, R., Almeida, D., Ammerman, N. C., Stringer, K. A., Capparelli, E. V., & Rosania, G. R. (2022). Quantitative Analysis of the Phase Transition Mechanism Underpinning the Systemic Self-Assembly of a Mechanopharmaceutical Device. Pharmaceutics, 14(1), 15. https://doi.org/10.3390/pharmaceutics14010015