
Anticancer and Structure Activity Relationship of Non-Symmetrical Choline Kinase Inhibitors

 , , ,
, , ,  and
and 
Abstract
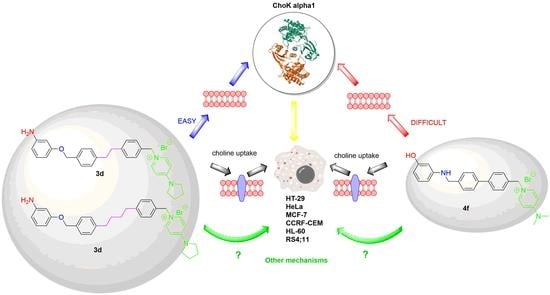
:
1. Introduction
2. Materials and Methods
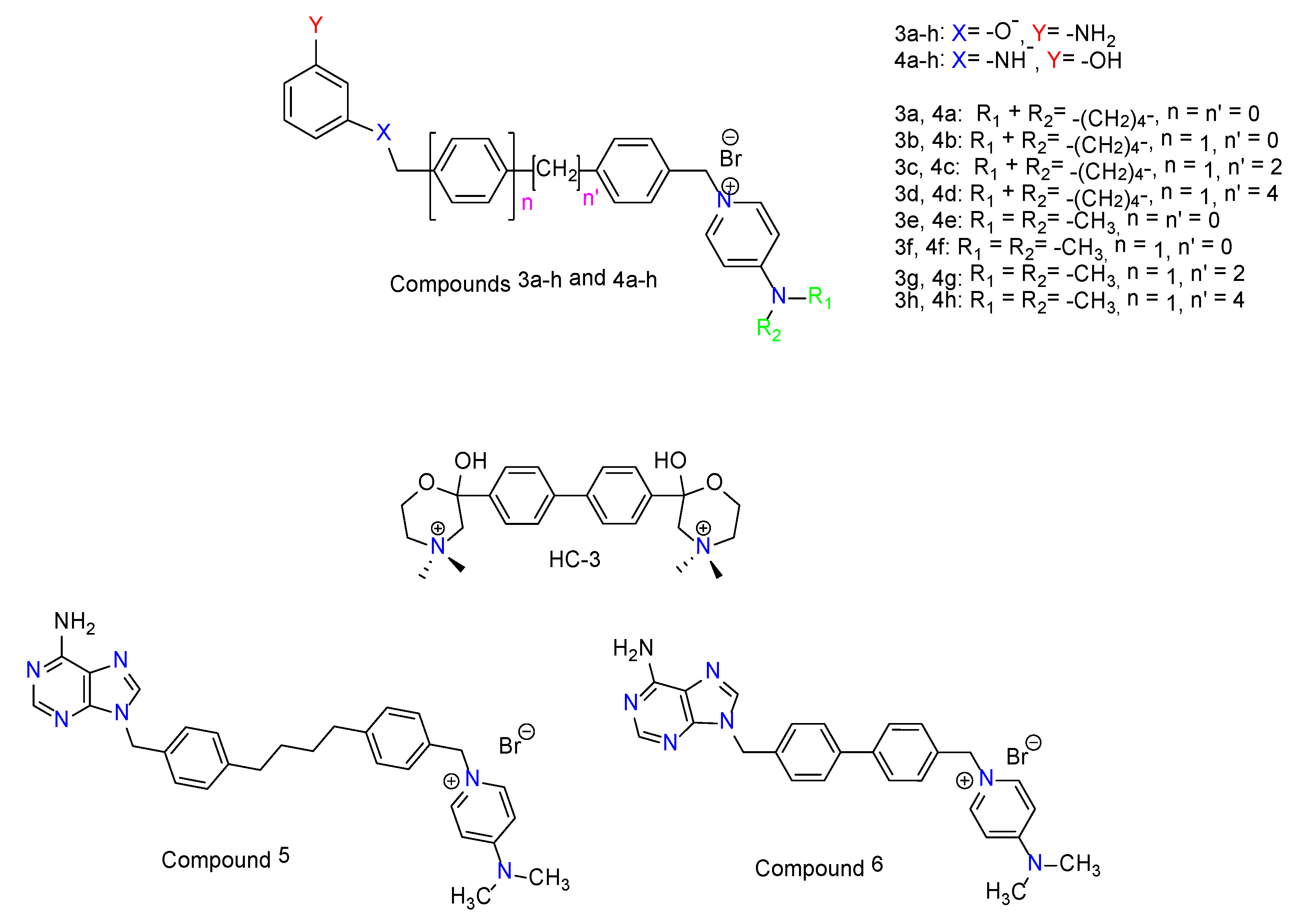
2.1. Chemistry
2.2. Cloning, Purification of CK, and Inhibition of Choline Kinase α1 by Compounds 3a–h and 4a–h
2.3. Molecular-Modeling Studies
2.4. Antiproliferative Assays in Cancer Cells
2.5. Antiproliferative Activity in Peripheral Blood Lymphocytes (PBL)
2.6. Cell Cycle Analysis
2.7. Measurement of Apoptosis by Flow Cytometry
2.8. Choline Uptake Assay
3. Results and Discussion
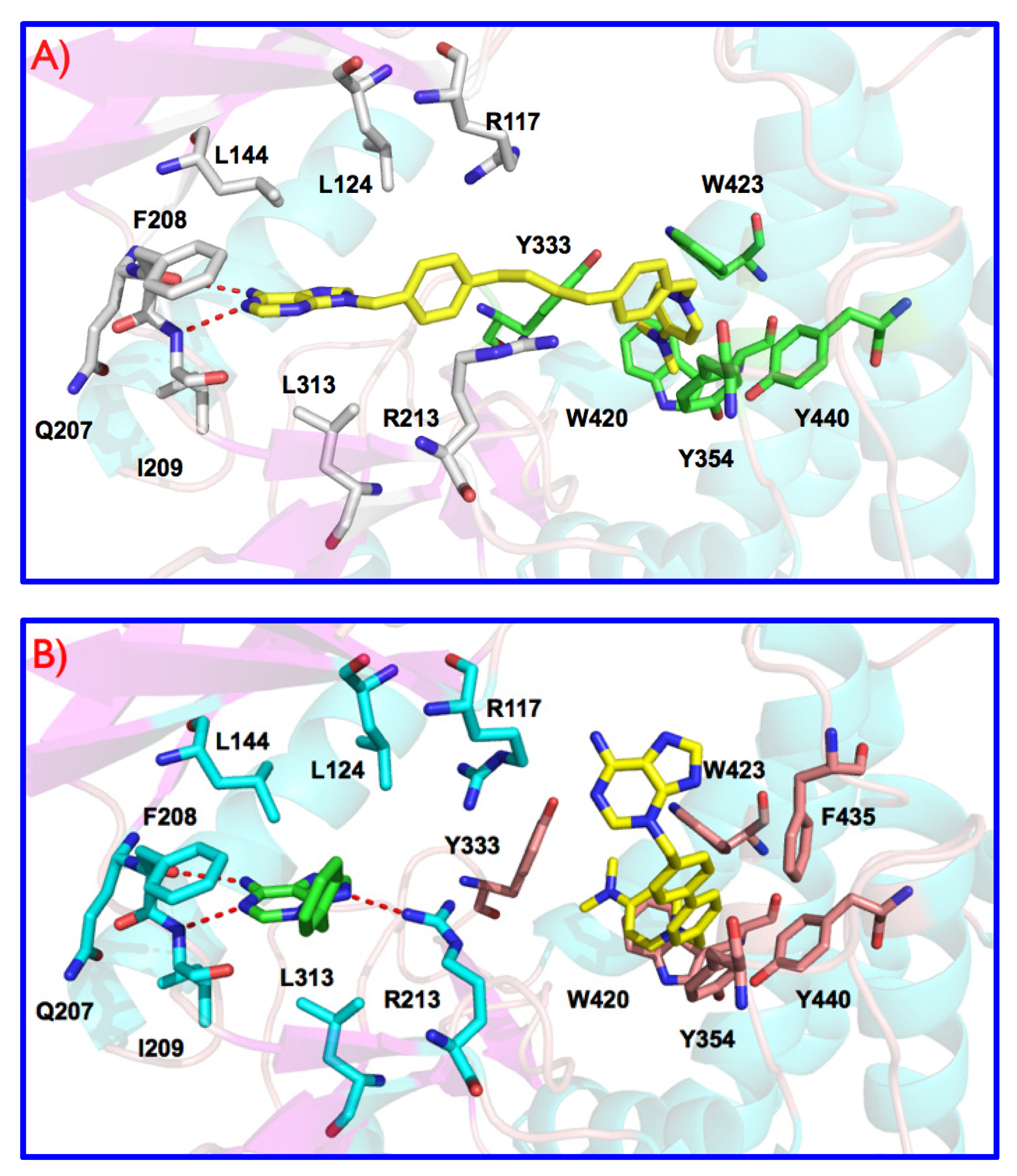
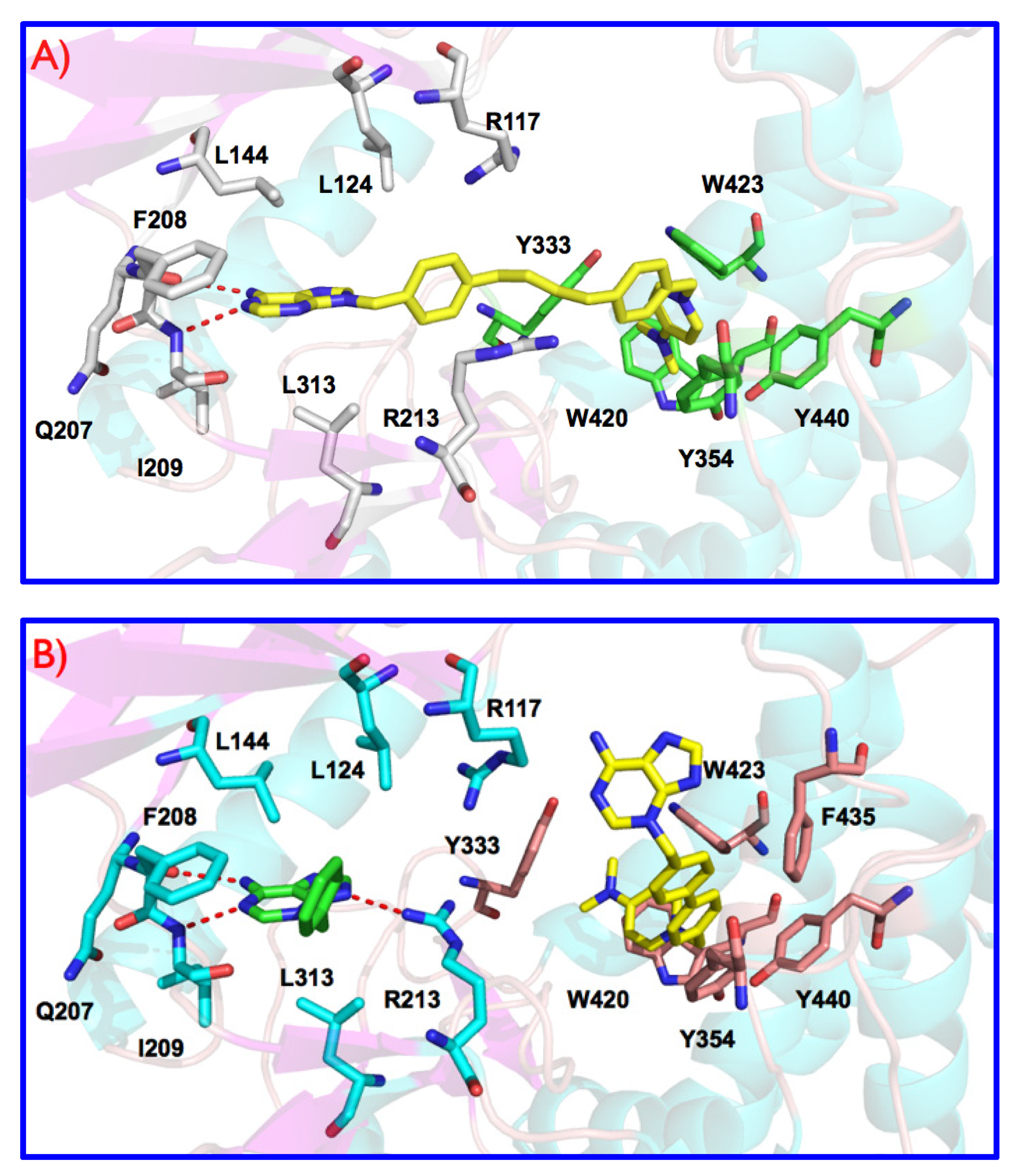
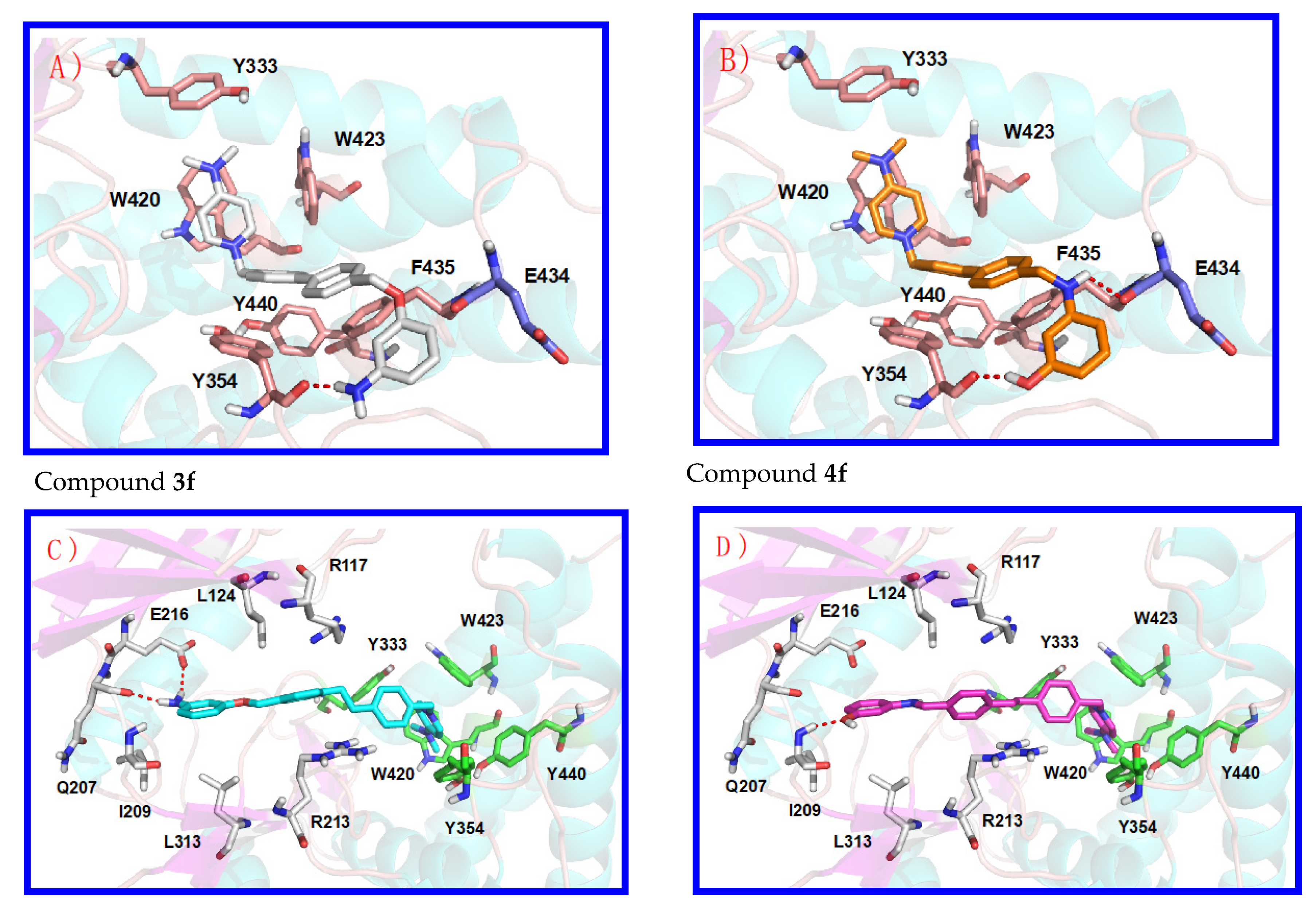
3.1. Preliminary Docking Studies
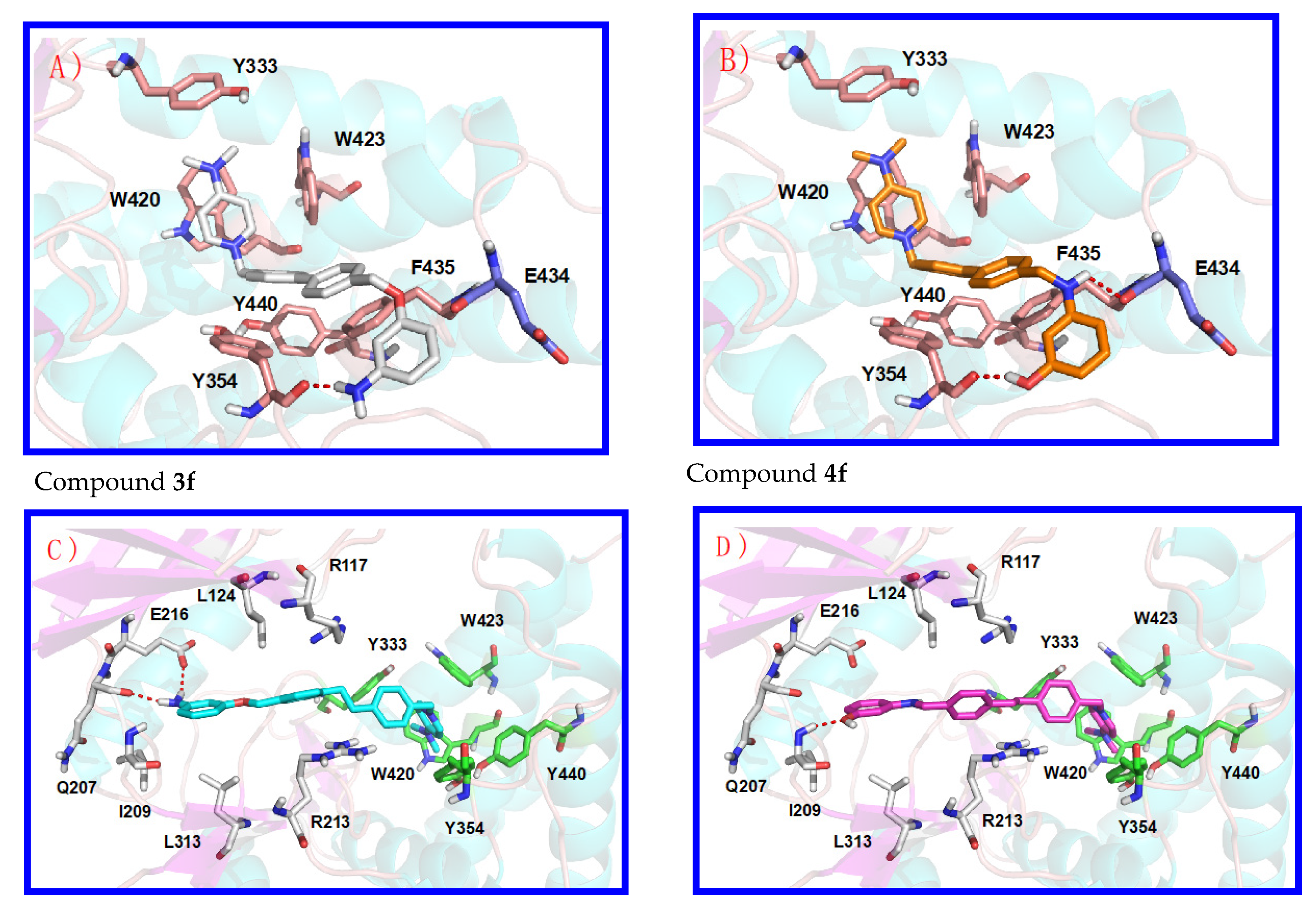
3.2. Inhibition of ChoKα1 by Compounds 3a–h and 4a–h as well as Docking Studies
3.3. In Vitro Antiproliferative Activities
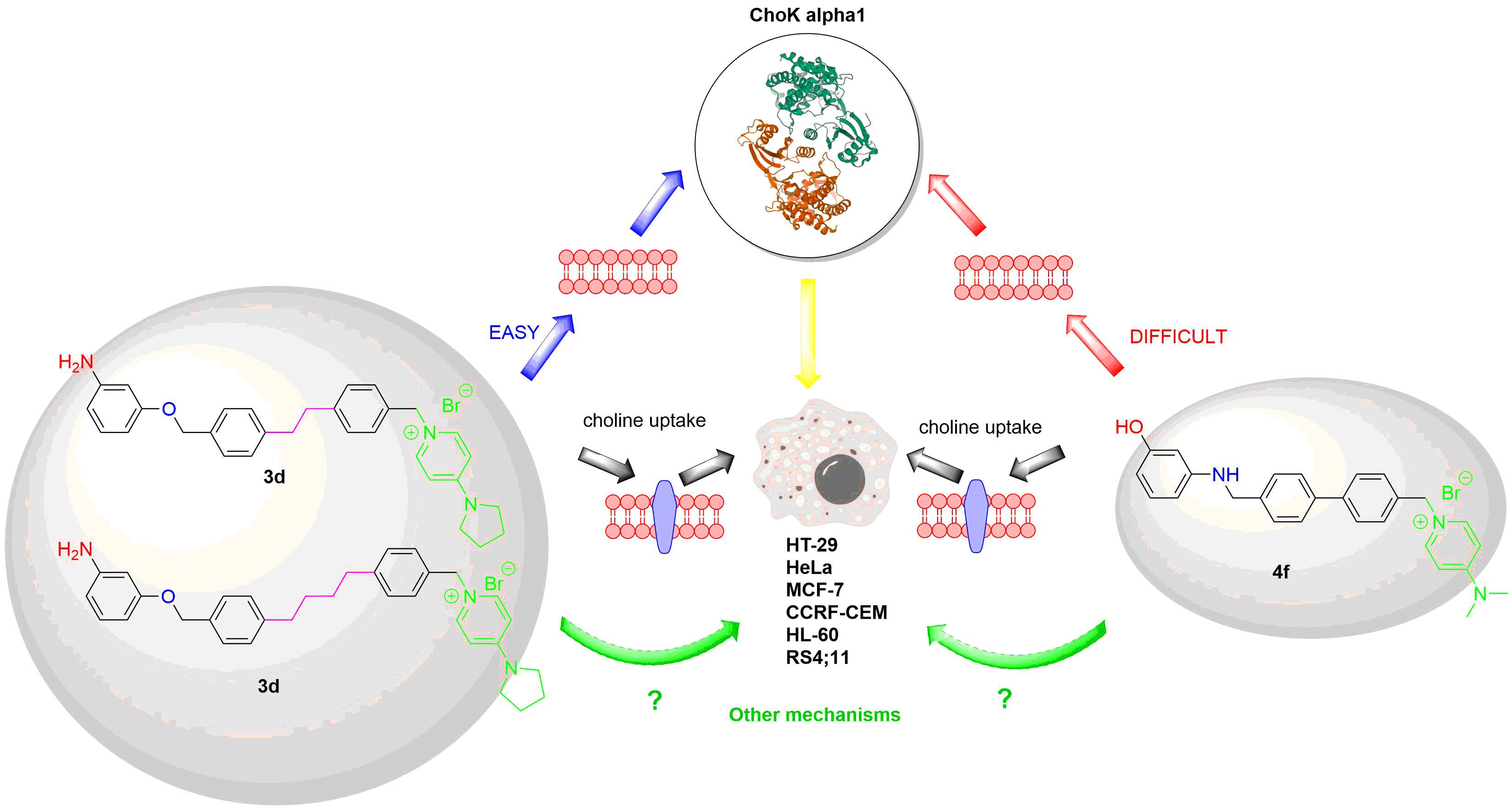
3.4. Effects of Compound 3c and 4f in Non-Tumor Cells
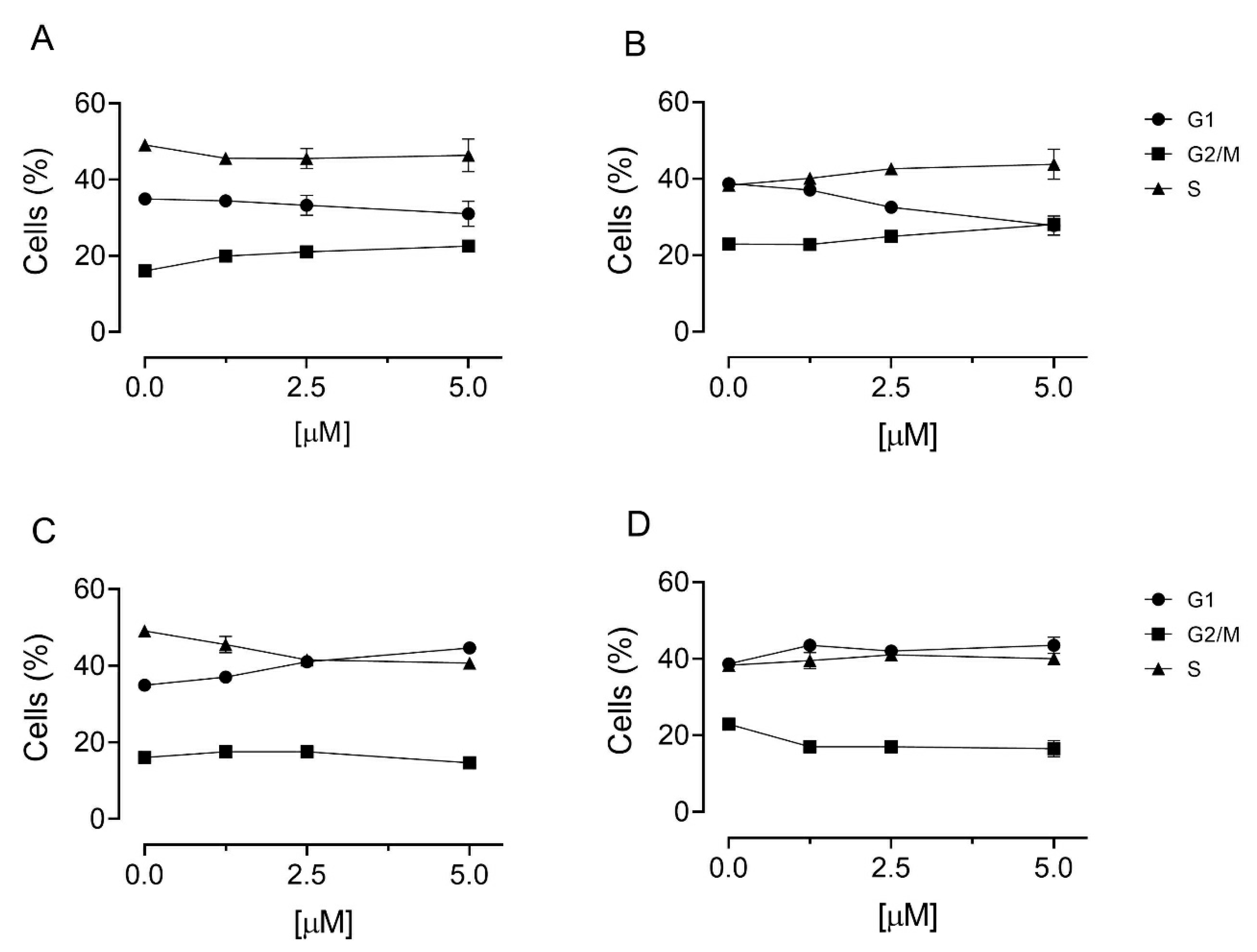
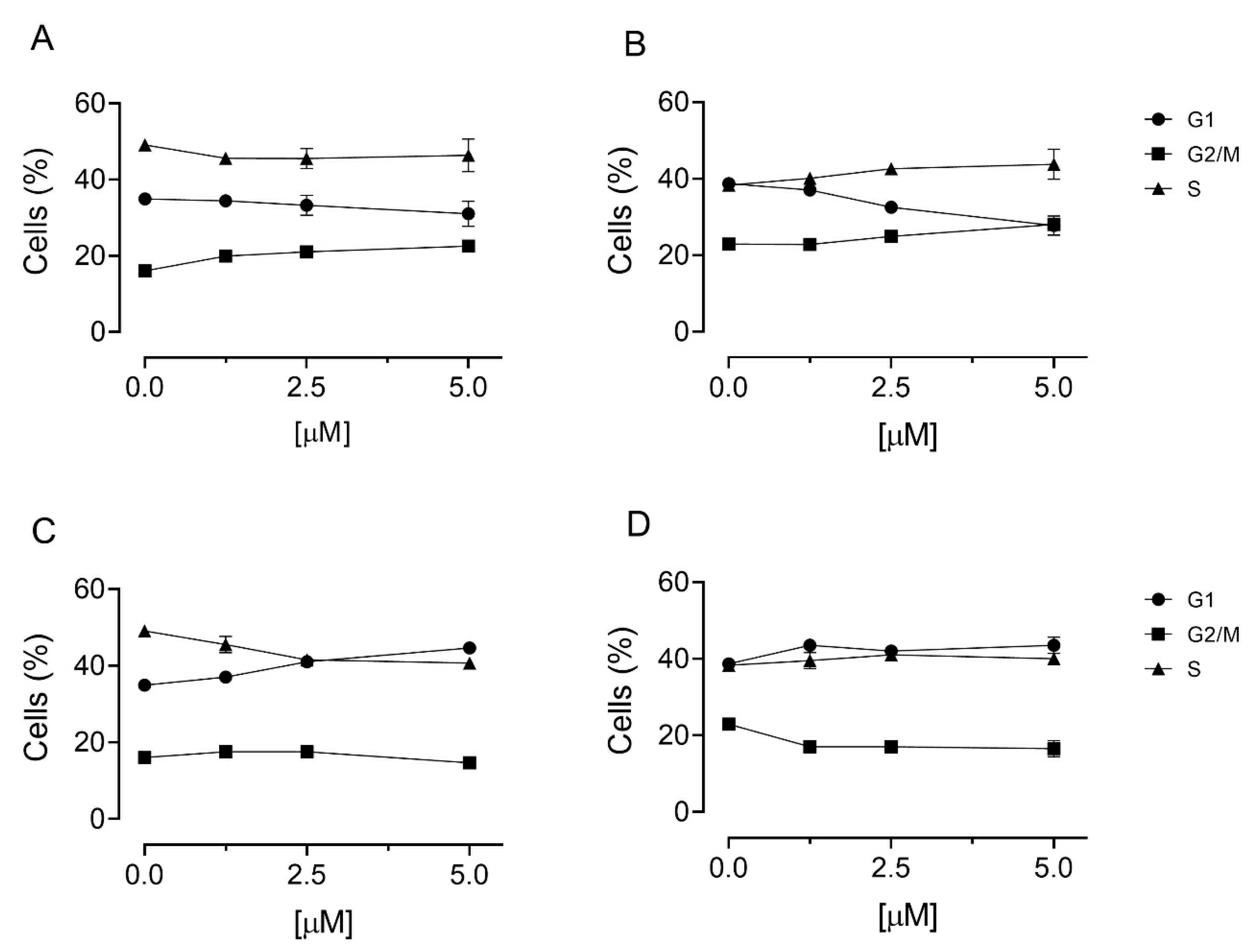
3.5. Cell Cycle Analysis
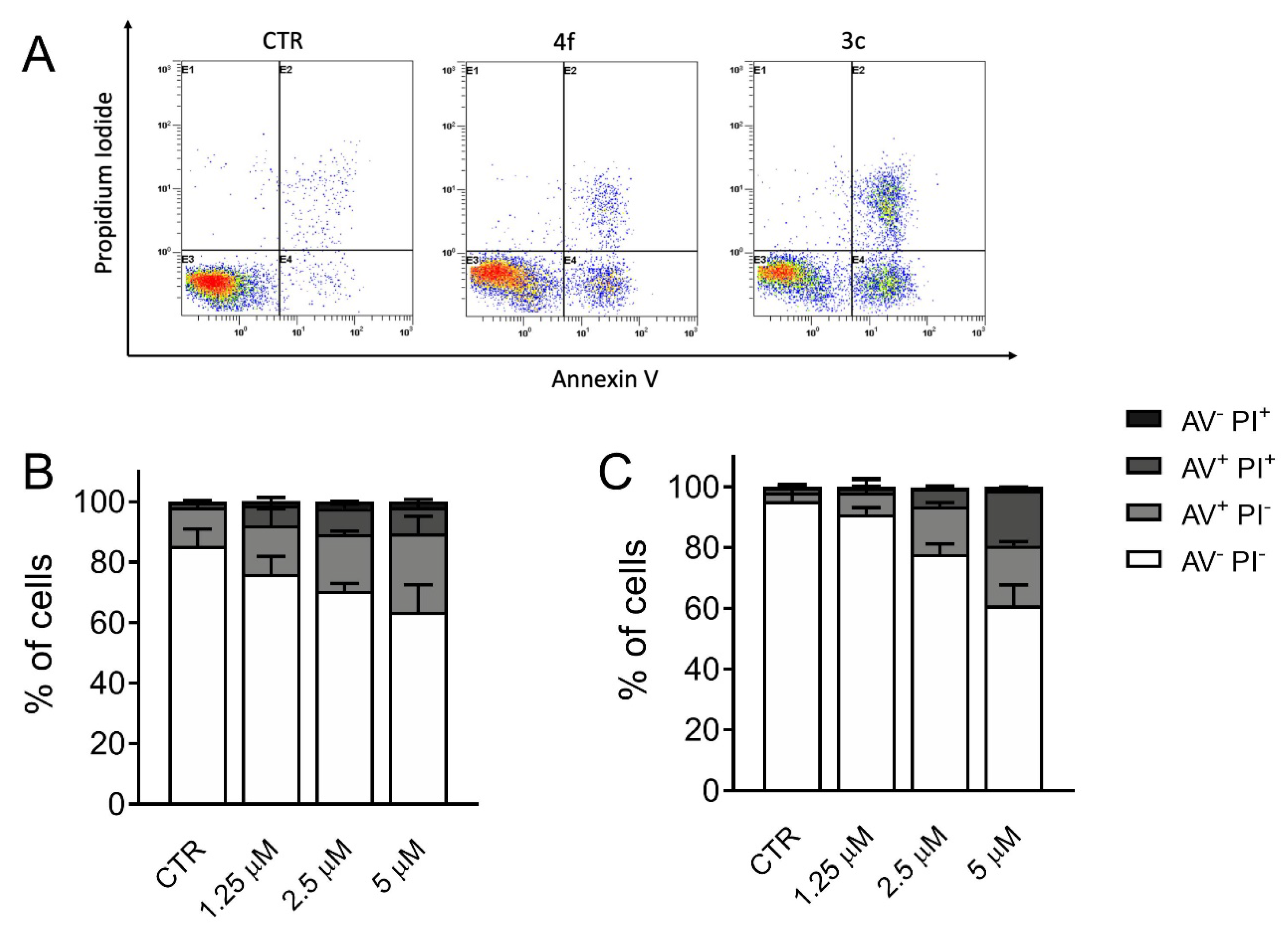
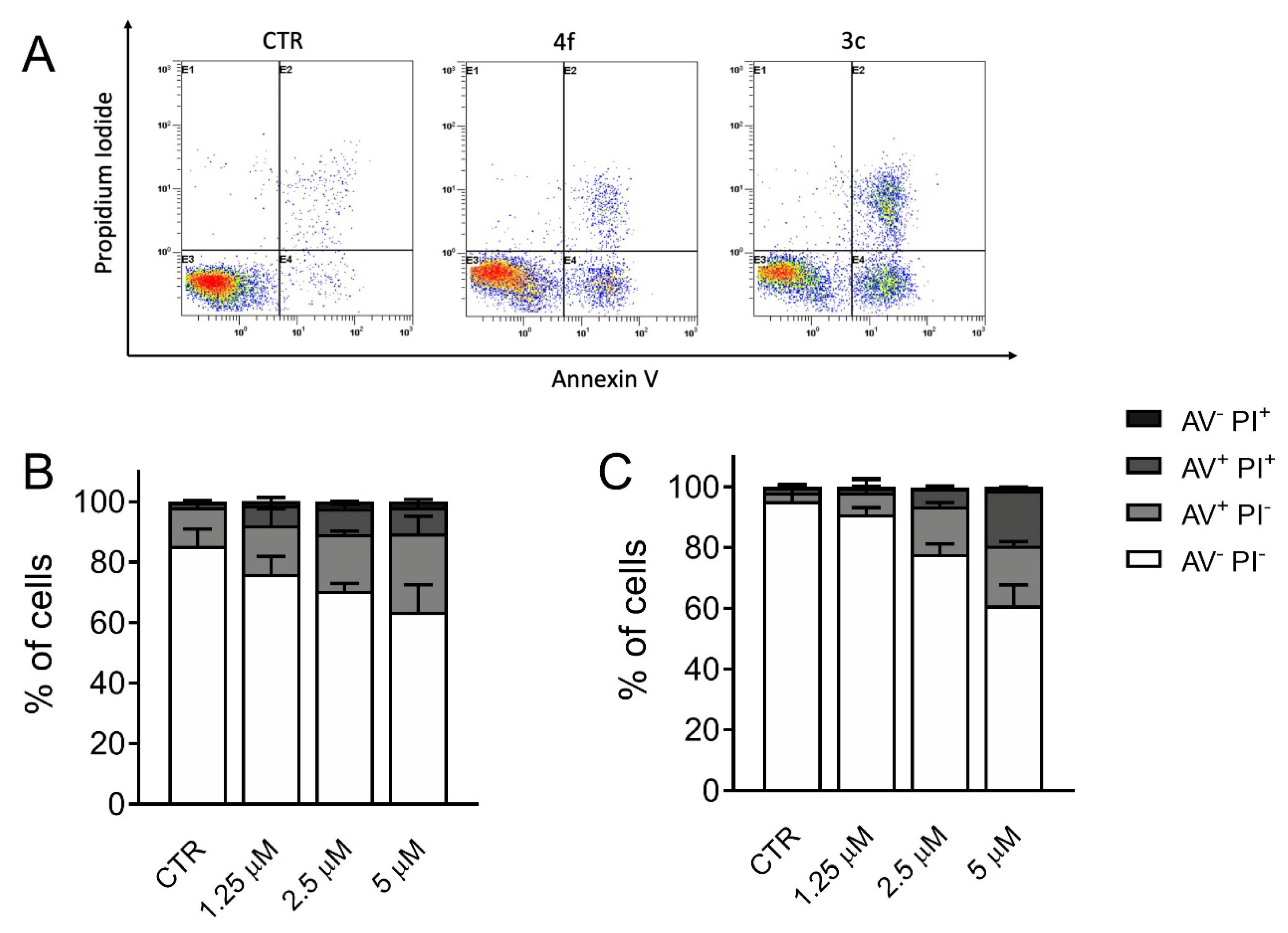
3.6. Measurement of Apoptosis by Flow Cytometry
3.7. Choline Uptake Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, F.; Zafar, H.; Zhang, S.; Kamal, Z.; Su, J.; Yuan, W.; Mingfeng, Q. Recent Advances in Cell Membrane-Derived Biomimetic Nanotechnology for Cancer Immunotherapy. Adv. Healthc. Mater. 2021, 10, 2002081. [Google Scholar] [CrossRef] [PubMed]
- Zafar, H.; Raza, F.; Ma, S.; Wei, Y.; Zhang, J.; Shen, Q. Recent progress on nanomedicine-induced ferroptosis for cancer therapy. Biomater. Sci. 2021, 9, 5092–5115. [Google Scholar] [CrossRef] [PubMed]
- Aquib, M.; Zhang, H.; Raza, F.; Banerjee, P.; Bavi, R.; Kesse, S.; Boakye-Yiadom, K.O.; Filli, M.S.; Farooq, M.A.; Wang, B. Delivery of repurposed disulfiram by aminated mesoporous silica nanoparticles for anticancer therapy. J. Mol. Liq. 2021, 117065. [Google Scholar] [CrossRef]
- Gibellini, F.; Smith, T.K. The Kennedy Pathway—De Novo Synthesis of Phosphatidylethanolamine and Phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Kanno, K.; Wu, M.K.; Scapa, E.F.; Roderick, S.L.; Cohen, D.E. Structure and Function of Phosphatidylcholine Transfer Protein (PC-TP)/StarD2. Biochim. Biophys. Acta 2007, 1771, 654–662. [Google Scholar] [CrossRef] [Green Version]
- Castro-Navas, F.F.; Schiaffino-Ortega, S.; Carrasco-Jimenez, M.P.; Rios Marco, P.; Marco, C.; Espinosa, A.; Gallo, M.A.; Mariotto, E.; Basso, G.; Viola, G.; et al. New more polar symmetrical bipyridinic compounds: New strategy for the inhibition of choline kinase alpha 1. Future Med. Chem. 2015, 7, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Estévez-Braun, A.; Ravelo, A.G.; Pérez-Sacau, E.; Lacal, J.C. A new family of choline kinase inhibitors with antiproliferative and antitumor activity derived from natural products. Clin. Transl. Oncol. 2015, 17, 74–84. [Google Scholar] [CrossRef]
- Rodríguez-González, A.; Ramírez de Molina, A.; Fernández, F.; Lacal, J.C. Choline kinase inhibition induces the increase in ceramides resulting in a highly specific and selective cytotoxic antitumoral strategy as a potential mechanism of action. Oncogene 2004, 23, 8247–8259. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, S.; Xie, P. ChoK-Full of Potential: Choline Kinase in B Cell and T Cell Malignancies. Pharmaceutics 2021, 13, 911. [Google Scholar] [CrossRef] [PubMed]
- Lacal, J.C.; Zimmerman, T.; Campos, J.M. Choline Kinase: An Unexpected Journey for a Precision Medicine Strategy in Human Diseases. Pharmaceutics 2021, 13, 788. [Google Scholar] [CrossRef]
- Rubio-Ruiz, B.; Serran-Aguilera, L.; Hurtado-Guerrero, R.; Conejo-Garcia, A. Recent advances in the design of choline kinase alpha inhibitors and the molecular basis of their inhibition. Med. Res. Rev. 2021, 41, 902–927. [Google Scholar] [CrossRef]
- Serrán Aguilera, L.; Mariotto, E.; Rubbini, G.; Franci Castro Navas, F.F.; Marco, C.; Carrasco-Jiménez, M.P.; Ballarotto, M.; Macchiarulo, A.; Hurtado-Guerrero, R.; Viola, G.; et al. Synthesis, Biological evaluation, in silico modeling and Crystallisation of novel small monocationic molecules with potent antiproliferative activity by dual mechanism. Eur. J. Med. Chem. 2020, 207, 112797. [Google Scholar] [CrossRef] [PubMed]
- Sola-Leyva, A.; López-Cara, L.C.; Ríos-Marco, P.; Ríos, A.; Marco, C.; Carrasco-Jimenez, M.P. Choline kinase inhibitors EB-3D and EB-3P interferes with lipid homeostasis in HepG2 cells. Sci. Rep. 2019, 9, 5109. [Google Scholar] [CrossRef] [Green Version]
- Jabalera, Y.; Sola-Leyva, A.; Peigneux, A.; Vurro, F.; Iglesias, G.R.; Vilchez-Garcia, J.; Inmaculada Pérez-Prieto, I.; Aguilar-Troyano, F.J.; López-Cara, L.C.; Carrasco-Jiménez, M.P.; et al. Biomimetic Magnetic Nanocarriers Drive Choline Kinase Alpha Inhibitor inside Cancer Cells for Combined Chemo-Hyperthermia Therapy. Pharmaceutics 2019, 12, 408. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, E.; Bortolozzi, R.; Volpin, I.; Carta, D.; Serafin, V.; Accordi, B.; Basso, G.; Luque Navarro, P.; López-Cara, L.C.; Viola, G. EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR Pathway and apoptosis in leukemia T-cells. Biochem. Pharmacol. 2018, 155, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, E.; Viola, G.; Ronca, R.; Persano, L.; Aveic, S.; Bhujwalla, Z.M.; Mori, N.; Accordi, B.; Serafin, V.; López-Cara, L.C.; et al. Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers 2018, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Janardhan, S.; Srivani, P.; Sastry, G.N. Choline Kinase: An Important Target for Cancer. Curr. Med. Chem. 2006, 13, 1169–1186. [Google Scholar] [CrossRef]
- Schiaffino-Ortega, S.; Lopez-Cara, L.C.; Rios-Marco, P.; Carrasco-Jimenez, M.P.; Gallo, M.A.; Espinosa, A.; Marco, C.; Entrena, A. New non-symmetrical choline kinase inhibitors. Bioorg. Med. Chem. 2013, 21, 7146–7154. [Google Scholar] [CrossRef] [PubMed]
- Sahun-Roncero, M.; Rubio-Ruiz, B.; Saladino, G.; Conejo-Garcia, A.; Espinosa, A.; Velazquez-Campoy, A.; Gervasio, F.L.; Entrena, A.; Hurtado-Guerrero, R. The Mechanism of Allosteric Coupling in Choline Kinase1 Revealed by the Action of a Rationally Designed Inhibitor. Angew. Chem. Int. Ed. 2013, 52, 4582–4684. [Google Scholar] [CrossRef]
- Sahún-Roncero, M.; Rubio-Ruiz, B.; Conejo-Garcia, A.; Velázquez-Campoy, A.; Entrena, A.; Hurtado-Guerrero, R. Determination of Potential Scaffolds for Human Choline Kinase α1 by Chemical Deconvolution Studies. ChemBioChem 2013, 14, 1291–1295. [Google Scholar] [CrossRef]
- Rubio-Ruiz, B.; Conejo-Garcia, A.; Rios-Marco, P.; Carrasco-Jimenez, M.P.; Segovia, J.; Marco, C.; Gallo, M.A.; Espinosa, A.; Entrena, A. Design, synthesis, theoretical calculations and biological evaluation of new non-symmetrical choline kinase inhibitors. Eur. J. Med. Chem. 2012, 20, 154–162. [Google Scholar] [CrossRef]
- Schiaffino-Ortega, S.; Baglioni, E.; Mariotto, E.; Bortolozzi, R.; Serrán-Aguilera, L.; Ríos-Marco, P.; Carrasco-Jimenez, M.P.; Gallo, M.A.; Hurtado-Guerrero, R.; Marco, C.; et al. Design, synthesis, crystallization and biological evaluation of new symmetrical biscationic compounds as selective inhibitors of human Choline Kinase α1 (ChoKα1). Sci. Rep. 2016, 6, 23793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbini, G.; Buades-Martín, A.B.; Kimatrai-Salvador, M.; Entrena, A.; Gallo-Mezo, M.A.; Ríos-Marco, P.; Marco, C.; Mattiuzzo, E.; Bortolozzi, R.; Mariotto, E.; et al. Lead optimization-hit expansion of new asymmetrical pyridinium/quinolinium compounds as choline kinase α1 inhibitors. Future Med. Chem. 2018, 10, 1769. [Google Scholar] [CrossRef] [PubMed]
- SYBYL-X 2.0, Tripos International, 1699 South Hanley Rd., St. Louis, MI 63144, USA. Available online: http://www.tripos.com (accessed on 28 August 2021).
- Conejo-Garcia, A.; Campos, J.M.; Entrena, A.; Sanchez-Martin, R.M.; Gallo, M.A.; Espinosa, A. Conformational dynamics of a bispyridinium cyclophane. J. Org. Chem. 2003, 68, 8697–8699. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, version 1.4; Schrodinger, LLC.: New York, NY, USA, 2021.
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Schiaffino Ortega, S.; Prencipe, F.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Mitola, S.; Ronca, R.; et al. Design, Synthesis, in Vitro and in Vivo Anticancer and Antiangiogenic Activity of Novel 3-Arylamino Benzofuran Derivatives Targeting the Colchicine Site on Tubulin. J. Med. Chem. 2015, 58, 3209–3222. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | clogP a | IC50 b (μM) hChoKα1 | GI50 c (μM) | |||||
|---|---|---|---|---|---|---|---|---|
| HT-29 | HeLa | MCF-7 | CCRF-CEM | HL-60 | RS4;11 | |||
| 3a | 0.68 | >50 | 36.0 ± 7.4 | 4.8 ± 0.58 | 22.8 ± 4.3 | 3.4 ± 0.7 | 4.8 ± 0.4 | 16.8 ± 4.8 |
| 3b | 2.57 | 17.55 ± 1.24 | nd | nd | nd | nd | nd | nd |
| 3c | 3.13 | 8.56 ± 0.94 | 0.63 ± 0.14 | 0.068 ± 0.028 | 0.3 ± 0.1 | 0.03 ± 0.003 | 0.011 ± 0.005 | 0.15 ± 0.012 |
| 3d | 4.18 | 6.79 ± 0.47 | 0.58 ± 0.17 | 0.16 ± 0.048 | 0.5 ± 0.2 | 0.015 ± 0.005 | 0.12 ± 0.091 | 0.29 ± 0.053 |
| 3e | 0.57 | 6.33 ± 0.80 | 26.1 ± 6.7 | 8.2 ± 2.4 | 23.6 ± 5.1 | 1.2 ± 0.5 | 38.7 ± 6.2 | 53.6 ± 13.4 |
| 3f | 2.45 | 6.39 ± 0.46 | 4.4 ± 0.7 | 4.2 ± 0.5 | 7.6 ± 1.6 | 0.54 ± 0.21 | 2.4 ± 0.7 | 2.0 ± 0.5 |
| 3g | 3.01 | 4.29± 0.12 | 2.2 ± 0.9 | 1.2 ± 0.2 | 4.3 ± 0.9 | 0.40 ± 0.26 | 1.6 ± 0.9 | 2.6 ± 0.2 |
| 3h | 4.07 | 7.01 ± 0.13 | nd | nd | nd | nd | nd | nd |
| 4a | 0.33 | 3.34 ± 0.34 | 49.7 ± 11.4 | 12.8 ± 2.6 | 17.8 ± 5.3 | 7.3 ± 2.5 | 4.1 ± 1.2 | 24.5 ± 2.9 |
| 4b | 2.22 | 3.85 ± 0.10 | 6.6 ± 2.3 | 2.1 ± 0.4 | 4.4 ± 0.5 | 0.5 ± 0.1 | 1.1 ± 0.3 | 2.8 ± 0.6 |
| 4c | 2.78 | 3.83 ± 0.91 | 5.3 ± 2.1 | 0.86 ± 0.26 | 1.7 ± 0.4 | 0.14 ± 0.03 | 0.065 ± 0.023 | 0.46 ± 0.061 |
| 4d | 3.84 | 3.36 ± 0.17 | 5.8 ± 1.9 | 0.39 ± 0.12 | 2.8 ± 0.5 | 0.086 ± 0.03 | 0.33 ± 0.074 | 0.71 ± 0.16 |
| 4e | 0.22 | 2.82 ±0.16 | 23.8 ± 8.2 | 12.42 ± 1.8 | 6.7 ± 1.5 | 8.4 ± 1.6 | 4.0 ± 0.9 | 31.0 ± 2.6 |
| 4f | 2.10 | 0,99 ± 0,17 | 13.2 ± 5.0 | 3.6 ± 0.4 | 5.0 ± 1.0 | 1.1 ± 0.4 | 1.3 ± 0.4 | 3.7 ± 0.4 |
| 4g | 2.66 | 3.66 ± 0.11 | 5.2 ± 1.7 | 1.7 ± 0.2 | 2.4 ± 0.2 | 0.7 ± 0.1 | 0.25 ± 0.066 | 1.1 ± 0.3 |
| 4h | 3.72 | 6.39 ± 0.46 | 3.6 ± 1.4 | 0.7 ± 0.1 | 2.2 ± 1.0 | 0.16 ± 0.034 | 0.37 ± 0.079 | 0.5 ± 0.08 |
| GI50 (µM) a | ||
|---|---|---|
| 3c | 4f | |
| PBLresting b | >10 | 7.6 ± 1.1 |
| PBLPHA c | >10 | 3.6 ± 0.8 |
| Compounds | IC50 (Choline Uptake, μM) |
|---|---|
| 3d | 15.8 ± 0.24 |
| 4f | 3.5 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiaffino-Ortega, S.; Mariotto, E.; Luque-Navarro, P.M.; Kimatrai-Salvador, M.; Rios-Marco, P.; Hurtado-Guerrero, R.; Marco, C.; Carrasco-Jimenez, M.P.; Viola, G.; López-Cara, L.C. Anticancer and Structure Activity Relationship of Non-Symmetrical Choline Kinase Inhibitors. Pharmaceutics 2021, 13, 1360. https://doi.org/10.3390/pharmaceutics13091360
Schiaffino-Ortega S, Mariotto E, Luque-Navarro PM, Kimatrai-Salvador M, Rios-Marco P, Hurtado-Guerrero R, Marco C, Carrasco-Jimenez MP, Viola G, López-Cara LC. Anticancer and Structure Activity Relationship of Non-Symmetrical Choline Kinase Inhibitors. Pharmaceutics. 2021; 13(9):1360. https://doi.org/10.3390/pharmaceutics13091360
Chicago/Turabian StyleSchiaffino-Ortega, Santiago, Elena Mariotto, Pilar María Luque-Navarro, María Kimatrai-Salvador, Pablo Rios-Marco, Ramon Hurtado-Guerrero, Carmen Marco, María Paz Carrasco-Jimenez, Giampietro Viola, and Luisa Carlota López-Cara. 2021. "Anticancer and Structure Activity Relationship of Non-Symmetrical Choline Kinase Inhibitors" Pharmaceutics 13, no. 9: 1360. https://doi.org/10.3390/pharmaceutics13091360
APA StyleSchiaffino-Ortega, S., Mariotto, E., Luque-Navarro, P. M., Kimatrai-Salvador, M., Rios-Marco, P., Hurtado-Guerrero, R., Marco, C., Carrasco-Jimenez, M. P., Viola, G., & López-Cara, L. C. (2021). Anticancer and Structure Activity Relationship of Non-Symmetrical Choline Kinase Inhibitors. Pharmaceutics, 13(9), 1360. https://doi.org/10.3390/pharmaceutics13091360