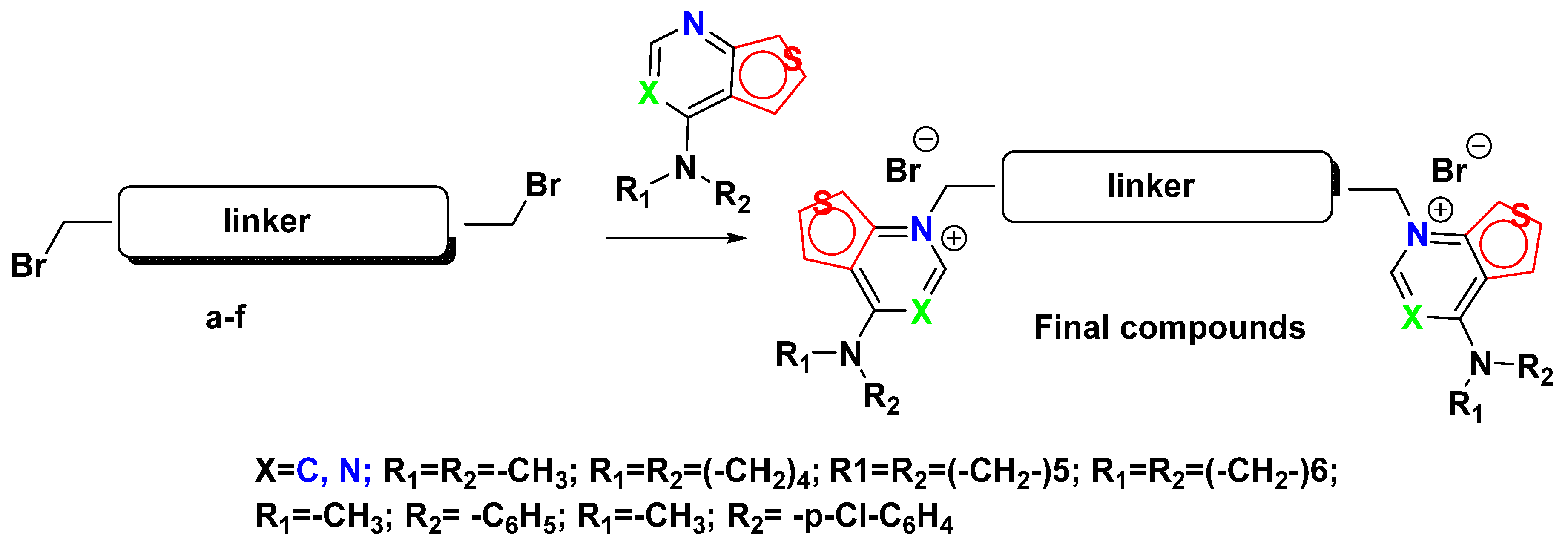
3.1.2. General Procedure for the Synthesis of the Target Compounds
A suspension of 1 equivalent of the linker and 2 equivalents of the corresponding cationic head in CH
3CN was prepared in a sealed tube. The reaction mixture was stirred at 100 °C for 5 days. Afterwards, the cooled reaction was recrystallized by diethyl ether, filtered to obtain the final product as a solid without purification (
Scheme 1).
4-([1,1’-biphenyl]-4-ylmethyl)-7-(pyrrolidin-1-yl)thieno[3,2-b]pyridin-4-ium bromide FM-a2. Following the general procedure, after workup as described previously, compound FM-a2 was isolated as a light-brown solid. Yield: 82%, mp: 271–273 °C. 1H NMR (400 MHz, CD3OD) δ 2.14 (dd, J = 16.1, 9.0 Hz, 4H), 3.71 (s, 2H), 4.25 (s, 2H), 5.70 (s, 2H), 6.73 (d, J = 7.4 Hz, 1H), 7.32 (pt, 1H), 7.32–7.35 (m, 2H), 7.38 (d, J = 7.3Hz, 2H), 7.55 (d, J = 7.4 Hz, 2H), 7.61 (d, J = 8.2 Hz, 2H), 7.64 (d, J = 5.8 Hz, 1H), 8.28 (d, J = 5.8 Hz, 1H), 8.35 (d, J = 7.4 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 24.12 (1C), 25.79 (1C), 49.89 (1C), 50.69 (1C), 57.92 (1C), 101.94 (1C), 117.29 (1C), 120.82 (1C), 126.49 (2C), 127.32 (1C), 127.34 (2C), 127.39 (2C), 128.51 (2C), 133.39 (1C), 136.01 (1C), 139.92 (1C), 141.58 (1C), 141.67 (1C), 145.90 (1C), 152.08 (1C). HRMS-m/z (M)+ calculated for C24H23N2S: 371.1582; found: 371.1585.
1-([1,1’-biphenyl]-4-ylmethyl)-4-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-1-ium bromide FM-a1. Following the general procedure, after workup as described previously, compound FM-a1 was isolated as a yellow solid. Yield: 69%, mp: 295–298 °C. 1H NMR (500 MHz, CD3OD) δ 2.09–2.14 (m, 2H), 2.26–2.30 (m, 2H), 3.98–4.04 (m, 2H), 4.19–4.22 (m, 2H), 5.72 (s, 2H), 7.35 (ddd, J = 1.5, 2.0, 7.0 Hz, 1H), 7.43 (m, 2H), 7.47 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 7.3 Hz, 2H), 7.63 (d, J = 5.7 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 8.45 (d, J = 5.7 Hz, 1H), 8.90 (s, 1H). 13C NMR (126 MHz, CD3OD) δ 23.64 (1C), 25.80 (1C), 49.30 (1C), 50.28 (1C), 55.23 (1C), 116.82 (1C), 117.19 (1C), 126.54 (2C), 127.41 (1C), 127.45 (2C), 127.62 (2C), 128.56 (2C), 132.72 (1C), 138.71 (1C), 139.90 (1C), 141.81 (1C), 146.19 (1C), 149.89 (1C), 155.30 (1C). HRMS-m/z (M)+ calculated for C23H22N3S: 372.1534; found, 372.1564.
1-([1,1’-biphenyl]-4-ylmethyl)-4-(pyrrolidin-1-yl)thieno[2,3-d]pyrimidin-1-ium bromide FMa3. Following the general procedure, after workup as described previously, compound FMa3 was isolated as a white-grey solid. Yield: 48%, mp: 275–278 °C. 1H NMR (400 MHz, DMSO) δ 1.95–197 (m, 2H), 2.05–2.08 (m, 2H), 3.88 (t, J = 6.8 Hz, 2H), 4.03 (t, J = 6.8 Hz, 2H), 5.70 (s, 2H), 7.34 (t, J = 7.3 Hz, 1H), 7.43 (t, J = 7.5 Hz, 2H), 7.52 (d, J = 8.1, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.69 (d, J = 8.2 Hz, 2H), 7.83 (dd, J = 26.1, 5.9 Hz, 2H), 9.20 (s, 1H). 13C NMR (101 MHz, DMSO) δ 23.91 (1C), 26.24 (1C), 50.72 (1C), 51.15 (1C), 56.96 (1C), 117.39 (1C), 123.85 (1C), 124.76 (1C), 127.14 (2C), 127.61 (2C), 128.25 (1C), 129.25 (2C), 129.42 (2C), 132.22 (1C), 139.62 (1C), 141.15 (1C), 149.62 (1C), 153.39 (1C), 154.39 (1C). HRMS-m/z (M)+ calculated for C23H22N3S: 372.1534; found: 372.1551.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(7-(pyrrolidin-1-yl)thieno[3,2-b]pyridin-4-ium) bromideFg-9. Following the general procedure, after workup as described previously, compound Fg-9 was isolated as a yellow solid. Yield: 40%, mp: >290 °C. 1H NMR (500 MHz, CD3OD) δ 2.20 (d, J = 18.4 Hz, 8H), 3.75 (s, 4H), 4.28 (s, 4H), 5.73 (s, 4H), 6.76 (d, J = 7.4 Hz, 2H), 7.38 (d, J = 8.5 Hz, 4H), 7.62 (s, 4H), 7.65 (d, J = 5.8 Hz, 2H), 8.30 (d, J = 5.8 Hz, 2H), 8.38 (d, J = 7.4 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 24.27 (4C), 25.81 (4C), 57.85 (2C), 101.96 (2C), 117.26 (2C), 120.81 (2C), 127.33 (4C), 127.46 (4C), 133.98 (2C), 136.02 (2C), 140.45 (2C), 141.70 (2C), 145.88 (2C), 152.09 (2C). HRMS-m/z (M)+ calculated for C36H36N4S2: 588.237; found: 588.2347.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fa-21. Following the general procedure, after workup as described previously, compound Fa-21 was isolated as a yellow-lemon solid. Yield: 75%, mp: 312–315 °C. 1H NMR (500 MHz, CD3OD) δ 2.12 (q, J = 6.9 Hz, 4H), 2.27 (q, J = 6.9 Hz, 4H), 4.01 (t, J = 7.0 Hz, 4H), 4.21 (t, J = 6.9 Hz, 4H), 5.73 (s, 4H), 7.47 (d, J = 8.2 Hz, 4H), 7.61 (d, J = 5.7 Hz, 2H), 7.65 (d, J = 8.3 Hz, 4H), 8.45 (d, J = 5.7 Hz, 2H), 8.91 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 23.64 (2C), 25.81 (2C), 49.32 (2C), 50.29 (2C), 55.15 (2C), 116.83 (2C), 117.18 (2C), 127.44 (4C), 127.76 (4C), 133.32 (2C), 138.74 (2C), 140.59 (2C), 146.16 (2C), 149.90 (2C), 155.29 (2C). HRMS-m/z (M)2+ calculated for C34H34N6S2: 590.2286; found: 590.2299.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(pyrrolidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-14. Following the general procedure, after workup as described previously, compound Fg-14 was isolated as a white solid. Yield: 70%, mp: 300–302 °C. 1H NMR (400 MHz, DMSO) δ 1.95 (dd, J = 13.4, 6.7 Hz, 4H), 2.09 (dt, J = 13.8, 6.8 Hz, 4H), 3.87 (t, J = 6.7 Hz, 4H), 4.03 (t, J = 6.6 Hz, 4H), 5.69 (s, 4H), 7.59 (dd, J = 69.1, 8.1 Hz, 8H), 7.83 (dd, J = 27.0, 5.9 Hz, 4H), 9.19 (s, 2H). 13C NMR (101 MHz, DMSO) δ 154.38 (2C), 153.38 (2C), 149.63 (2C), 140.17 (2C), 132.65 (2C), 129.27 (4C), 124.29 (4C), 124.72 (2C), 123.86 (2C), 117.37 (2C), 56.90 (2C), 51.16 (2C), 50.65 (2C), 26.24 (2C), 23.91 (2C). HRMS-m/z (M)+ calculated for C34H34N6S2: 590.2275; found: 590.2261.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(piperidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromideFa-24. Following the general procedure, after workup as described previously, compound Fa-24 was isolated as a yellow-lemon solid. Yield: 80.36%, mp: 312–315 °C. 1H NMR (400 MHz, CD3OD) δ 1.83–186 (m, 12H), 4.24 (d, J = 19.3 Hz, 8H), 5.72 (s, 4H), 7.45 (d, J = 8.1 Hz, 4H), 7.61 (d, J = 5.9 Hz, 2H), 7.63 (d, J = 8.2 Hz, 4H), 8.40 (d, J = 5.8 Hz, 2H), 8.90 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 23.44 (2C), 25.61 (2C), 26.34 (2C), 47.77 (2C), 50.11 (2C), 55.22 (2C), 115.04 (2C), 117.47 (2C), 127.44 (4C), 127.74 (4C), 133.24 (2C), 138.09 (2C), 140.60 (2C), 147.50 (2C), 149.51 (2C), 155.82 (2C). HRMS–m/z (M)+ calculated for C36H38N6S2: 618.2599, found: 618.2609.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(piperidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-30. Following the general procedure, after workup as described previously, compound Fg-30 was isolated as a white solid. Yield: 57%, mp: 388–290 °C. 1H NMR (500 MHz, CD3OD) δ 1.95–1.97 (m, 12H), 4.17 (s, 4H), 4.29 (s, 4H), 5.70 (s, 4H), 7.63 (d, J = 8.3 Hz, 8H), 7.78 (d, J = 6.0 Hz, 4H), 8.93 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 23.39 (6C), 50.35 (2C), 56.99 (4C), 116.26 (2C), 123.04 (2C), 124.42 (2C), 127.49 (4C), 128.69 (4C), 131.62 (2C), 141.02 (2C), 148.22 (2C), 154.83 (2C), 155.99 (2C). HRMS-m/z (M)+ calculated for C36H38N8S2: 618.2588, found: 618.2568.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(7-(azepan-1-yl)thieno[3,2-b]pyridin-4-ium) bromide Fg-10. Following the general procedure, after workup as described previously, compound Fg-10 was isolated as a white solid. Yield: 53%, mp: 220–222 °C. 1H NMR (600 MHz, CD3OD) δ 1.68 (s, 8H), 2.01 (s, 8H), 4,08 (s, 8H), 5.76 (s, 4H), 6.98 (d, J = 7.5 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 5.9 Hz, 2H), 8.31 (d, J = 5.9 Hz, 2H), 8.40 (d, J = 7.5 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 26.11 (4C), 32.23 (4C), 52.40 (4C), 58.08 (2C), 101.75 (2C), 117.41 (2C), 119.20 (2C), 126.62 (2C), 127.39 (2C), 127.50 (2C), 129.37 (2C), 133.64 (2C), 135.90 (2C), 137.88 (2C), 139.89 (2C), 140.90 (2C), 141,71 (2C), 146.55 (2C), 154,18 (2C). HRMS-m/z (M)+ calculated for C40H44N4S2: 644.2996; found: 644.3033.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(azepan-1-yl) thieno[2,3-d]pyrimidin-1-ium) bromide Fa-22. Following the general procedure, after workup as described previously, compound Fa-22 was isolated as a yellow-brown solid. Yield: 47.22%, mp: 304–306 °C. 1H NMR (500 MHz, CD3OD) δ 1.69–171 (m, 8H), 1.96–197 (m, 4H), 2.06–2.08 (m, 4H), 4.21–4.23 (m, 4H), 4.24–4.27 (m, 4H), 5.74 (s, 4H), 7.48 (d, J = 8.3 Hz, 4H), 7.63 (d, J = 5.8 Hz, 2H), 7.67 (d, J = 8.3 Hz, 4H), 8.46 (d, J = 5.8 Hz, 2H), 8.92 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 26.08 (2C), 26.20 (2C), 26.31 (2C), 28.09 (2C), 50.34 (2C), 51.35 (2C), 55.24 (2C), 115.28 (2C), 117.28 (2C), 127.49 (4C), 127.77 (4C), 133.23 (2C), 138.73 (2C), 140.66 (2C), 147.05 (2C), 149.45 (2C), 157.11 (2C). HRMS-m/z (M)+ calculated for C38H42N6S2: 646.2912, found: 646.2885.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(azepan-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fg-18. Following the general procedure, after workup as described previously, compound Fg-18 was isolated as a white solid. Yield: 74%, mp: 303–305 °C. 1H NMR (400 MHz, CD3OD) δ 1.68–1.66 (m, 8H), 1.94 (s, 4H), 2.01 (s, 4H), 4.16–4.12 (m, 4H), 4.21–4.17 (m, 4H), 5.68 (s, 4H), 7.61 (d, J = 8.3 Hz, 8H), 7.76 (d, J = 6.0 Hz, 4H), 8.94 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 26.16 (2C), 26.37 (2C), 26.46 (2C), 27.35 (2C), 50.33 (2C), 51.56 (2C), 57.03 (2C), 116.37 (2C), 123.31 (2C), 123.67 (2C), 127.46 (4C), 128.69 (4C), 131.53 (2C), 140.9 (2C), 148.05 (2C), 154.54 (2C), 156.60 (2C). HRMS-m/z (M)+ calculated for C38H42N6S2: 646.2901; found: 646.2908.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-(methyl(phenyl)amino)thieno[3,2-d]pyrimidin-1-ium)bromide Fp-1. Following the general procedure, after workup as described previously, compound Fp-1 was isolated as a blue solid. Yield: 20%, mp: 185–187 °C. 1H NMR (400 MHz, CD3OD) δ 3.84 (s, 6H). 5.80 (s, 4H), 7.71–7.44 (m, 20H), 8.14 (d, J = 5.7 Hz, 2H), 9.16 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 41.78 (2C), 57.05 (2C), 117.92 (2C), 128.23 (4C), 128.87 (4C), 129.20 (4C), 130.36 (4C), 131.84 (2C), 134.26 (2C), 138.96 (2C), 141.25 (2C), 141.77 (2C), 142.52 (2C), 148.62 (2C), 151.66 (2C), 159.00 (2C). HRMS-m/z (M)+ calculated for C40H34N6S2: 662.2286, found: 662.2267.
1,1’-([1,1’-biphenyl]-4,4’-diylbis(methylene))bis(4-((4-chlorophenyl)(methyl)amino)thieno[2,3-d]pyrimidin-1-ium) bromide Fp-8. Following the general procedure, after workup as described previously, compound Fp-8 was isolated as a white solid. Yield: 40%, mp: 264–265 °C. 1H NMR (400 MHz, DMSO) δ 3.76 (s, 6H), 5.81 (s, 4H), 7.74–7.57 (m, 20H), 9.44 (s, 2H). 13C NMR (126 MHz, DMSO) δ 41.85 (2C), 56.94 (2C), 117.33 (4C), 125.60 (2C), 127.27 (6C), 129.00 (8C), 130.74 (4C), 132.00 (2C), 139.82 (4C), 149.66 (2C), 154.25 (2C), 156.60 (2C). HRMS-m/z (M)+ calculated for C40H32N6S2Cl2: 730.1507, found: 730.1520.
4,4’-([2,2’-bipypidine]-5,5’-diylbis(methylene))bis(7-(pyrrolidin-1-yl)thieno[3,2-b]pyridin-4-ium bromide).Fg-12. Following the general procedure, after workup as described previously, compound Fg-12 was isolated as a purple solid. Yield: 60%, mp: >290 °C. 1H NMR (400 MHz, DMSO) δ 2.05 (s, 4H), 3.63 (s, 4H), 4.09 (d, J = 28.6 Hz, 8H), 5.83 (s, 4H), 6.80 (d, J = 7.4 Hz, 2H), 7.78 (dd, J = 8.4, 2.0 Hz, 2H), 7.82 (d, J = 5.8 Hz, 2H), 8.28 (d, J = 8.3 Hz, 2H), 8.49 (d, J = 5.8 Hz, 2H), 8.61 (d, J = 7.4 Hz, 2H), 8.69 (d, J = 1.4 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 24.41 (2C), 26.36 (2C), 51.04 (2C), 51.47 (2C), 65.33 (2C) 102.99 (2C), 118.15 (2C), 120.55 (2C), 120.98 (2C), 132.09 (2C), 136.65 (2C), 137.66 (2C), 142.59 (2C), 145.78 (2C), 148.85 (2C), 151.85 (2C), 155.07 (2C). HRMS-m/z (M)+ calculated for C34H34N6S2: 590.2275; found: 590.2247.
1,1’-([2,2’-bipyridine]-5,5’-diylbis(methylene))bis(4-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fg-17. Following the general procedure, after workup as described previously, compound Fg-17 was isolated as a yellow solid. Yield: 62%, mp: 330–332 °C. 1H NMR (400 MHz, CD3OD) δ 2.09 (q, J = 6.8 Hz, 4H), 2.25 (q, J = 6.8 Hz, 4H), 3.99 (t, J = 6.9 Hz, 4H), 4.19 (t, J = 6.9 Hz, 4H), 5.79 (s, 4H), 7.63 (d, J = 5.7 Hz, 2H), 7.90 (dd, J = 8.3, 2.0 Hz, 2H), 8.35 (d, J = 8.3 Hz, 2H), 8.45 (d, J = 5.7 Hz, 2H), 8.72 (d, J = 1.5 Hz, 2H), 8.91 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 23.61 (2C), 25.77 (2C), 49.35 (2C), 50.33 (2C), 52.77 (2C), 116.87 (2C), 121.13 (2C), 130.57 (2C), 136.20 (4C), 138.98 (2C), 145.89 (2C), 148.16 (2C), 149.96 (2C), 155.29 (2C), 155.35 (2C). HRMS-m/z (M)+ calculated for C32H32N8S2: 592.2180; found: 592.2235.
1,1’-([2,2’-bipyridine]-5,5’-diylbis(methylene))bis(4-(pyrrolidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-13. Following the general procedure, after workup as described previously, compound Fg-13 was isolated as a white solid. Yield: 71%, mp: 238–240 °C. 1H NMR (400 MHz, DMSO) δ 2.14–1.91 (m, 8H), 3.87 (t, J = 6.9 Hz, 4H), 4.03 (t, J = 6.9 Hz, 4H), 5.79 (s, 2H), 7.80 (d, J = 5.9 Hz, 2H), 7.87 (d, J = 5.9 Hz, 2H), 8.01 (dd, J = 8.3, 2.3 Hz, 2H), 8.35 (d, J = 8.3 Hz, 2H), 8.83 (d, J = 2.3 Hz, 2H), 9.21 (s, 2H). 13C NMR (101 MHz, DMSO) δ 23.91 (2C), 26.24 (2C), 50.75 (2C), 51.18 (2C), 54.67 (2C), 117.43 (2C), 121.04 (2C), 123.96 (2C), 124.62 (2C), 129.78 (2C), 137.72 (2C), 149.75 (2C), 155.44 (2C), 149.86 (2C), 153.25 (2C), 154.41 (2C). HRMS-m/z (M)+ calculated for C32H32N8S2: 592.2180; found: 592.2200.
1,1’-([2,2’-bipyridine]-5,5’-diylbis(methylene))bis(4-(piperidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fa-27. Following the general procedure, after workup as described previously, compound Fa-27 was isolated as a yellow-brown solid. Yield: 58%, mp: 307–309 °C. 1H NMR (500 MHz, CD3OD) δ 1.86–189 (m, 12H), 4.29–4.27 (m, 8H), 5.84 (s, 4H), 7.68 (d, J = 5.8 Hz, 2H), 7.93 (dd, J = 8.3, 2.2 Hz, 2H), 8.38 (d, J = 8.3 Hz, 2H), 8.46 (d, J = 5.8 Hz, 2H), 8.76 (d, J = 2.0 Hz, 2H), 8.96 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 23.45 (2C), 25.62 (2C), 26.35 (2C), 47.81 (2C), 50.19 (2C), 52.90 (2C), 115.16 (2C), 117.24 (2C), 121.18 (2C), 130.55 (2C), 136.28 (2C), 138.38 (2C), 147.30 (2C), 148.22 (2C), 149.62 (2C), 155.50 (2C), 155.85 (2C). HRMS-m/z (M)+ calculated for C34H36N8S2: 620.2504, found: 620.2446.
1,1’-([2,2’-bipyridine]-5,5’-diylbis(methylene))bis(4-(piperidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-32. Following the general procedure, after workup as described previously, compound Fg-32 was isolated as a white solid. Yield: 64%, mp: 267–269C°. 1H NMR (400 MHz, CD3OD) δ 8.94 (s, 2H), 8.81 (s, 2H), 8.41 (d, J = 8.2 Hz, 2H), 8.01 (d, J = 8.2 Hz, 2H), 7.76 (d, J 5.9 Hz, 4H), 5.76 (s, 4H), 4.27 (s, 4H), 4.14 (s, 4H), 1.84 (s, 12H). 13C NMR (101 MHz, CD3OD) δ 155.95 (2C), 155.80 (2C), 154.55 (2C), 149.07 (2C), 148.31 (2C), 137.05 (2C), 129.01 (2C), 123.36 (2C), 123.23 (2C), 121.20 (2C), 116.29 (2C), 54.53 (2C), 50.23 (2C), 24.93 (2C), 23.34 (6C). HRMS-m/z (M)+ calculated for C34H36N8S2: 620.2493; found: 620.2459.
1,1’-([2,2’-bipyridine]-5,5’-diylbis(methylene))bis(4-(azepan-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fa-26. Following the general procedure, after workup as described previously, compound Fa-26 was isolated as a light brown solid. Yield: 20%, mp: 310–312 °C. 1H NMR (400 MHz, CD3OD) δ 1.67–1.69 (m, 8H), 1.96–198 (m, 4H), 2.02–2.04 (m, 4H), 4.20–4.22 (m, 8H), 5.80 (s, 4H), 7.64 (d, J = 5.8 Hz, 2H), 7.91 (d, J = 8.0 Hz, 2H), 8.36 (d, J = 8.3 Hz, 2H), 8.46 (d, J = 5.8 Hz, 2H), 8.73 (d, J = 1.3 Hz, 2H), 8.92 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 26.07 (2C), 26.19 (2C), 26.31 (2C), 28.04 (2C), 50.38 (2C), 51.40 (2C), 52.89 (2C), 115.39 (2C), 117.02 (2C), 121.18 (2C), 130.51 (2C), 136.29 (2C), 139.00 (2C), 146.82 (2C), 148.23 (2C), 149.54 (2C), 155.51 (2C), 157.12 (2C). HRMS–m/z (M)+ calculated for C36H40N8S2: 648.2817, found: 648.2833.
1,1’-([2,2’-bipyridine]-5,5’-diylbis(methylene))bis(4-(azepan-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-20. Following the general procedure, after workup as described previously, compound Fg-20 was isolated as a white solid. Yield: 68%, mp: 289–291 °C. 1H NMR (500 MHz, CD3OD) δ 1.74–1.66 (m, 8H), 2.00–1.94 (m, 4H), 2.08–2.01 (m, 4H), 4.19–4.16 (m, 4H), 4.24–4.21 (m, 4H), 5.81 (s, 4H), 7.75 (d, J = 6.0 Hz, 2H), 7.84 (d, J = 6.0 Hz, 2H), 8.04 (dd, J = 8.3, 2.3 Hz, 2H), 8.44 (d, J = 8.3 Hz, 2H), 8.84 (d, J = 2.1 Hz, 2H), 8.96 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 26.40 (4C), 26.48 (2C), 27.35 (2C), 50.39 (2C), 51.63 (2C), 54.63 (2C), 116.48 (2C), 121.24 (2C), 123.16 (2C), 123.92 (2C), 128.97 (2C), 137.10 (2C), 148.20 (2C), 149.12 (2C), 154.33 (2C), 155.83 (2C), 156.64 (2C). HRMS-m/z (M)+ calculated for C36H40N8S2: 648.2806; found: 648.2794.
4,4’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(7-(pyrrolidin-1-yl)thieno[3,2-b]pyridin-4-ium) bromide Fg-11. Following the general procedure, after workup as described previously, compound Fg-11 was isolated as a white solid. Yield: 68%, mp: 253–255 °C. 1H NMR (400 MHz, CD3OD) δ 2.17 (s, 8H), 3.29 (dt, J = 3.2, 1.6 Hz, 2H), 3.33 (s, 2H), 3,71 (s, 4H), 4.25 (s, 4H), 5.61 (s, 4H), 6.71 (d, J = 7.4 Hz, 2H), 7.15 (s, 8H), 7.58 (d, J = 5.8 Hz, 2H), 8.27 (d, J = 5.8 Hz, 2H), 8.29 (d, J = 7.4 Hz, 2H). 13C NMR (126 MHz, CD3OD) δ 24.05 (4C), 25.69 (4C), 36.88 (1C), 50.71 (1C), 58.04 (2C), 101.90 (2C), 117.29 (2C), 120.81 (2C), 126.92 (4C), 129.00 (4C), 132.08 (2C), 135.92 (2C), 141.63 (2C), 141.96 (2C), 145.91 (2C), 152.08 (2C). HRMS-m/z (M)+ calculated for C38H40N4S2: 616.2683; found: 616.2717.
1,1’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fg-16. Following the general procedure, after workup as described previously, compound Fg-16 was isolated as a yellow solid. Yield: 72%, mp: 231–233 °C. 1H NMR (400 MHz, CD3OD) δ 2.09 (d, J = 6.8 Hz, 4H), 2.24 (d, J = 6.8 Hz, 4H), 2.85 (s, 2H), 3.29 (dt, J = 3.0, 1.5 Hz, 2H), 3.98 (t, J = 7.0 Hz, 4H), 4.18 (t, J = 6.9 Hz, 4H), 5.60 (s, 4H), 7.20 (d, J = 8.1 Hz, 4H), 7,24 (d, J = 8,1 Hz, 4H), 7.54 (d, J = 5.7 Hz, 2H), 8.42 (d, J = 5.7 Hz, 2H), 8.81 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 23.61 (4C), 25.78 (2C), 36.81 (2C), 49.28 (1C), 50.23 (1C), 55.31 (2C), 116.77 (2C), 117.16 (2C), 127.15 (4C), 129.09 (4C), 131.31 (2C), 138.62 (2C), 142.42 (2C), 146.16 (2C), 149.75 (2C), 155.25 (2C). HRMS-m/z (M)+ calculated for C36H38N6S2: 618.2588; found: 618.2586.
1,1’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-15. Following the general procedure, after workup as described previously, compound Fg-15 was isolated as a white solid. Yield: 80%, mp: 293–295 °C. 1H NMR (400 MHz, CD3OD) δ 2.10 (q, J = 6.9 Hz, 2H), 2.23 (q, J = 6.9 Hz, 2H), 2.88 (s, 4H), 4.00 (t, J = 6.9 Hz, 4H), 4.11 (t, J = 6.9 Hz, 4H), 5.56 (s, 4H), 7.19 (d, J = 8.0 Hz, 4H), 7.30 (d, J = 8.0 Hz, 4H), 7.79 (d, J = 6.0Hz, 2H), 7.88 (d, J = 6.0 Hz, 2H), 8.86 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 23.43 (4C), 25.88 (2C), 36.90 (2C), 50.33 (1C), 50.78 (1C), 57.19 (2C), 117.34 (2C), 123.08 (2C), 123.29 (2C), 128.06 (4C), 129.16 (4C), 129.57 (2C), 142.98 (2C), 148.42 (2C), 153.48 (2C), 154.51 (2C). HRMS-m/z (M)+ calculated for C36H38N6S2: 618.2588; found: 618.2586.
1,1’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(4-(piperidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fa-25. Following the general procedure, after workup as described previously, compound Fa-25 was isolated as a yellow-brown solid. Yield: 76%, mp: 314–316 °C. 1H NMR (400 MHz, CD3OD) δ 1.82–1.85 (m, 12H), 2.84–2.87 (s, 4H), 4.24–4.27 (m, 8H), 5.62 (s, 4H), 7.19 (d, J = 8.1 Hz, 4H), 7.25 (d, J = 8.1 Hz, 4H), 7.57 (d, J = 5.8 Hz, 2H), 8.40 (d, J = 5.7 Hz, 2H), 8.83 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 23.48 (2C), 25.70 (2C), 26.42 (2C), 36.88 (2C), 47.75 (2C), 50.11 (2C), 55.52 (2C), 114.99 (2C), 117.56 (2C), 127.27 (4C), 129.18 (4C), 131.10 (2C), 138.17 (2C), 142.50 (2C), 147.55 (2C), 149.34 (2C), 155.78 (2C). HRMS-m/z (M)+ calculated for C38H42N6S2: 646.2912, found: 646.2922.
1,1’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(4-(piperidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-31. Following the general procedure, after workup as described previously, compound Fg-31 was isolated as a white solid. Yield: 85%, mp: 295–297 °C. 1H NMR (400 MHz, CD3OD) δ 1.83–1.85 (s, 12H), 2.88 (s, 2H), 3.29 (s, 2H), 4.13 (s, 4H), 4.25 (s, 4H), 5.56 (s, 4H), 7.32 (dd, J = 7.9 Hz, 8H), 7.75 (dd, J = 19.1, 5.9 Hz, 4H), 8.84 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 23.36 (6C), 25.90 (2C), 36.87 (2C), 50.17 (2C) 57.17 (2C), 116.22 (2C), 122.95 (2C), 123.57 (2C), 128.10 (4C), 129.14 (4C), 129.53 (2C), 142.97 (2C), 148.07 (2C), 154.79 (2C), 155.95 (2C). HRMS-m/z (M)+ calculated for C38H42N6S2: 646.2901; found: 646.2946.
1,1’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(4-(azepan-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fa-23. Following the general procedure, after workup as described previously, compound Fa-23 was isolated as a yellow-orange solid. Yield: 81%, mp: 321–323 °C. 1H NMR (500 MHz, CD3OD) δ 1.67–1.70 (m, 8H), 1.95–1.98 (m, 4H), 2.05–2.07 (m, 4H), 2.89 (s, 4H), 4.21 (t, J = 6 Hz, 4H), 4.25 (t, J = 6 Hz, 4H), 5.65 (s, 4H), 7.22 (d, J = 8.2 Hz, 4H), 7.29 (d, J = 8.2 Hz, 4H), 7.59 (d, J = 5.8 Hz, 2H), 8.47 (t, J = 5.9 Hz, 2H), 8.87 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 26.09 (2C), 26.21 (2C), 26.32 (2C), 28.11 (2C), 36.86 (2C), 50.31 (2C), 51.34 (2C), 55.44 (2C), 115.25 (2C), 117.33 (2C), 127.24 (4C), 129.14 (4C), 131.26 (2C), 138.66 (2C), 142.52 (2C), 147.07 (2C), 149.34 (2C), 157.09 (2C). HRMS-m/z (M)+ calculated for C40H46N6S2: 674.3225, found: 674.3286.
1,1’-((ethane-1,2-diylbis(4,1-phenylene))bis(methylene))bis(4-(azepan-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Fg-19. Following the general procedure, after workup as described previously, compound Fg-19 was isolated as a white solid. Yield: 78%, mp: 298–300 °C. 1H NMR (500 MHz, CD3OD) δ 1.69–1.71 (m, 8H), 2.02–2.06 (s, 8H), 2.92 (s, 4H), 4.18–4.15 (m, 4H), 4.23–4.19 (m, 4H), 5.60 (s, 4H), 7.24 (d, J = 8.1 Hz, 4H), 7.35 (d, J = 8.1 Hz, 4H), 7.79 (d, J = 6.0 Hz, 2H), 7.81 (d, 6.0 Hz, 2H), 8.89 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 26.18 (2C), 26.39 (2C), 26.48 (2C), 27.38 (2C), 36.92 (2C), 50.34 (2C), 51.56 (2C), 57.24 (2C), 116.38 (2C), 123.33 (2C), 123.64 (2C), 128.13 (4C), 129.16 (4C), 129.49 (2C), 143.04 (2C), 147.96 (2C), 154.56 (2C), 156.62 (2C). HRMS-m/z (M)+ calculated for C40H46N6S2: 646.3214; found: 646.3174.
4,4’-((butane-1,4-diylbis(4,1-phenylene))bis(methylene))bis(7-(pyrrolidin-1-yl)thieno[3,2-b]pyridin-4-ium) bromideFf-1. Following the general procedure, after workup as described previously, compound Ff-1 was isolated as a white solid. Yield: 17%, mp: 195–197 °C. 1H NMR (400 MHz, CD3OD) δ 1.61 (s, 4H), 2.14–2.16 (m, 8H), 2.62 (s, 4H), 3.75–3.78 (m, 4H), 4.29–4.32 (m, 4H), 5.67 (s, 4H), 6.77 (d, J = 7.4 Hz, 2H), 7.20 (d, J = 8.3 Hz, 4H), 7.23 (d, J = 8.3 Hz, 4H), 7.66 (d, J = 5.8 Hz, 2H), 8.32 (d, J = 5.8 Hz, 2H), 8.36 (d, J = 7.4 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 25.11(2C), 26.80 (2C), 31.60 (2C), 35.79 (2C), 51.49 (2C), 51.70 (2C), 59.04 (2C), 102.91 (2C), 118.30 (2C), 121.77 (2C), 127.95 (4C), 129.85 (4C), 132.79 (2C), 136.92 (2C), 142.60 (2C), 144.24 (2C), 146.86 (2C), 153.06 (2C). HRMS-m/z (M)+ calculated for C40H44N4S2: 644.2996; found: 648.2584.
1,1’-((butane-1,4-diylbis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Ff-7. Following the general procedure, after workup as described previously, compound Ff-7 was isolated as a yellow solid. Yield: 19%, mp: 215–217 °C. 1H NMR (400 MHz, CD3OD) δ 1.62 (s, 4H), 2.13–2.15 (m, 4H), 2.29–2.32 (m, 4H), 2.64 (s, 4H), 4.01–4.04 (m, 4H), 4.21–4.23 (m, 4H), 5.66 (s, 4H), 7.22 (d, J = 8.0 Hz, 4H), 7.32 (d, J = 8.0 Hz, 4H), 7.62 (d, J = 5.7 Hz, 2H), 8.46 (d, J = 5.7 Hz, 2H), 8.88 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 24.41 (2C), 26.58 (2C), 31.37 (2C), 35.58 (2C), 50.06 (2C), 51.02 (2C), 56.12 (2C), 117.56 (2C), 117.96 (2C), 127.95 (4C), 129.73 (4C), 131.85 (2C), 139.41 (2C), 144.25 (2C), 146.96 (2C), 150.55 (2C), 156.05 (2C). HRMS-m/z (M)+ calculated for C38H42N6S2: 646.2901; found: 646.2936.
1,1’-((butane-1,4-diylbis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Ff-3. Following the general procedure, after workup as described previously, compound Ff-3 was isolated as a grey solid. Yield: 42%, mp: 288–290 °C. 1H NMR (500 MHz, CD3OD) δ 1.63 (s, 4H), 2.12–2.14 (m, 4H), 2.26–2.28 (m, 4H), 2.66 (s, 4H), 4.03–4.06 (m, 4H), 4.14–4.17 (m, 4H), 5.62 (s, 4H), 7.25 (d, J = 8.1 Hz, 4H), 7.37 (d, J = 8.1 Hz, 4H), 7.73 (d, J = 5.9 Hz, 2H), 7.92 (d, J = 6.0 Hz, 2H), 8.92 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 24.33 (2C), 26.78 (2C), 31.50 (2C), 35.76 (2C), 51.22 (2C), 51.67 (2C), 58.12 (2C), 118.25 (2C), 123.98 (2C), 124.17 (2C), 128.97 (4C), 129.87 (4C), 130.22 (2C), 144.97 (2C), 149.32 (2C), 154.40 (2C), 155.42 (2C). HRMS-m/z (M)+ calculated for C38H42N6S2: 646.2901; found: 646.2930.
1,1’-((butane-1,4-diylbis(4,1-phenylene))bis(methylene))bis(4-(piperidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Fa-33. Following the general procedure, after workup as described previously, compound Fa-33 was isolated as a light-yellow solid. Yield: 89%, mp: 276–278 °C. 1H NMR (400 MHz, CD3OD) δ 1.57 (m, 4H), 1.82–1.84 (m, 12H), 2.59 (t, J = 6.7 Hz, 4H), 4.21–4.24 (m, 8H), 5.62 (s, 4H), 7.18 (d, J = 8.2 Hz, 4H), 7.26 (d, J = 8.2 Hz, 4H), 7.58 (d, J = 5.8 Hz, 2H), 8.39 (d, J = 5.8 Hz, 2H), 8.84 (s, 2H). 13C NMR (101 MHz, CD3OD) δ 155.82 (2C), 149.38 (2C), 147.54 (2C), 143.51 (2C), 137.97 (2C), 130.99 (2C), 128.95 (4C), 127.15 (4C), 117.47 (2C), 115.00 (2C), 55.40 (2C), 50.09 (4C), 34.79 (2C), 30.61 (2C), 25.76 (4C), 23.44 (2C). HRMS-m/z (M)+ calculated for C40H46N6S2: 674.3225, found: 674.3190.
1,1’-((butane-1,4-diylbis(4,1-phenylene))bis(methylene))bis(4-(piperidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Ff-6. Following the general procedure, after workup as described previously, compound Ff-6 was isolated as a grey solid. Yield: 11%, mp: 282–284 °C. 1H NMR (400 MHz, CD3OD) δ 1.66 (s, 4H), 1.90–1.92 (m, 12H), 2.68 (s, 4H), 4.30–4.32 (m, 8H), 5.63 (s, 4H), 7.27 (d, J = 8.0 Hz, 4H), 7.40 (d, J = 8.5 Hz, 4H), 7.78 (d, J = 5.8 Hz, 2H), 7.84 (d, J = 6.2 Hz, 2H), 8.91 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 24.23 (4C), 26.39 (2C), 31.44 (2C), 35.70 (2C), 51.05 (4C), 58.06 (2C), 117.08 (2C), 123.81 (2C), 124.42 (2C), 128.97 (4C), 129.82 (4C), 130.89 (2C), 144.92 (2C), 148.92 (2C), 155.67 (2C), 156.82 (2C). HRMS-m/z (M)+ calculated for C40H46N6S2: 674.3225, found: 674.3239.
(1,1’((butane1,4diylbis(4,1phenylene))bis(methylene))bis(4-(azepan-1-yl)thieno[3,2-d]pyrimidin-1-ium)) bromide Fa-29. Following the general procedure, after workup as described previously, compound Fa-29 was isolated as a light-brown solid. Yield: 40%, mp: 268–270 °C. 1H NMR (500 MHz, CD3OD) δ 1.59–1.61 (m, 4H), 1.69–1.71 (m, 8H), 1.96–1.98 (m, 4H), 2.04–2.06 (m, 4H), 2.62 (t, J = 6.9 Hz, 4H), 4.20–4.22 (m, 4H), 4.24–4.26 (m, 4H), 5.65 (s, 4H), 7.21 (d, J = 7.9 Hz, 4H), 7.30 (d, J = 7.9 Hz, 4H), 7.61 (d, J = 5.8 Hz, 2H), 8.45 (d, J = 5.7 Hz, 2H), 8.87 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 26.09 (2C), 26.20 (2C), 26.31 (2C), 28.11 (2C), 30.65 (2C), 34.83 (2C), 50.30 (2C), 51.32 (2C), 55.45 (2C), 115.25 (2C), 117.31 (2C), 127.22 (4C), 128.99 (4C), 130.97 (2C), 138.63 (2C), 143.55 (2C), 147.08 (2C), 149.32 (2C), 157.10 (2C). HRMS-m/z (M)+ calculated for C42H50N6S2: 702.3538, found: 702.3601.
(1,1’((butane1,4diylbis(4,1phenylene))bis(methylene))bis(4-(azepan-1-yl)thieno[2,3-d]pyrimidin-1-ium)) bromide Ff-35. Following the general procedure, after workup as described previously, compound Ff-35 was isolated as a brown solid. Yield: 34%, mp: 117–119 °C. 1H NMR (500 MHz, CD3OD) δ 1.64 (s, 4H), 1.71–1.73 (m, 8H), 1.91–1.93 (m, 4H), 2.06–2.08 (m, 4H), 2.66 (s, 4H), 4.18–4.20 (m, 4H), 4.22–4.24 (m, 4H), 5.64 (s, 4H), 7.26 (d, J = 8.2 Hz, 4H), 7.40 (d, J = 8.1 Hz, 4H), 7.77 (d, J = 6.0 Hz, 2H), 7.85 (d, J = 6.0 Hz, 2H), 8.94 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 27.39 (4C), 28.29 (4C), 31.49 (2C), 35.76 (2C), 51.22 (2C), 52.47 (2C), 58.18 (2C), 117.27 (2C), 124.25 (2C), 124.53 (2C), 129.05 (4C), 129.89 (4C), 130.12 (2C), 144.98 (2C), 148.84 (2C), 155.47 (2C), 157.52 (2C). HRMS-m/z (M)+ calculated for C42H50N6S2: 702.3538, found: 702.3527.
4,4’-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(7-(pyrrolidin-1-yl)thieno[3,2-b]pyridin-4-ium) bromide Ff-2. Following the general procedure, after workup as described previously, compound Ff-2 was isolated as a brown solid. Yield: 21%, mp: 280–282 °C. 1H NMR (400 MHz, CD3OD) δ 2.19–2.21 (m, 8H), 3.74–3.76 (m, 4H), 4.30–4.32 (m, 8H), 5.63 (s, 4H), 6.74 (d, J = 7.4 Hz, 2H), 6.99 (d, J = 8.8 Hz, 4H), 7.29 (d, J = 8.8 Hz, 4H), 7.69 (d, J = 5.8 Hz, 2H), 8.32 (d, J = 5.7 Hz 2H), 8.33 (d, J = 7.5 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 24.12 (2C), 25.79 (2C), 50.65 (4C), 57.77 (2C), 66.50 (2C), 101.85 (2C), 114.86 (4C), 117.26 (2C), 120.79 (2C), 126.59 (2C), 128.60 (4C), 135.86 (2C), 141.38 (2C), 145.84 (2C), 152.02 (2C), 159.15 (2C). HRMS-m/z (M)+ calculated for C38H40N4O2S2: 648.2582, found: 648.2584.
1,1’-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Ff-8. Following the general procedure, after workup as described previously, compound Ff-8 was isolated as a white solid. Yield: 32%, mp: 217–219 °C. 1H NMR (500 MHz, CD3OD) δ 2.13–2.15 (m, 4H). 2.28–2.30 (m, 4H), 4.01–4.03 (m, 4H), 4.22–4.24 (m, 4H), 4.33 (s, 4H), 5.62 (s, 4H), 7.02 (d, J = 8.7 Hz, 4H), 7.38 (d, J = 8.6 Hz, 4H), 7.66 (d, J = 5.7 Hz, 2H), 8.47 (d, J = 5.7 Hz, 2H), 8.86 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 24.53 (2C), 26.70 (2C), 50.17 (2C), 51.13 (2C), 56.05 (2C), 67.44 (2C), 115.92 (4C), 117.70 (2C), 118.08 (2C), 126.76 (2C), 129.81 (4C), 139.56 (2C), 147.07 (2C), 150.55 (2C), 156.18 (2C), 160.20 (2C). HRMS-m/z (M)+ calculated for C36H38N6O2S2: 650.2487, found: 650.2497.
1,1’-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Ff-4. Following the general procedure, after workup as described previously, compound Ff-4 was isolated as a brown solid. Yield: 42%, mp: 193–195 °C. 1H NMR (500 MHz, CD3OD) δ 2.12–2.14 (m, 4H). 2.26–2.28 (m, 4H), 4.03–4.05 (m, 4H), 4.15–4.17 (m, 4H), 4.36 (s, 4H), 5.60 (s, 4H), 7.05 (d, J = 8.8 Hz, 4H), 7.45 (d, J = 8.8 Hz, 4H), 7.75 (d, J = 5.9 Hz, 2H), 7.93 (d, J = 5.9 Hz, 2H), 8.92 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 24.50 (2C), 26.95 (2C), 50.12 (2C), 50.86 (2C), 58.12 (2C), 67.63 (2C), 116.02 (4C), 118.43 (2C), 124.10 (2C), 124.40 (2C), 125.12 (2C), 130.90 (4C), 149.38 (2C), 154.48 (2C), 155.49 (2C), 160.76 (2C). HRMS-m/z (M)+ calculated for C36H38N6O2S2: 650.2487, found: 650.2514.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(piperidin-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Ff-28. Following the general procedure, after workup as described previously, compound Ff-28 was isolated as a grey solid. Yield: 25%, mp: 198–200 °C. 1H NMR (400 MHz, DMSO) δ 1.76–178-(m, 12H), 4.15–4.17 (m, 8H), 4.28 (s, 4H), 5.66 (s, 4H), 6.99 (d, J = 8.6 Hz, 4H), 7.44 (d, J = 8.5 Hz, 4H), 7.80 (d, J = 5.7 Hz, 2H), 8.65 (d, J = 5.7 Hz, 2H), 9.17 (s, 2H). 13C NMR (101 MHz, DMSO) δ 23.22 (2C), 25.94 (4C), 49.60 (4C), 54.51 (2C), 66.31 (2C), 114.58 (4C), 114.83 (2C), 117.95 (2C), 126.44 (2C), 129.35 (4C), 139.02 (2C), 147.11 (2C), 149.84 (2C), 155.36 (2C), 158.40 (2C). HRMS-m/z (M)+ calculated for C38H42N6O2S2: 678.2800, found: 678.2774.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(piperidin-1-yl)thieno[2,3-d]pyrimidin-1-ium) bromide Ff-5. Following the general procedure, after workup as described previously, compound Ff-5 was isolated as a brown solid. Yield: 41%, mp: >280 °C. 1H NMR (500 MHz, CD3OD) δ 1.89 (m, 12H), 4.25 (m, 8H), 4.36 (s, 4H), 5.60 (s, 4H), 7.06 (d, J = 8.8 Hz, 4H), 7.46 (d, J = 8.8 Hz, 4H), 7.78 (d, J = 6.0 Hz, 2H), 7.83 (d, J = 6.0 Hz, 2H), 8.89 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 24.29 (2C), 26.56 (4C), 51.06 (4C), 57.95 (2C), 67.46 (2C), 115.86 (4C), 117.16 (2C), 123.82 (2C), 124.54 (2C), 124.92 (2C), 130.79 (4C), 148.86 (2C), 155.65 (2C), 156.90 (2C), 160.61 (2C). HRMS-m/z (M)+ calculated for C38H42N6O2S2: 678.2811, found: 678.2839.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(azepan-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Ff-34. Following the general procedure, after workup as described previously, compound Ff-34 was isolated as a brown solid. Yield: 45%, mp: 201–202 °C. 1H NMR (500 MHz, CD3OD) δ 1.71–1.73 (m, 4H), 1.92–1.94 (m, 4H), 2.08–2.10 (m, 8H), 4.22–4.24 (m, 4H), 4.25–4.27 (m, 4H), 4.33 (s, 4H), 5.64 (s, 4H), 7.03 (d, J = 8.8 Hz, 4H), 7.39 (d, J = 8.7 Hz, 4H), 7.67 (d, J = 5.8 Hz, 2H), 8.48 (d, J = 5.8 Hz, 2H), 8.87 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 27.22 (4C), 29.02 (4C), 51.21 (2C), 52.22 (2C), 56.15 (2C), 67.45 (2C), 115.90 (4C), 116.16 (2C), 118.21 (2C), 126.68 (2C), 129.82 (4C), 139.51 (2C), 147.97 (2C), 150.11 (2C), 158.01 (2C), 160.26 (2C). HRMS-m/z (M)+ calculated for C40H46N6O2S2: 706.3124, found: 706.3068.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(azepan-1-yl)thieno[3,2-d]pyrimidin-1-ium) bromide Ff-36. Following the general procedure, after workup as described previously, compound Ff-36 was isolated as a brown solid. Yield: 27%, mp: 205–207 °C. 1H NMR (400 MHz, CD3OD) δ 1.72–1.73 (m, 8H), 2.00–2.03 (m, 4H), 2.04–2.06 (m, 4H), 4.19–4.22 (m, 4H), 4.22–4.24 (m, 4H), 4.37 (s, 4H), 5.61 (s, 4H), 7.07 (d, J = 8.7 Hz, 4H), 7.47 (d, J = 8.7 Hz, 4H), 7.78 (d, J = 6.0 Hz, 2H), 7.85 (d, J = 6.0 Hz, 2H), 8.91 (s, 2H). 13C NMR (101 MHz, DMSO) δ 26.31 (4C), 27.00 (4C), 49.82 (2C), 51.23 (2C), 56.51 (2C), 66.35 (2C), 114.84 (4C), 116.08 (2C), 123.83 (2C), 124.35 (2C), 124.45 (2C), 130.11 (4C), 148.46 (2C), 153.91 (2C), 156.10 (2C), 158.73 (2C). HRMS-m/z (M)+ calculated for C40H46N6O2S2: 706.3124, found: 706.3183.


 ,
, 




{kind=link}
{kind=link}
{kind=link}


















