
Drug Screening, Oral Bioavailability and Regulatory Aspects: A Need for Human Organoids



Abstract
:
1. Introduction
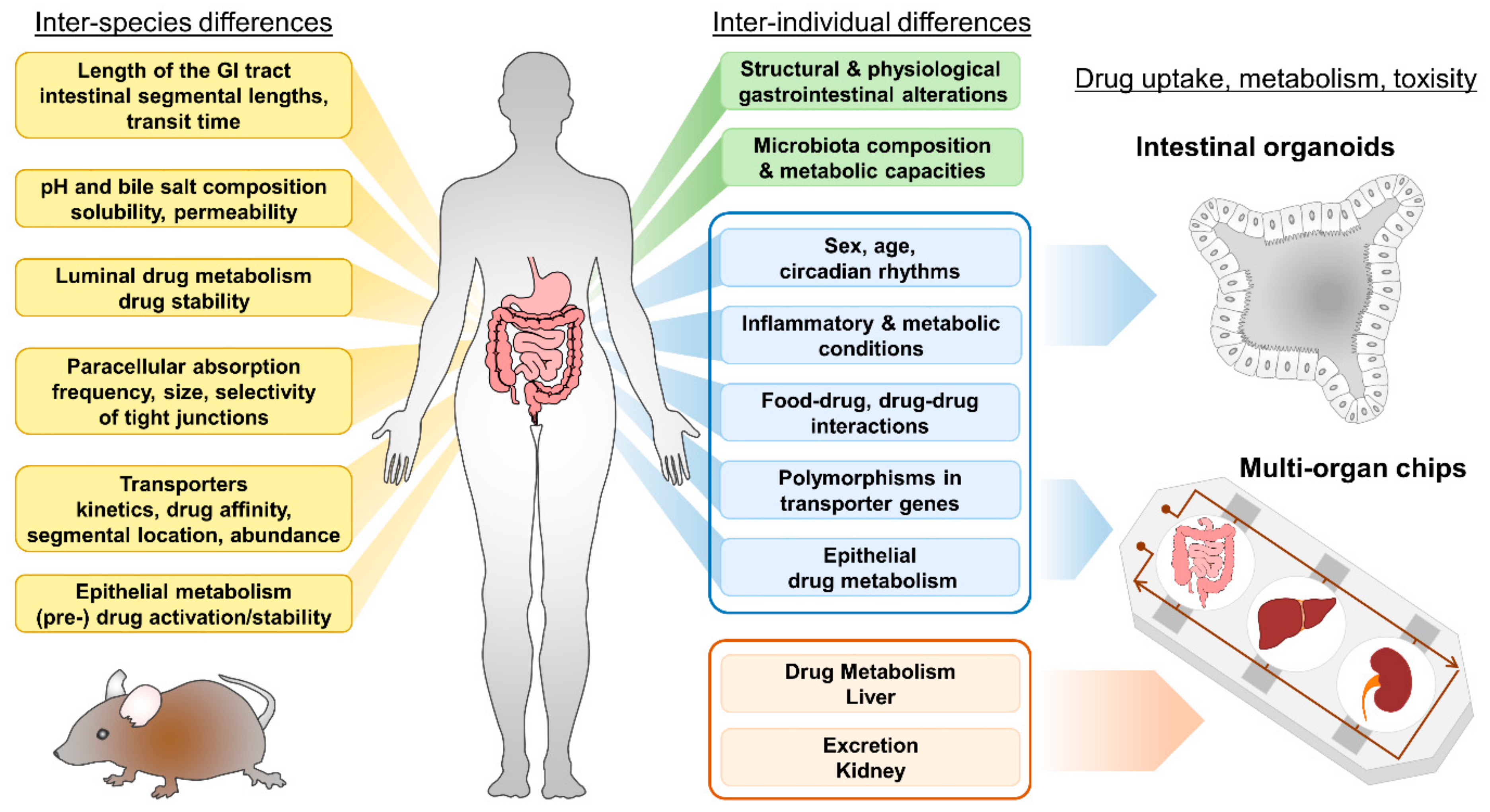
2. Bioavailability and the Intestinal Epithelium
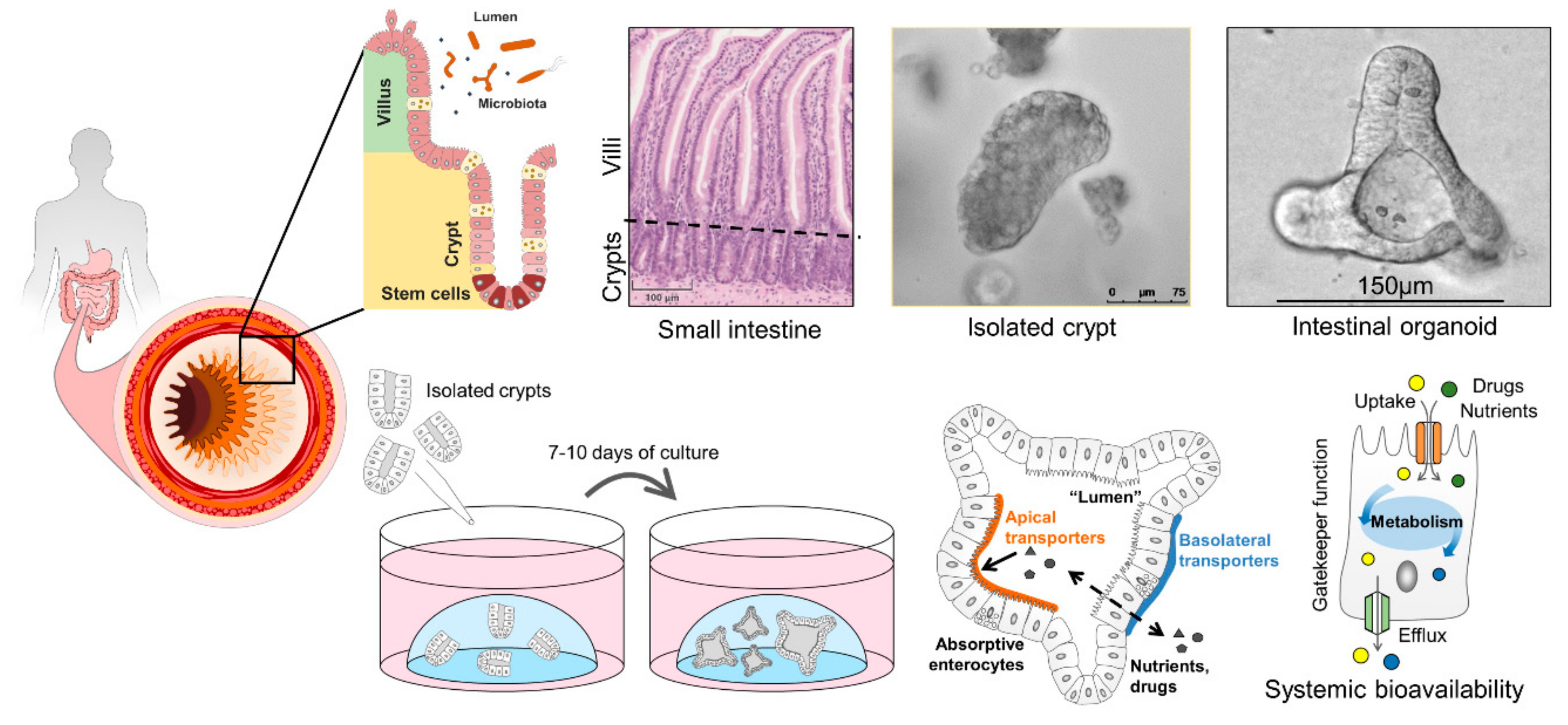
2.1. Anatomy and Function of the Intestinal Epithelium
2.2. Presystemic Luminal Drug Metabolism
3. Intestinal Nutrient and Drug Transport
3.1. Intestinal Nutrient Absorption
3.2. Intestinal Transporters and Drug Uptake
3.3. Intestinal Ion Pumps
4. Intestinal Epithelial Cell Metabolism
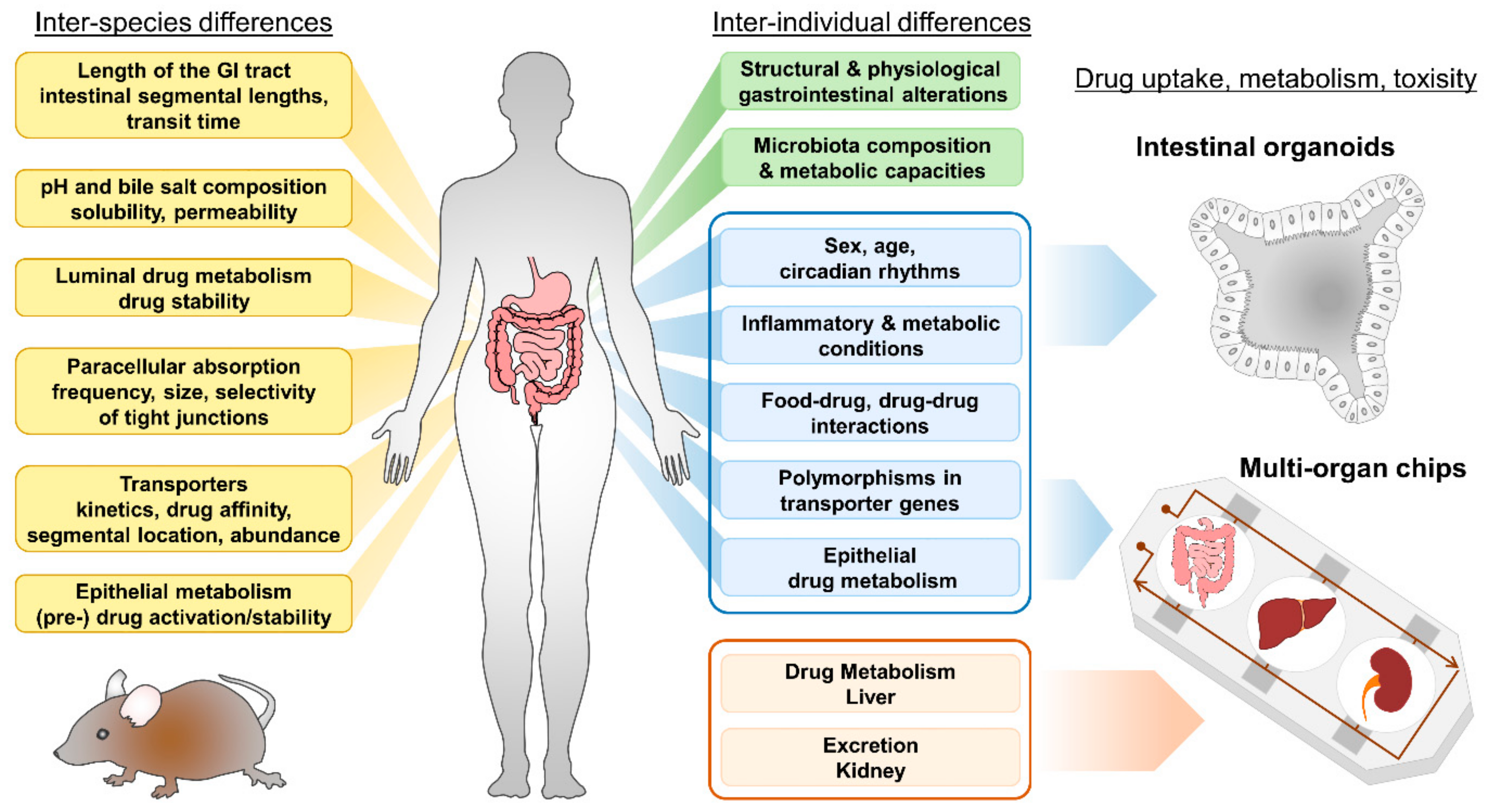
5. Currently Used Models
5.1. Cell Lines
5.2. Other Models
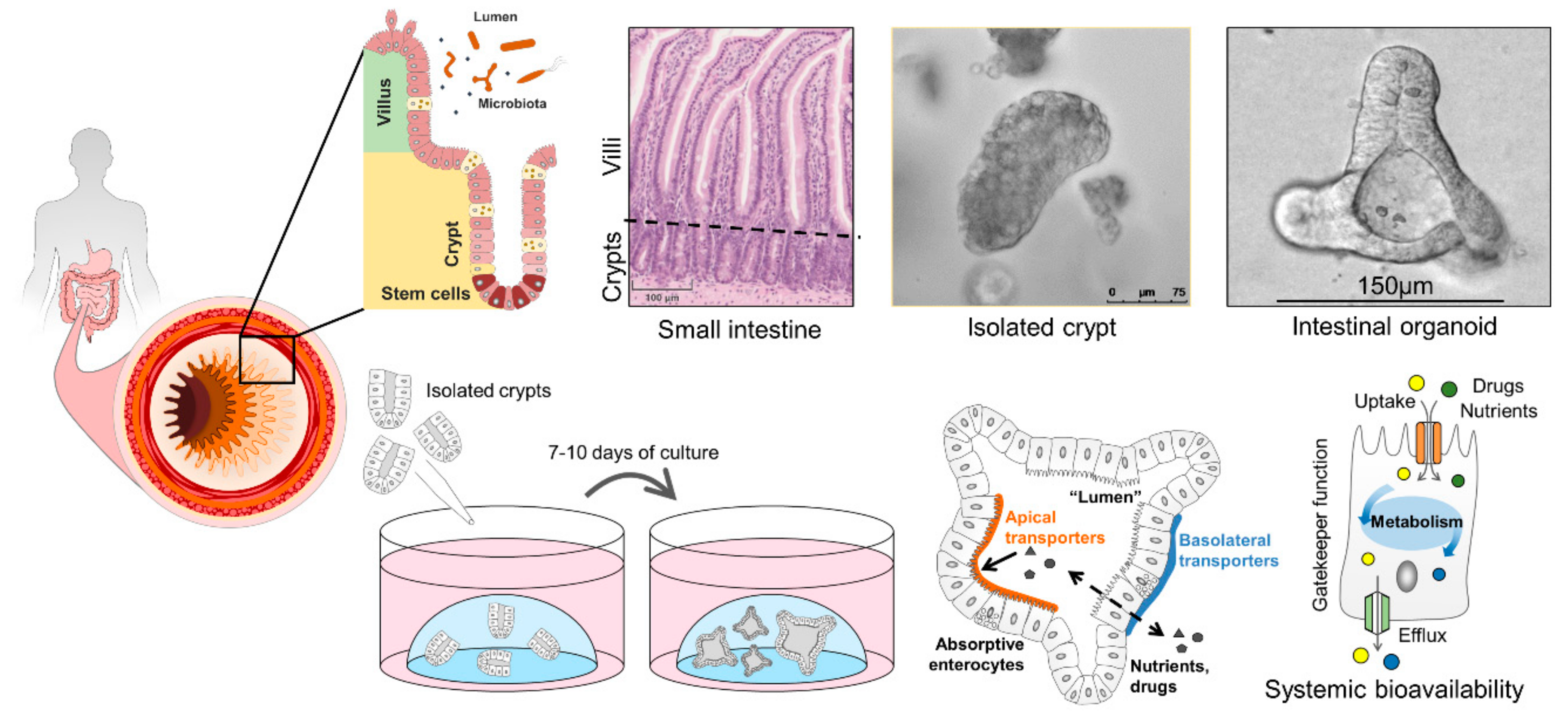
6. Human Intestinal Organoids and How They Can Improve Drug Screening
6.1. Properties of Intestinal Organoids
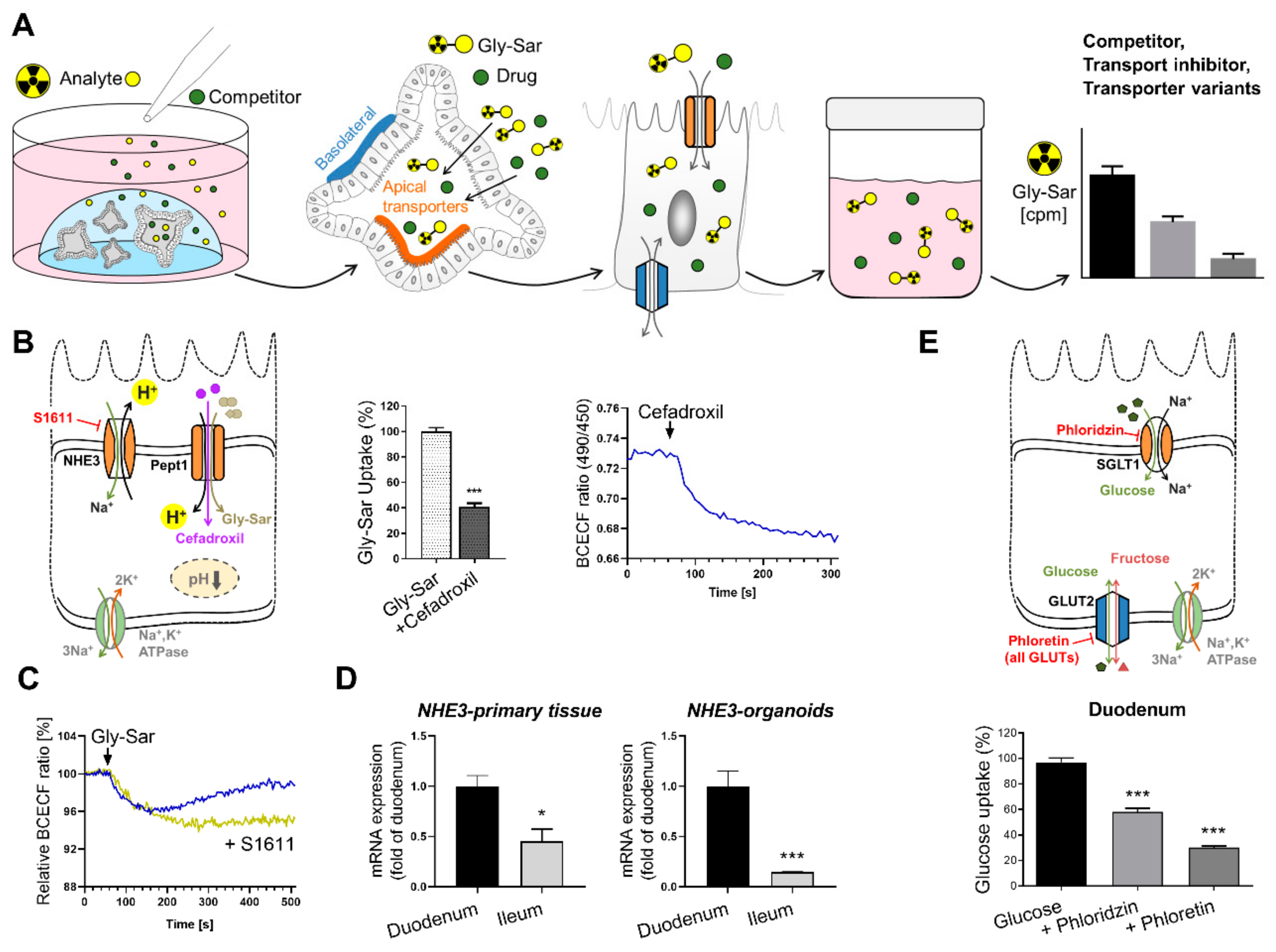
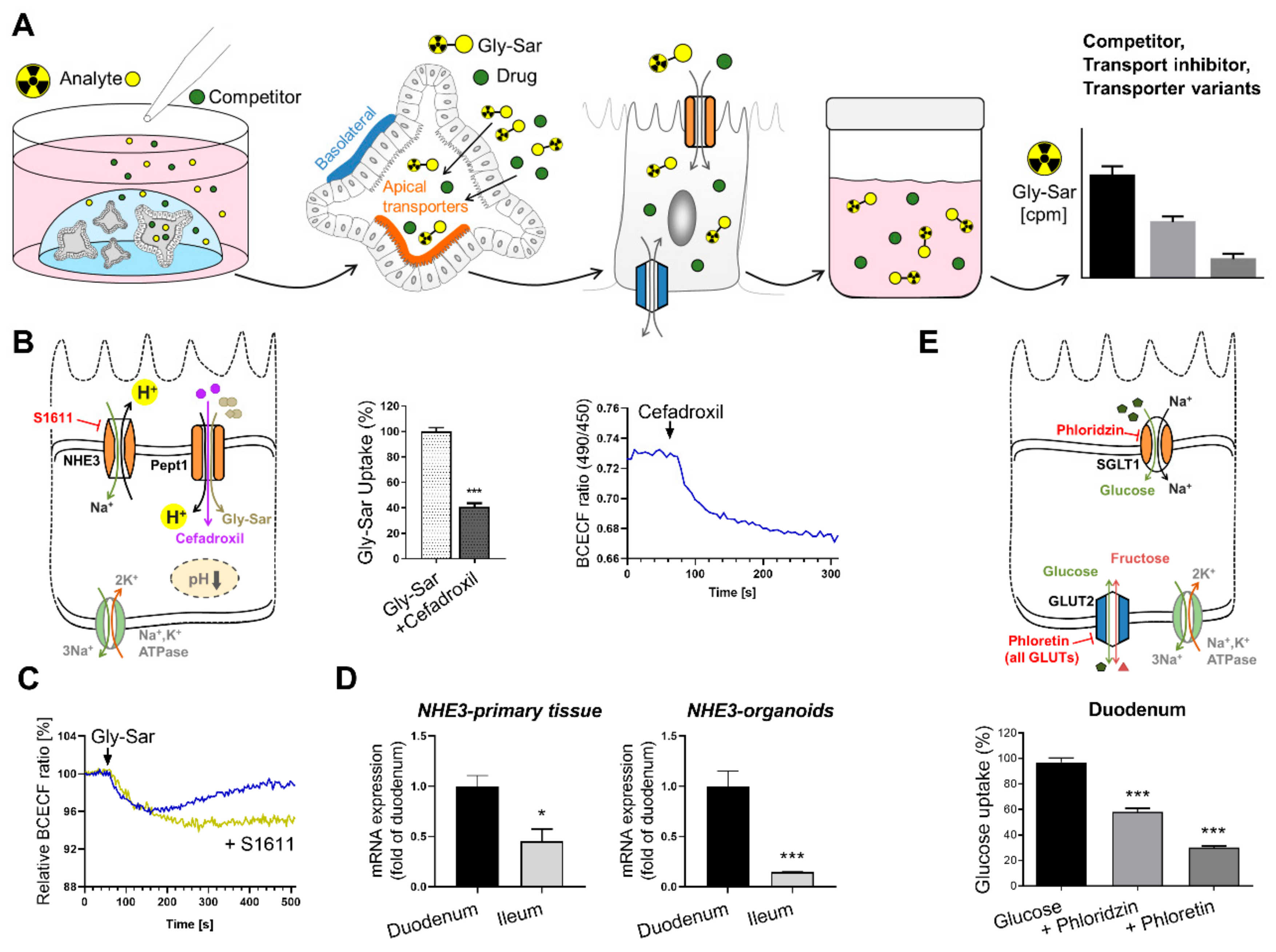
6.2. Intestinal Organoids to Assess Nutrient and Drug Uptake
6.3. Current and Future Strategies for Organoids in Pharmacological Research
7. Regulatory Aspects
8. Future Directions
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADME | (Drug) Absorption, Distribution, Metabolism, and Excretion |
| ECVAM | European Centre for the Validation of Alternative Methods |
| FDA | U.S. Food and Drug Administration |
| HTS | High-Throughput Screening |
| IECs | Intestinal Epithelial Cells |
| JRC | Joint Research Centre |
| MOC | Multi-Organ Chip |
| MPS | MicroPhysiological System |
| NAMs | New Approach Methods |
| NAT | Non-Animal Methods |
| OOC | Organ-On-a-Chip |
| PAMPA | Parallel Artificial Membrane Permeability Assay |
| PBKD | Physiologically Based Kinetic and Dynamic |
| QSAR | Quantitative Structure-Activity Relationship |
| VCBA | Virtual Cell Based Assay |
References
- Currie, G.M. Pharmacology, part 2: Introduction to pharmacokinetics. J. Nucl. Med. Technol. 2018, 46, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.G.; Geier, A.; Oude Elferink, R.P. Abc of oral bioavailability: Transporters as gatekeepers in the gut. Gut 2003, 52, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Kuntz, S.; Diestel, S.; Daniel, H. Pept1-mediated cefixime uptake into human intestinal epithelial cells is increased by Ca2+ channel blockers. Antimicrob. Agents Chemother. 2002, 46, 1375–1380. [Google Scholar] [CrossRef] [Green Version]
- Brocks, D.R.; Davies, N.M. Lymphatic drug absorption via the enterocytes: Pharmacokinetic simulation, modeling, and considerations for optimal drug development. J. Pharm. Pharm. Sci. 2018, 21, 254s–270s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhans, W.; Leitner, C.; Arnold, M. Dietary fat sensing via fatty acid oxidation in enterocytes: Possible role in the control of eating. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R554–R565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, D.; Clara, R.; Fedele, S.; Michel, L.; Burkard, J.; Kaufman, S.; Diaz, A.A.; Weissfeld, N.; De Bock, K.; Prip-Buus, C.; et al. Enhancing enterocyte fatty acid oxidation in mice affects glycemic control depending on dietary fat. Sci. Rep. 2018, 8, 10818. [Google Scholar] [CrossRef]
- Ebert, K.; Witt, H. Fructose malabsorption. Mol. Cell Pediatr. 2016, 3, 10. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Marinshaw, J.M.; Shaikh, N.; Miyoshi, H.; Moon, C.; Tarr, P.I.; Ciorba, M.A.; Stappenbeck, T.S. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 2015, 64, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Zietek, T.; Giesbertz, P.; Ewers, M.; Reichart, F.; Weinmüller, M.; Urbauer, E.; Haller, D.; Demir, I.E.; Ceyhan, G.O.; Kessler, H.; et al. Organoids to study intestinal nutrient transport, drug uptake and metabolism—Update to the human model and expansion of applications. Front. Bioeng. Biotechnol. 2020, 8, 1065. [Google Scholar] [CrossRef]
- Zietek, T.; Rath, E.; Haller, D.; Daniel, H. Intestinal organoids for assessing nutrient transport, sensing and incretin secretion. Sci. Rep. 2015, 5, 16831. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Zietek, T.; Rath, E. Chapter 3—Intestinal organoids: Mini-guts grown in the laboratory. In Organoids and Mini-Organs; Davies, J.A., Lawrence, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 43–71. [Google Scholar]
- Helander, H.F.; Fandriks, L. Surface area of the digestive tract—Revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef]
- Zietek, T.; Waldschmitt, N.; Rath, E. Role of incretin hormones in bowel diseases. Endocr. Dev. 2017, 32, 49–73. [Google Scholar] [PubMed]
- Daniel, H.; Zietek, T. Taste and move: Glucose and peptide transporters in the gastrointestinal tract. Exp. Physiol. 2015, 100, 1441–1450. [Google Scholar] [CrossRef]
- Middendorp, S.; Schneeberger, K.; Wiegerinck, C.L.; Mokry, M.; Akkerman, R.D.; van Wijngaarden, S.; Clevers, H.; Nieuwenhuis, E.E. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells 2014, 32, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, C.T.; Asano, S.; Sakai, H. Chapter 38—The cell biology of gastric acid secretion. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 831–867. [Google Scholar]
- Ortega, B.; Welling, P.A. Chapter 45—Molecular mechanisms of polarized protein trafficking in epithelial cells. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 1027–1050. [Google Scholar]
- Daugherty, A.L.; Mrsny, R.J. Transcellular uptake mechanisms of the intestinal epithelial barrier part one. Pharm. Sci. Technol. Today 1999, 4, 144–151. [Google Scholar] [CrossRef]
- Gaucher, G.; Satturwar, P.; Jones, M.C.; Furtos, A.; Leroux, J.C. Polymeric micelles for oral drug delivery. Eur. J. Pharm. Biopharm. 2010, 76, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Inoue, R.; Matsumoto, M.; Yajima, T.; Ushida, K.; Iwanaga, T. Comparative expression of hexose transporters (sglt1, glut1, glut2 and glut5) throughout the mouse gastrointestinal tract. Histochem. Cell Biol. 2011, 135, 183–194. [Google Scholar] [CrossRef]
- Wuensch, T.; Schulz, S.; Ullrich, S.; Lill, N.; Stelzl, T.; Rubio-Aliaga, I.; Loh, G.; Chamaillard, M.; Haller, D.; Daniel, H. The peptide transporter pept1 is expressed in distal colon in rodents and humans and contributes to water absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G66–G73. [Google Scholar] [CrossRef] [Green Version]
- Borde, A.S.; Karlsson, E.M.; Andersson, K.; Bjorhall, K.; Lennernas, H.; Abrahamsson, B. Assessment of enzymatic prodrug stability in human, dog and simulated intestinal fluids. Eur. J. Pharm. Biopharm. 2012, 80, 630–637. [Google Scholar] [CrossRef]
- Martinez, M.N.; El-Kattan, A.; Awji, E.; Papich, M. Reconciling human-canine differences in oral bioavailability: Looking beyond the biopharmaceutics classification system. AAPS J. 2019, 21, 99. [Google Scholar] [CrossRef]
- Ayo, J.A.; Agu, H.; Madaki, I. Food and drug interactions: Its side effects. Nutr. Food Sci. 2005, 35, 243–252. [Google Scholar] [CrossRef]
- Schmidt, L.E.; Dalhoff, K. Food-drug interactions. Drugs 2002, 62, 1481–1502. [Google Scholar] [CrossRef]
- Lown, K.S.; Bailey, D.G.; Fontana, R.J.; Janardan, S.K.; Adair, C.H.; Fortlage, L.A.; Brown, M.B.; Guo, W.; Watkins, P.B. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal cyp3a protein expression. J. Clin. Investig. 1997, 99, 2545–2553. [Google Scholar] [CrossRef]
- Pereira de Sousa, I.; Bernkop-Schnurch, A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release 2014, 192, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut reactions: Breaking down xenobiotic-microbiome interactions. Pharm. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of dietary and microbial vitamin b family in the regulation of host immunity. Front. Nutr. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, S.; Kim, S.; Cho, S.W. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp. Mol. Med. 2020, 52, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubert, J.; Schweiger, P.J.; Mattivi, F.; Tuohy, K.; Jensen, K.B.; Lunardi, A. Intestinal organoids: A tool for modelling diet-microbiome-host interactions. Trends Endocrinol. Metab. 2020, 31, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 2018, 24, 833–846.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, L.; Casciano, F.; Gianotti, A. Intestinal fermentation in vitro models to study food-induced gut microbiota shift: An updated review. FEMS Microbiol. Lett. 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. Gut reaction: Impact of systemic diseases on gastrointestinal physiology and drug absorption. Drug Discov. Today 2019, 24, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Klatt, S.; Fromm, M.F.; Konig, J. Transporter-mediated drug-drug interactions with oral antidiabetic drugs. Pharmaceutics 2011, 3, 680–705. [Google Scholar] [CrossRef] [Green Version]
- Soldin, O.P.; Mattison, D.R. Sex differences in pharmacokinetics and pharmacodynamics. Clin. Pharm. 2009, 48, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Rajman, I.; Knapp, L.; Hanna, I. Genetic diversity in drug transporters: Impact in african populations. Clin. Transl. Sci. 2020, 13, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Price, G.; Patel, D.A. Drug Bioavailability; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Almeqdadi, M.; Mana, M.D.; Roper, J.; Yilmaz, O.H. Gut organoids: Mini-tissues in culture to study intestinal physiology and disease. Am. J. Physiol. Cell Physiol. 2019, 317, C405–C419. [Google Scholar] [CrossRef]
- Rader, A.F.B.; Reichart, F.; Weinmuller, M.; Kessler, H. Improving oral bioavailability of cyclic peptides by n-methylation. Bioorg. Med. Chem. 2018, 26, 2766–2773. [Google Scholar] [CrossRef]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef]
- Schumacher-Klinger, A.; Fanous, J.; Merzbach, S.; Weinmuller, M.; Reichart, F.; Rader, A.F.B.; Gitlin-Domagalska, A.; Gilon, C.; Kessler, H.; Hoffman, A. Enhancing oral bioavailability of cyclic rgd hexa-peptides by the lipophilic prodrug charge masking approach: Redirection of peptide intestinal permeability from a paracellular to transcellular pathway. Mol. Pharm. 2018, 15, 3468–3477. [Google Scholar] [CrossRef]
- Pappenheimer, J.R.; Reiss, K.Z. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J. Membr. Biol. 1987, 100, 123–136. [Google Scholar] [CrossRef]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.M. Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 2014, 25, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, J.R.; Dere, S.G.; Awasthi, R.; Dua, K. Efavirenz loaded mixed polymeric micelles: Formulation, optimization, and in vitro characterization. Assay Drug Dev. Technol. 2021, 19, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Kotake-Nara, E.; Komba, S.; Hase, M. Uptake of vitamins d2, d3, d4, d5, d6, and d7 solubilized in mixed micelles by human intestinal cells, caco-2, an enhancing effect of lysophosphatidylcholine on the cellular uptake, and estimation of vitamins d’ biological activities. Nutrients 2021, 13, 1126. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Carrier proteins and active membrane transport. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (mfs). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Perland, E.; Fredriksson, R. Classification systems of secondary active transporters. Trends Pharm. Sci. 2017, 38, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Mueckler, M.; Thorens, B. The slc2 (glut) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st century. Am. J. Physiol Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [Green Version]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Mutations in glut2, the gene for the liver-type glucose transporter, in patients with fanconi-bickel syndrome. Nat. Genet. 1997, 17, 324–326. [Google Scholar] [CrossRef]
- Martin, M.G.; Turk, E.; Lostao, M.P.; Kerner, C.; Wright, E.M. Defects in na+/glucose cotransporter (sglt1) trafficking and function cause glucose-galactose malabsorption. Nat. Genet. 1996, 12, 216–220. [Google Scholar] [CrossRef]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar]
- Brandsch, M. Drug transport via the intestinal peptide transporter pept1. Curr. Opin. Pharm. 2013, 13, 881–887. [Google Scholar] [CrossRef]
- Ganapathy, M.E.; Brandsch, M.; Prasad, P.D.; Ganapathy, V.; Leibach, F.H. Differential recognition of beta -lactam antibiotics by intestinal and renal peptide transporters, pept 1 and pept 2. J. Biol. Chem. 1995, 270, 25672–25677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottra, G.; Spanier, B.; Verri, T.; Daniel, H. Peptide transporter isoforms are discriminated by the fluorophore-conjugated dipeptides beta-ala- and d-ala-lys-n-7-amino-4-methylcoumarin-3-acetic acid. Physiol Rep. 2013, 1, e00165. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Shen, H.; Hopfer, U.; Smith, D.E. Mechanism of intestinal absorption and renal reabsorption of an orally active ace inhibitor: Uptake and transport of fosinopril in cell cultures. Drug Metab. Dispos. 2001, 29, 1307–1315. [Google Scholar] [PubMed]
- Minhas, G.S.; Newstead, S. Structural basis for prodrug recognition by the slc15 family of proton-coupled peptide transporters. Proc. Natl. Acad. Sci. USA 2019, 116, 804–809. [Google Scholar] [CrossRef] [Green Version]
- Minhas, G.S.; Newstead, S. Recent advances in understanding prodrug transport through the slc15 family of proton-coupled transporters. Biochem. Soc. Trans. 2020, 48, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Marshall, G.R.; Ballante, F. Limiting assumptions in the design of peptidomimetics. Drug Dev. Res. 2017, 78, 245–267. [Google Scholar] [CrossRef]
- Giannis, A.; Kolter, T. Peptidomimetics for receptor ligands-discovery, development, and medical perspectives. Angew. Chem. Int. Ed. 1993, 32, 1244–1267. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, R.M.; Peyton, M.; Larsen, J.E.; Girard, L.; Xie, Y.; Gazdar, A.F.; Harran, P.; Wang, L.; Brekken, R.A.; Wang, X.; et al. Smac mimetic (jp1201) sensitizes non-small cell lung cancers to multiple chemotherapy agents in an iap-dependent but tnf-alpha-independent manner. Cancer Res. 2011, 71, 7640–7648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled bh3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [Green Version]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmuller, M.; Rader, A.; Steiger, K.; Kessler, H. Exploring the role of rgd-recognizing integrins in cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef]
- Becker, A.; von Richter, O.; Kovar, A.; Scheible, H.; van Lier, J.J.; Johne, A. Metabolism and disposition of the alphav-integrin ss3/ss5 receptor antagonist cilengitide, a cyclic polypeptide, in humans. J. Clin. Pharm. 2015, 55, 815–824. [Google Scholar] [CrossRef]
- Ovadia, O.; Greenberg, S.; Chatterjee, J.; Laufer, B.; Opperer, F.; Kessler, H.; Gilon, C.; Hoffman, A. The effect of multiple n-methylation on intestinal permeability of cyclic hexapeptides. Mol. Pharm. 2011, 8, 479–487. [Google Scholar] [CrossRef]
- Rader, A.F.B.; Weinmuller, M.; Reichart, F.; Schumacher-Klinger, A.; Merzbach, S.; Gilon, C.; Hoffman, A.; Kessler, H. Orally active peptides: Is there a magic bullet? Angew. Chem. Int. Ed. Engl. 2018, 57, 14414–14438. [Google Scholar] [CrossRef]
- Chen, M.; Singh, A.; Xiao, F.; Dringenberg, U.; Wang, J.; Engelhardt, R.; Yeruva, S.; Rubio-Aliaga, I.; Nassl, A.M.; Kottra, G.; et al. Gene ablation for pept1 in mice abolishes the effects of dipeptides on small intestinal fluid absorption, short-circuit current, and intracellular pH. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G265–G274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulke-Abel, J.; In, J.; Yin, J.; Zachos, N.C.; Kovbasnjuk, O.; Estes, M.K.; de Jonge, H.; Donowitz, M. Human enteroids as a model of upper small intestinal ion transport physiology and pathophysiology. Gastroenterology 2016, 150, 638–649.e8. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.K. The slc22 transporter family: A paradigm for the impact of drug transporters on metabolic pathways, signaling, and disease. Annu. Rev. Pharm. Toxicol. 2018, 58, 663–687. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. Slc transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Transporter, C.; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Zietek, T.; Rath, E. Inflammation meets metabolic disease: Gut feeling mediated by glp-1. Front. Immunol. 2016, 7, 154. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile acids as hormones: The fxr-fgf15/19 pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Le Drean, G.; Segain, J.P. Connecting metabolism to intestinal barrier function: The role of leptin. Tissue Barriers 2014, 2, e970940. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, O.H.; Katajisto, P.; Lamming, D.W.; Gultekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. Mtorc1 in the paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Corrigendum: Short chain fatty acids (scfas)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 1486. [Google Scholar] [CrossRef] [Green Version]
- Varga, V.; Muranyi, Z.; Kurucz, A.; Marcolongo, P.; Benedetti, A.; Banhegyi, G.; Margittai, E. Species-specific glucose-6-phosphatase activity in the small intestine-studies in three different mammalian models. Int. J. Mol. Sci. 2019, 20, 5039. [Google Scholar] [CrossRef] [Green Version]
- Potts, A.; Uchida, A.; Deja, S.; Berglund, E.D.; Kucejova, B.; Duarte, J.A.; Fu, X.; Browning, J.D.; Magnuson, M.A.; Burgess, S.C. Cytosolic phosphoenolpyruvate carboxykinase as a cataplerotic pathway in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G249–G258. [Google Scholar] [CrossRef]
- Sinha, N.; Suarez-Diez, M.; van Schothorst, E.M.; Keijer, J.; Martins Dos Santos, V.A.P.; Hooiveld, G. Predicting the murine enterocyte metabolic response to diets that differ in lipid and carbohydrate composition. Sci. Rep. 2017, 7, 8784. [Google Scholar] [CrossRef] [Green Version]
- Schober, G.; Arnold, M.; Birtles, S.; Buckett, L.K.; Pacheco-Lopez, G.; Turnbull, A.V.; Langhans, W.; Mansouri, A. Diacylglycerol acyltransferase-1 inhibition enhances intestinal fatty acid oxidation and reduces energy intake in rats. J. Lipid Res. 2013, 54, 1369–1384. [Google Scholar] [CrossRef] [Green Version]
- Wacher, V.J.; Salphati, L.; Benet, L.Z. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv. Drug Deliv. Rev. 2001, 46, 89–102. [Google Scholar] [CrossRef]
- Jones, C.R.; Hatley, O.J.; Ungell, A.L.; Hilgendorf, C.; Peters, S.A.; Rostami-Hodjegan, A. Gut wall metabolism: Application of pre-clinical models for the prediction of human drug absorption and first-pass elimination. AAPS J. 2016, 18, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharm. 2012, 165, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, F.; Habib, A.M.; Tolhurst, G.; Parker, H.E.; Rogers, G.J.; Gribble, F.M. Glucose sensing in l cells: A primary cell study. Cell Metab. 2008, 8, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogh, J.; Fogh, J.M.; Orfeo, T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst. 1977, 59, 221–226. [Google Scholar] [CrossRef]
- Vincent, M.L.; Russell, R.M.; Sasak, V. Folic acid uptake characteristics of a human colon carcinoma cell line, caco-2. A newly-described cellular model for small intestinal epithelium. Hum. Nutr. Clin. Nutr. 1985, 39, 355–360. [Google Scholar]
- Yee, S. In vitro permeability across caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man—Fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef]
- Pinto, M.; Robineleon, S.; Appay, M.D.; Kedinger, M.; Triadou, N.; Dussaulx, E.; Lacroix, B.; Simonassmann, P.; Haffen, K.; Fogh, J.; et al. Enterocyte-like differentiation and polarization of the human-colon carcinoma cell-line caco-2 in culture. Biol. Cell 1983, 47, 323–330. [Google Scholar]
- Calcagno, A.M.; Ludwig, J.A.; Fostel, J.M.; Gottesman, M.M.; Ambudkar, S.V. Comparison of drug transporter levels in normal colon, colon cancer, and caco-2 cells: Impact on drug disposition and discovery. Mol. Pharm. 2006, 3, 87–93. [Google Scholar] [CrossRef]
- Matsson, P.; Bergstrom, C.A.; Nagahara, N.; Tavelin, S.; Norinder, U.; Artursson, P. Exploring the role of different drug transport routes in permeability screening. J. Med. Chem. 2005, 48, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Jezyk, N.; Li, C.; Stewart, B.H.; Wu, X.; Bockbrader, H.N.; Fleisher, D. Transport of pregabalin in rat intestine and caco-2 monolayers. Pharm. Res. 1999, 16, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Harwood, M.D.; Achour, B.; Neuhoff, S.; Russell, M.R.; Carlson, G.; Warhurst, G.; Amin, R.-H. In vitro-in vivo extrapolation scaling factors for intestinal p-glycoprotein and breast cancer resistance protein: Part I: A cross-laboratory comparison of transporter-protein abundances and relative expression factors in human intestine and caco-2 cells. Drug Metab. Dispos. 2016, 44, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Mobbs, C.L.; Gonzalez-Hau, A.L.; Freer, M.; Przyborski, S. Bioengineering novel in vitro co-culture models that represent the human intestinal mucosa with improved caco-2 structure and barrier function. Front. Bioeng. Biotechnol. 2020, 8, 992. [Google Scholar] [CrossRef]
- Beduneau, A.; Tempesta, C.; Fimbel, S.; Pellequer, Y.; Jannin, V.; Demarne, F.; Lamprecht, A. A tunable caco-2/ht29-mtx co-culture model mimicking variable permeabilities of the human intestine obtained by an original seeding procedure. Eur. J. Pharm. Biopharm. 2014, 87, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Bujard, A.; Petit, C.; Carrupt, P.A.; Rudaz, S.; Schappler, J. Hdm-pampa to predict gastrointestinal absorption, binding percentage, equilibrium and kinetics constants with human serum albumin and using 2 end-point measurements. Eur. J. Pharm. Sci. 2017, 97, 143–150. [Google Scholar] [CrossRef]
- Roder, P.V.; Geillinger, K.E.; Zietek, T.S.; Thorens, B.; Koepsell, H.; Daniel, H. The role of sglt1 and glut2 in intestinal glucose transport and sensing. PLoS ONE 2014, 9, e89977. [Google Scholar] [CrossRef]
- Bermejo, M.; Avdeef, A.; Ruiz, A.; Nalda, R.; Ruell, J.A.; Tsinman, O.; Gonzalez, I.; Fernandez, C.; Sanchez, G.; Garrigues, T.M.; et al. Pampa—A drug absorption in vitro model 7: Comparing rat in situ, caco-2, and pampa permeability of fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [Google Scholar] [CrossRef] [PubMed]
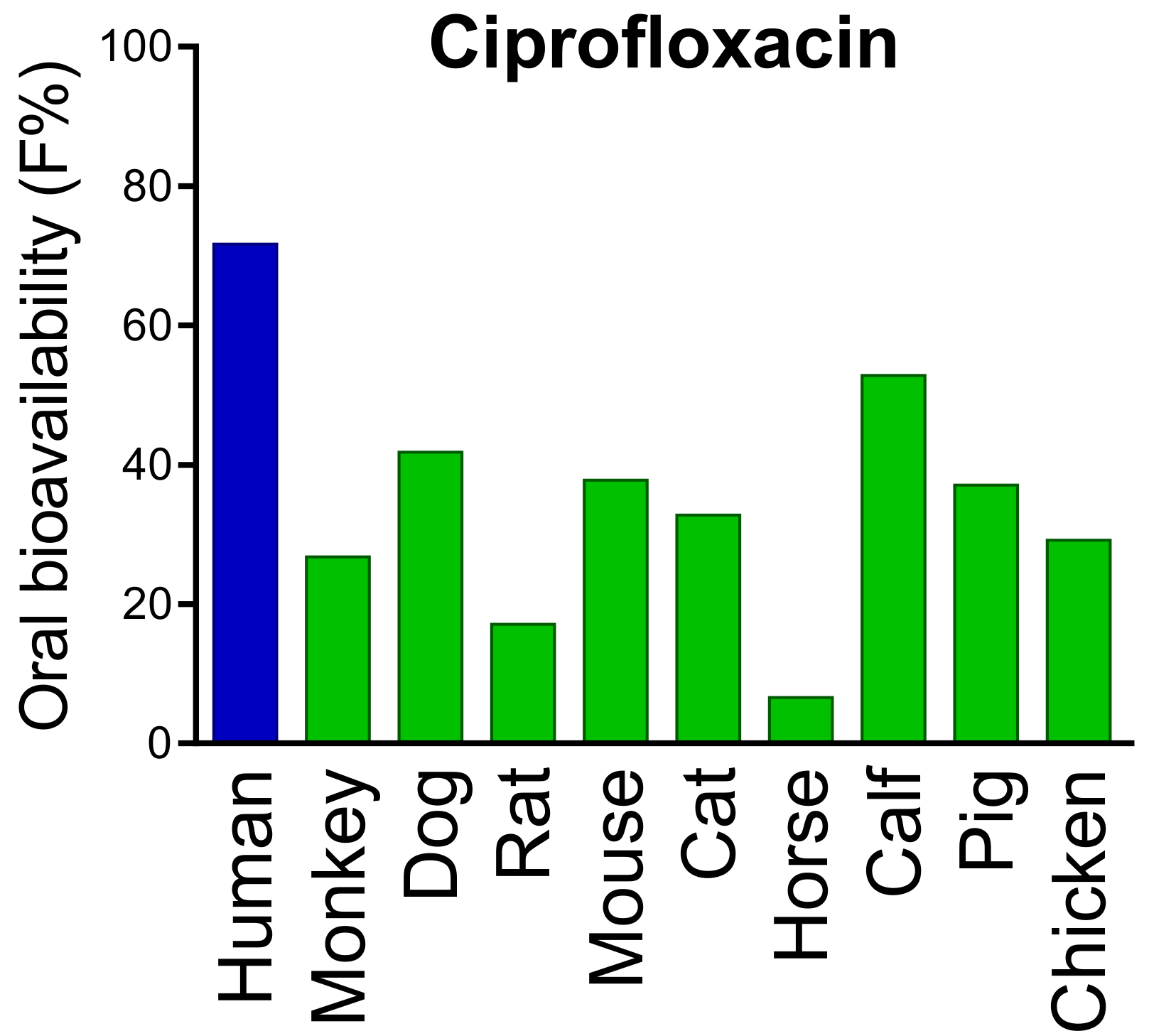
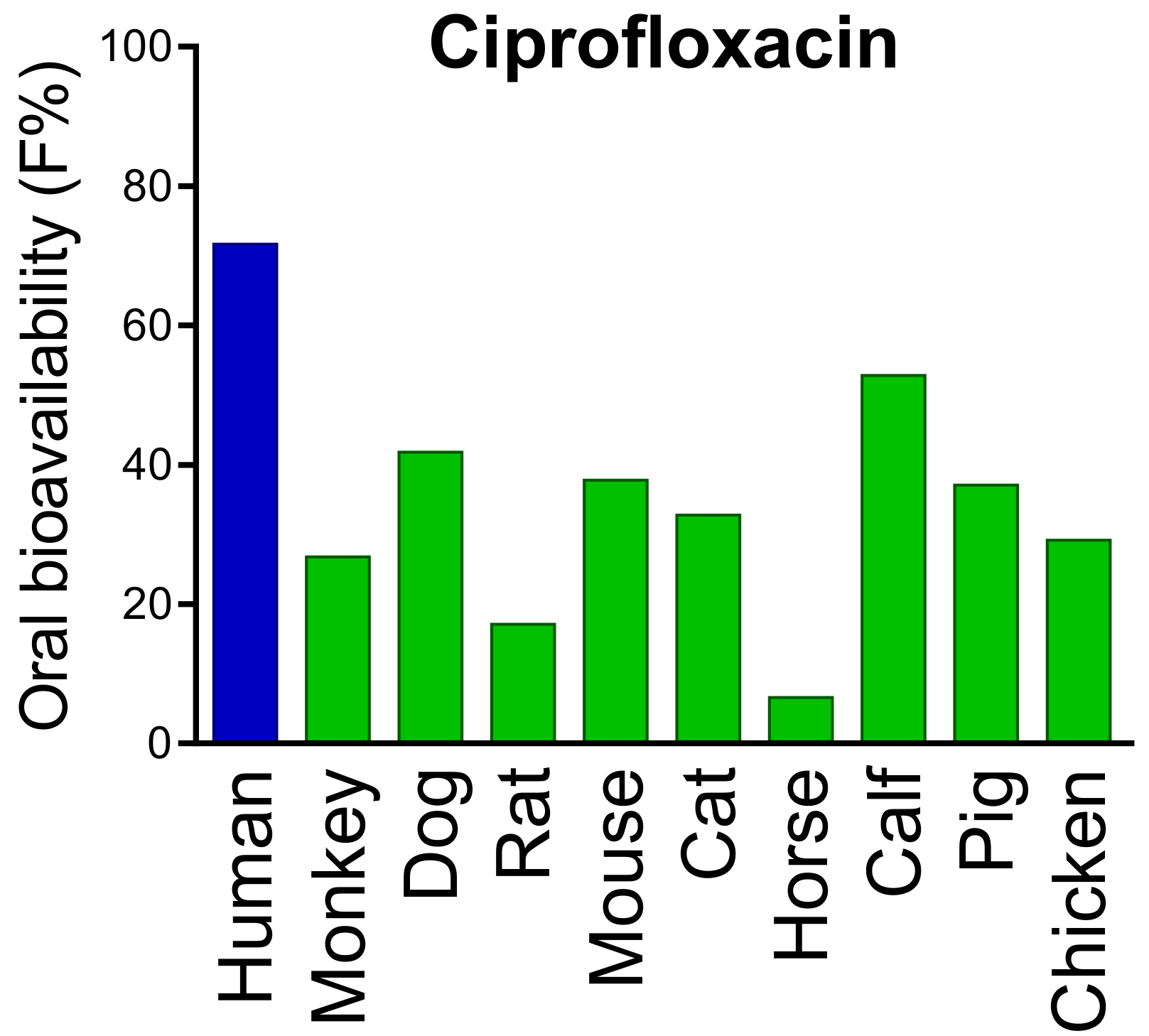
- Kararli, T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar] [CrossRef]
- Fagerholm, U.; Johansson, M.; Lennernäs, H. Comparison between permeability coefficients in rat and human jejunum. Pharm. Res. 1996, 13, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Youhanna, S.; Lauschke, V.M. The past, present and future of intestinal in vitro cell systems for drug absorption studies. J. Pharm. Sci. 2021, 110, 50–65. [Google Scholar] [CrossRef]
- PharmaInformatic. Blockbuster Drugs 2021. Available online: www.pharmainformatic.com/html/blockbuster_drugs.html (accessed on 1 August 2021).
- Ali, Z.; Chandrasekera, P.C.; Pippin, J.J. Animal research for type 2 diabetes mellitus, its limited translation for clinical benefit, and the way forward. Altern. Lab. Anim. 2018, 46, 13–22. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Arrowsmith, J. A decade of change. Nat. Rev. Drug Discov. 2012, 11, 17–18. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical Development Success Rates 2006–2015. Nat. Biotechnol. 2016, 32, 40–51. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of astrazeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Arrowsmith, J.; Miller, P. Trial watch: Phase ii and phase iii attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef]
- Mullard, A. Parsing clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 447–448. [Google Scholar] [CrossRef]
- Fagerholm, U.; Hellberg, S.; Spjuth, O. Advances in predictions of oral bioavailability of candidate drugs in man with new machine learning methodology. Molecules 2021, 26, 2572. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Capuzzi, S.J.; Muratov, E.; Braga, R.C.; Thornton, T.; Fourches, D.; Strickland, J.; Kleinstreuer, N.; Andrade, C.H.; Tropsha, A. Qsar models of human data can enrich or replace llna testing for human skin sensitization. Green Chem. 2016, 18, 6501–6515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, M.; Ashikaga, T.; Kouzuki, H. Development of an artificial neural network model for risk assessment of skin sensitization using human cell line activation test, direct peptide reactivity assay, keratinosens and in silico structure alert parameter. J. Appl. Toxicol. 2018, 38, 514–526. [Google Scholar] [CrossRef] [PubMed]
- PharmaInformatic. Impact-f. Available online: www.pharmainformatic.com/html/impact-f.html (accessed on 1 August 2021).
- Daina, A.; Michielin, O.; Zoete, V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- PharmaInformatic. Partnerships 2021. Available online: www.pharmainformatic.com/html/partnerships.html (accessed on 1 August 2021).
- Cabrera-Perez, M.A.; Pham-The, H. Computational modeling of human oral bioavailability: What will be next? Expert Opin. Drug Discov. 2018, 13, 509–521. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharmacol. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Smietana, K.; Siatkowski, M.; Moller, M. Trends in clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Thomas, D.; Burns, J.; Audette, J.; Carroll, A.; Dow-Hygelund, C.; Hay, M. Clinical Development Success Rates 2006–2015; Biotechnology Innovation Organization (BIO): Washington, DC, USA, 2016; pp. 1–16. [Google Scholar]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Lensink, M.A.; Boers, S.N.; Jongsma, K.R.; Carter, S.E.; van der Ent, C.K.; Bredenoord, A.L. Organoids for personalized treatment of cystic fibrosis: Professional perspectives on the ethics and governance of organoid biobanking. J. Cyst. Fibros. 2021, 20, 443–451. [Google Scholar] [CrossRef]
- Renner, H.; Grabos, M.; Becker, K.J.; Kagermeier, T.E.; Wu, J.; Otto, M.; Peischard, S.; Zeuschner, D.; TsyTsyura, Y.; Disse, P.; et al. A fully automated high-throughput workflow for 3d-based chemical screening in human midbrain organoids. eLife 2020, 9, e52904. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botti, G.; Di Bonito, M.; Cantile, M. Organoid biobanks as a new tool for pre-clinical validation of candidate drug efficacy and safety. Int. J. Physiol. Pathophysiol. Pharm. 2021, 13, 17–21. [Google Scholar]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Pistollato, F.; Bernasconi, C.; McCarthy, J.; Campia, I.; Desaintes, C.; Wittwehr, C.; Deceuninck, P.; Whelan, M. Alzheimer’s disease, and breast and prostate cancer research: Translational failures and the importance to monitor outputs and impact of funded research. Animals 2020, 10, 1194. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, J.C. Is parkinson’s disease a neurodevelopmental disorder and will brain organoids help us to understand it? Stem Cells Dev. 2018, 27, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Huldt, C.W.; Kanebratt, K.P.; Durieux, I.; Gunne, D.; Andersson, S.; Ewart, L.; Haynes, W.G.; Maschmeyer, I.; Winter, A.; et al. Functional coupling of human pancreatic islets and liver spheroids on-a-chip: Towards a novel human ex vivo type 2 diabetes model. Sci. Rep. 2017, 7, 14620. [Google Scholar] [CrossRef]
- Angus, H.C.K.; Butt, A.G.; Schultz, M.; Kemp, R.A. Intestinal organoids as a tool for inflammatory bowel disease research. Front. Med. 2020, 6, 334. [Google Scholar] [CrossRef]
- Nawroth, J.C.; Lucchesi, C.; Cheng, D.; Shukla, A.; Ngyuen, J.; Shroff, T.; Varone, A.; Karalis, K.; Lee, H.H.; Alves, S.; et al. A microengineered airway lung chip models key features of viral-induced exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 2020, 63, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.J.; Cook, R.S.; Sanders, M.E.; Arteaga, C.L.; Skala, M.C. Drug response in organoids generated from frozen primary tumor tissues. Sci. Rep. 2016, 6, 18889. [Google Scholar] [CrossRef] [Green Version]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to egfr inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartfeld, S.; Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. J. Mol. Med. 2017, 95, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional cftr assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Chakradhar, S. Put to the test: Organoid-based testing becomes a clinical tool. Nat. Med. 2017, 23, 796–799. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef] [Green Version]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [Green Version]
- Calandrini, C.; Schutgens, F.; Oka, R.; Margaritis, T.; Candelli, T.; Mathijsen, L.; Ammerlaan, C.; van Ineveld, R.L.; Derakhshan, S.; de Haan, S.; et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 2020, 11, 1310. [Google Scholar] [CrossRef]
- Halfter, K.; Mayer, B. Bringing 3d tumor models to the clinic—Predictive value for personalized medicine. Biotechnol. J. 2017, 12, 1600295. [Google Scholar] [CrossRef]
- Grabinger, T.; Luks, L.; Kostadinova, F.; Zimberlin, C.; Medema, J.P.; Leist, M.; Brunner, T. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis. 2014, 5, e1228. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T. Organoids for drug discovery and personalized medicine. Annu. Rev. Pharm. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Mahe, M.M.; Sundaram, N.; Watson, C.L.; Shroyer, N.F.; Helmrath, M.A. Establishment of human epithelial enteroids and colonoids from whole tissue and biopsy. J. Vis. Exp. 2015, 97, e52483. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.M.; Spence, J.R. How to make an intestine. Development 2014, 141, 752–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzner, M.; Helmrath, M.; Dunn, J.C.; Henning, S.J.; Houchen, C.W.; Kuo, C.; Lynch, J.; Li, L.; Magness, S.T.; Martin, M.G.; et al. A nomenclature for intestinal in vitro cultures. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1359–G1363. [Google Scholar] [CrossRef] [Green Version]
- Koo, B.K.; Stange, D.E.; Sato, T.; Karthaus, W.; Farin, H.F.; Huch, M.; van Es, J.H.; Clevers, H. Controlled gene expression in primary lgr5 organoid cultures. Nat. Methods 2011, 9, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of cftr by crispr/cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Berger, E.; Rath, E.; Yuan, D.; Waldschmitt, N.; Khaloian, S.; Allgäuer, M.; Staszewski, O.; Lobner, E.M.; Schöttl, T.; Giesbertz, P.; et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016, 7, 13171. [Google Scholar] [CrossRef] [Green Version]
- Petersen, N.; Reimann, F.; van Es, J.H.; van den Berg, B.M.; Kroone, C.; Pais, R.; Jansen, E.; Clevers, H.; Gribble, F.M.; de Koning, E.J. Targeting development of incretin-producing cells increases insulin secretion. J. Clin. Investig. 2015, 125, 379–385. [Google Scholar] [CrossRef] [Green Version]
- De Lau, W.; Kujala, P.; Schneeberger, K.; Middendorp, S.; Li, V.S.; Barker, N.; Martens, A.; Hofhuis, F.; DeKoter, R.P.; Peters, P.J.; et al. Peyer’s patch m cells derived from lgr5(+) stem cells require spib and are induced by rankl in cultured “miniguts”. Mol. Cell Biol. 2012, 32, 3639–3647. [Google Scholar] [CrossRef] [Green Version]
- Lindeboom, R.G.; van Voorthuijsen, L.; Oost, K.C.; Rodriguez-Colman, M.J.; Luna-Velez, M.V.; Furlan, C.; Baraille, F.; Jansen, P.W.; Ribeiro, A.; Burgering, B.M.; et al. Integrative multi-omics analysis of intestinal organoid differentiation. Mol. Syst. Biol. 2018, 14, e8227. [Google Scholar] [CrossRef]
- Xu, J.; Zeug, A.; Riederer, B.; Yeruva, S.; Griesbeck, O.; Daniel, H.; Tuo, B.; Ponimaskin, E.; Dong, H.; Seidler, U. Calcium-sensing receptor regulates intestinal dipeptide absorption via ca(2+) signaling and ikca activation. Physiol. Rep. 2020, 8, e14337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellett, G.L.; Brot-Laroche, E.; Mace, O.J.; Leturque, A. Sugar absorption in the intestine: The role of glut2. Annu. Rev. Nutr. 2008, 28, 35–54. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome inhibitor drugs. Annu. Rev. Pharm. Toxicol 2020, 60, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Co, J.Y.; Margalef-Catala, M.; Li, X.; Mah, A.T.; Kuo, C.J.; Monack, D.M.; Amieva, M.R. Controlling epithelial polarity: A human enteroid model for host-pathogen interactions. Cell Rep. 2019, 26, 2509–2520.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chiang, I.L.; Ohara, T.E.; Fujii, S.; Cheng, J.; Muegge, B.D.; Ver Heul, A.; Han, N.D.; Lu, Q.; Xiong, S.; et al. Long-term culture captures injury-repair cycles of colonic stem cells. Cell 2019, 179, 1144–1159.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweinlin, M.; Wilhelm, S.; Schwedhelm, I.; Hansmann, J.; Rietscher, R.; Jurowich, C.; Walles, H.; Metzger, M. Development of an advanced primary human in vitro model of the small intestine. Tissue Eng. Part C Methods 2016, 22, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Clevers, H. Organoid culture systems to study host-pathogen interactions. Curr. Opin. Immunol. 2017, 48, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bijvelds, M.; Dang, W.; Xu, L.; van der Eijk, A.A.; Knipping, K.; Tuysuz, N.; Dekkers, J.F.; Wang, Y.; de Jonge, J.; et al. Modeling rotavirus infection and antiviral therapy using primary intestinal organoids. Antivir. Res. 2015, 123, 120–131. [Google Scholar] [CrossRef]
- Ingber, D.E. Is it time for reviewer 3 to request human organ chip experiments instead of animal validation studies? Adv. Sci. 2020, 7, 2002030. [Google Scholar] [CrossRef] [PubMed]
- Edington, C.D.; Chen, W.L.K.; Geishecker, E.; Kassis, T.; Soenksen, L.R.; Bhushan, B.M.; Freake, D.; Kirschner, J.; Maass, C.; Tsamandouras, N.; et al. Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep. 2018, 8, 4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, U.; Akabane, T.; Andersson, T.B.; Baker, E.; Beilmann, M.; Beken, S.; Brendler-Schwaab, S.; Cirit, M.; David, R.; Dehne, E.-M.; et al. Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. ALTEX Altern. Anim. Exp. 2020, 37, 365. [Google Scholar]
- Skardal, A.; Aleman, J.; Forsythe, S.; Rajan, S.; Murphy, S.; Devarasetty, M.; Pourhabibi Zarandi, N.; Nzou, G.; Wicks, R.; Sadri-Ardekani, H.; et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication 2020, 12, 025017. [Google Scholar] [CrossRef]
- Fitzpatrick, S.; Sprando, R. Advancing regulatory science through innovation: In vitro microphysiological systems. Cell. Mol. Gastroenterol. Hepatol. 2018, 7, 239–240. [Google Scholar] [CrossRef] [Green Version]
- Ministerie van Landbouw, Natuur en Voedselkwaliteit. NCAD Opinion Transition to Non-Animal Research—Rapport—Nationaal Comité Advies Dierproevenbeleid; NCAD: The Hague, The Netherlands, 2016. [Google Scholar]
- Forsøksdyrkomitéen. Behov for Utredning om Forskning uten Forsøksdyr og et Nasjonalt 3r-Senter. 2020. Available online: https://www.forsoksdyrkomiteen.no/behov-for-utredning-om-forskning-uten-forsoksdyr-og-et-nasjonalt-3r-senter/ (accessed on 1 August 2021).
- Eurogroup for Animals. Norwegian National Experimental Animals Committee Proposes Steps to Develop a Concrete Plan for a Transition to Non-Animal Science; Eurogroup for Animals: Brussels, Belgium, 2020. [Google Scholar]
- FDA. Fda’s Predictive Toxicology Roadmap. 2017. Available online: https://www.fda.gov/media/109634/download (accessed on 1 August 2021).
- FDA. Partnership to Apply Lung Chips to Safety Evaluation of COVID-19 Vaccines and Therapies: Altex—Alternatives to Animal Experimentation; FDA: Silver Spring, MD, USA, 2020.
- Ribeiro, A.J.S.; Yang, X.; Patel, V.; Madabushi, R.; Strauss, D.G. Liver microphysiological systems for predicting and evaluating drug effects. Clin. Pharm. 2019, 106, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Rooney, J.P.; Choksi, N.Y.; Ceger, P.; Daniel, A.B.; Truax, J.; Allen, D.; Kleinstreuer, N. Analysis of variability in the rabbit skin irritation assay. Regul. Toxicol. Pharm. 2021, 122, 104920. [Google Scholar] [CrossRef]
- Hoffmann, S. Llna variability: An essential ingredient for a comprehensive assessment of non-animal skin sensitization test methods and strategies. ALTEX 2015, 32, 379–383. [Google Scholar] [PubMed] [Green Version]
- Clippinger, A.J.; Raabe, H.A.; Allen, D.G.; Choksi, N.Y.; van der Zalm, A.J.; Kleinstreuer, N.C.; Barroso, J.; Lowit, A.B. Human-relevant approaches to assess eye corrosion/irritation potential of agrochemical formulations. Cutan. Ocul. Toxicol. 2021, 40, 145–167. [Google Scholar] [CrossRef]
- Hoffmann, S.; Kleinstreuer, N.; Alepee, N.; Allen, D.; Api, A.M.; Ashikaga, T.; Clouet, E.; Cluzel, M.; Desprez, B.; Gellatly, N.; et al. Non-animal methods to predict skin sensitization (i): The cosmetics europe database. Crit. Rev. Toxicol. 2018, 48, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Braeuning, C.; Braeuning, A.; Mielke, H.; Holzwarth, A.; Peiser, M. Evaluation and improvement of qsar predictions of skin sensitization for pesticides. SAR QSAR Environ. Res. 2018, 29, 823–846. [Google Scholar] [CrossRef] [PubMed]
- FDA. Advancing Alternative Methodologies at FDA. 2021. Available online: https://www.fda.gov/media/144891/download (accessed on 1 August 2021).
- Hynes, J.; Marshall, L.; Adcock, I.; Novotny, T.; Nic, M.; Dibusz, K.; Gribaldo, L.; Whelan, M. Advanced Non-Animal Models in Biomedical Research: Respiratory Tract Diseases; Publications Office of the European Union: Luxembourg, 2020. [Google Scholar]
- Hynes, J.; Marshall, L.; Adcock, I.; Novotny, T.; Nic, M.; Dibusz, K.; Gribaldo, L.; Whelan, M. Advanced Non-Animal Models in Biomedical Research: Breast Cancer; Publications Office of the European Union: Luxembourg, 2020. [Google Scholar]
- Berg, J.; Kurreck, J. Clean bioprinting—Fabrication of 3d organ models devoid of animal components. ALTEX Altern. Anim. Exp. 2020, 38, 269–288. [Google Scholar]
- Ng, W.L.; Wang, S.; Yeong, W.Y.; Naing, M.W. Skin bioprinting: Impending reality or fantasy? Trends Biotechnol. 2016, 34, 689–699. [Google Scholar] [CrossRef]
- Isaacson, A.; Swioklo, S.; Connon, C.J. 3D bioprinting of a corneal stroma equivalent. Exp. Eye Res. 2018, 173, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Raasch, M.; Fritsche, E.; Kurtz, A.; Bauer, M.; Mosig, A.S. Microphysiological systems meet hipsc technology—New tools for disease modeling of liver infections in basic research and drug development. Adv. Drug Deliv. Rev. 2019, 140, 51–67. [Google Scholar] [CrossRef]
- Hartung, T. Predicting toxicity of chemicals: Software beats animal testing. EFSA J. 2019, 17, e170710. [Google Scholar] [CrossRef] [PubMed]
- Noorden, R.V. Software beats animal tests at predicting toxicity of chemicals. Nature 2018, 559, 163. [Google Scholar] [CrossRef] [Green Version]
- Comenges, J.M.Z.; Joossens, E.; Benito, J.V.S.; Worth, A.; Paini, A. Theoretical and mathematical foundation of the virtual cell based assay—A review. Toxicol. In Vitro 2017, 45, 209–221. [Google Scholar] [CrossRef]
- Herrmann, K.; Pistollato, F.; Stephens, M.L. Beyond the 3rs: Expanding the use of human-relevant replacement methods in biomedical research. ALTEX Altern. Anim. Exp. 2019, 36, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pound, P.; Ram, R. Are researchers moving away from animal models as a result of poor clinical translation in the field of stroke? An analysis of opinion papers. BMJ Open Sci. 2020, 4, e100041. [Google Scholar] [CrossRef]
- Smirnova, L.; Kleinstreuer, N.; Corvi, R.; Levchenko, A.; Fitzpatrick, S.C.; Hartung, T. 3s—Systematic, systemic, and systems biology and toxicology. ALTEX Altern. Anim. Exp. 2018, 35, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Urani, C.; Bruschi, M.; Casati, S.; Gribaldo, L. Use of alternative methods: From fundamental to industrial research. ALTEX Altern. Anim. Exp. 2019, 36, 320–321. [Google Scholar] [CrossRef]
- Veening-Griffioen, D.H.; Ferreira, G.S.; van Meer, P.J.K.; Boon, W.P.C.; Gispen-de Wied, C.C.; Moors, E.H.M.; Schellekens, H. Are some animal models more equal than others? A case study on the translational value of animal models of efficacy for alzheimer’s disease. Eur. J. Pharmacol. 2019, 859, 172524. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Failure Rate | No of Drugs Tested | Years | Reference |
|---|---|---|---|
| 81–84% | 1.738 | 1993–2004 | DiMasi et al., 2010 [125] |
| 88.6% | >9.200 | 1996–2014 | Smietana et al., 2016 [126] |
| 86.2% | 21.143 | 2000–2015 | Wong et al., 2019 [127] |
| 90–95% | >850 | 2002–2008 | Arrowsmith, 2012 [113] |
| 89.6% | >7.300 | 2003–2011 | Hay et al., 2014 [128] |
| 90.4% | 7.455 | 2006–2015 | Thomas et al., 2016 [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zietek, T.; Boomgaarden, W.A.D.; Rath, E. Drug Screening, Oral Bioavailability and Regulatory Aspects: A Need for Human Organoids. Pharmaceutics 2021, 13, 1280. https://doi.org/10.3390/pharmaceutics13081280
Zietek T, Boomgaarden WAD, Rath E. Drug Screening, Oral Bioavailability and Regulatory Aspects: A Need for Human Organoids. Pharmaceutics. 2021; 13(8):1280. https://doi.org/10.3390/pharmaceutics13081280
Chicago/Turabian StyleZietek, Tamara, Wolfgang A. D. Boomgaarden, and Eva Rath. 2021. "Drug Screening, Oral Bioavailability and Regulatory Aspects: A Need for Human Organoids" Pharmaceutics 13, no. 8: 1280. https://doi.org/10.3390/pharmaceutics13081280
APA StyleZietek, T., Boomgaarden, W. A. D., & Rath, E. (2021). Drug Screening, Oral Bioavailability and Regulatory Aspects: A Need for Human Organoids. Pharmaceutics, 13(8), 1280. https://doi.org/10.3390/pharmaceutics13081280