
PBPK Modeling as a Tool for Predicting and Understanding Intestinal Metabolism of Uridine 5′-Diphospho-glucuronosyltransferase Substrates

Abstract
:1. Introduction
2. Incorporating Intestinal Metabolism in PBPK Models
3. UGT Expression in the GI Tract
4. In Vitro Methods to Measure UGT-Mediated Drug Metabolism
5. PBPK Modeling of UGT Intestinal Metabolism
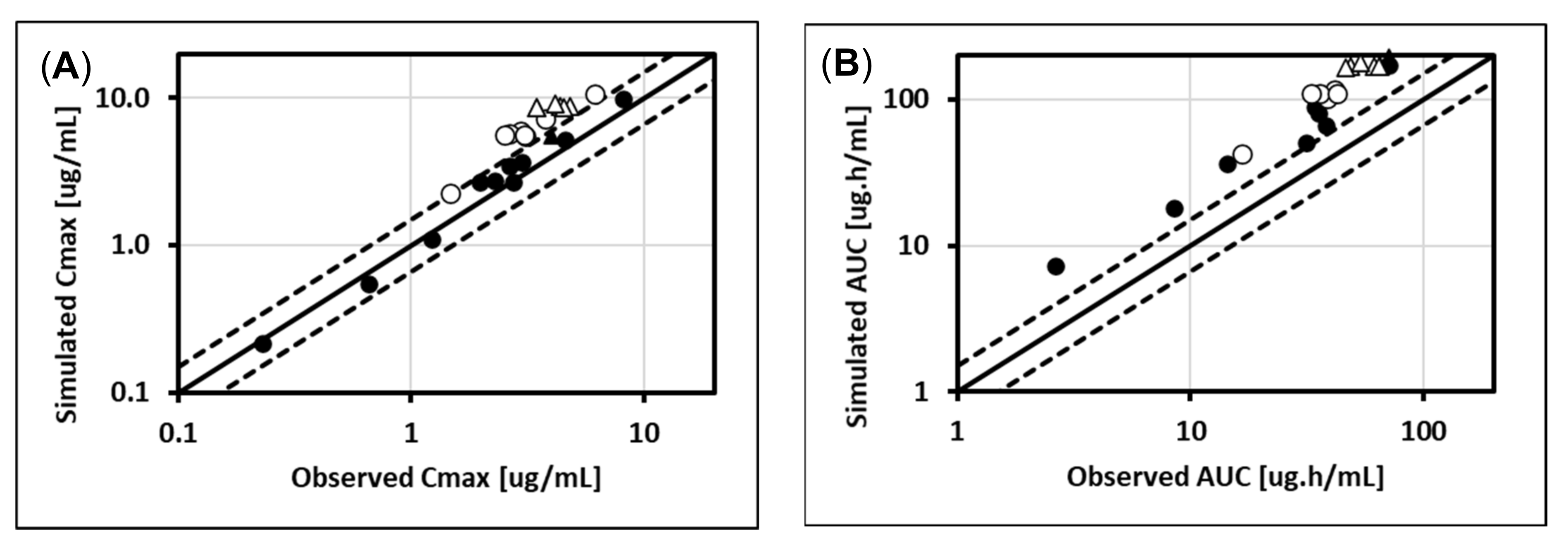
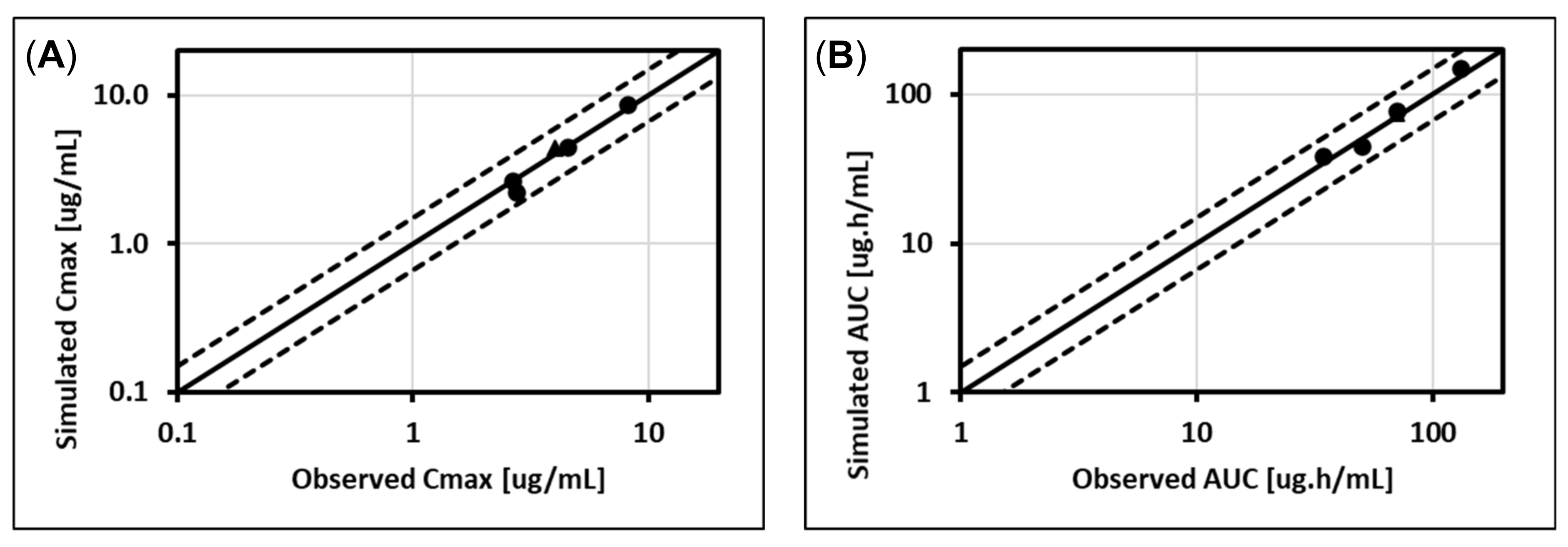
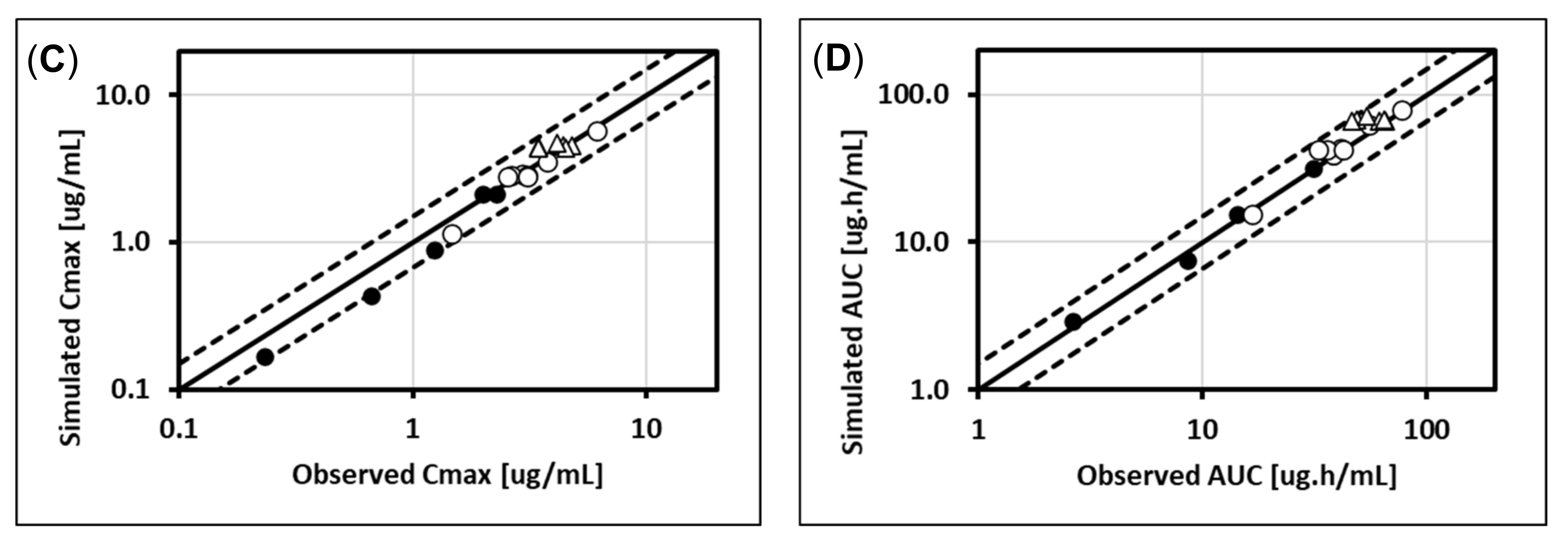
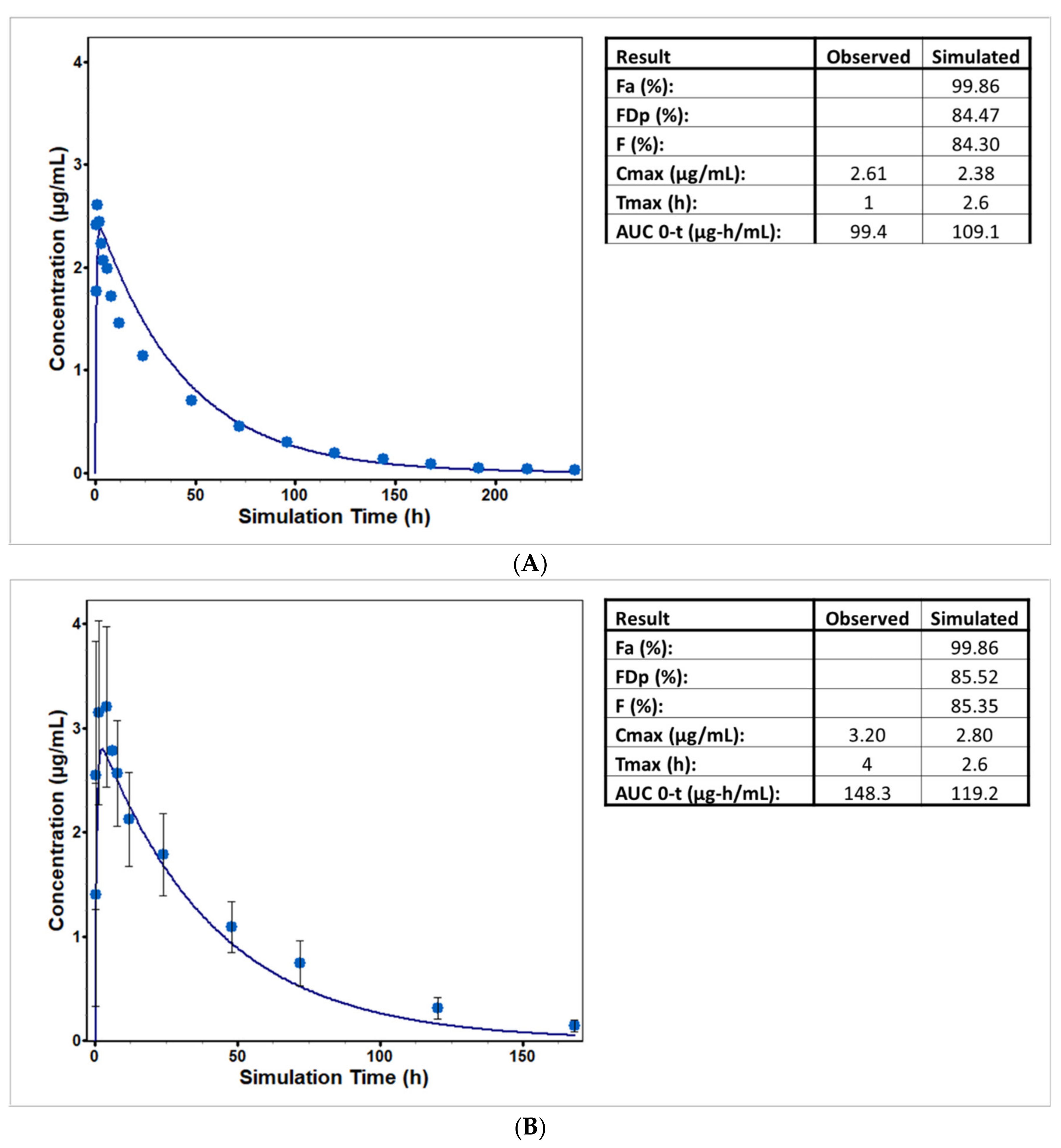
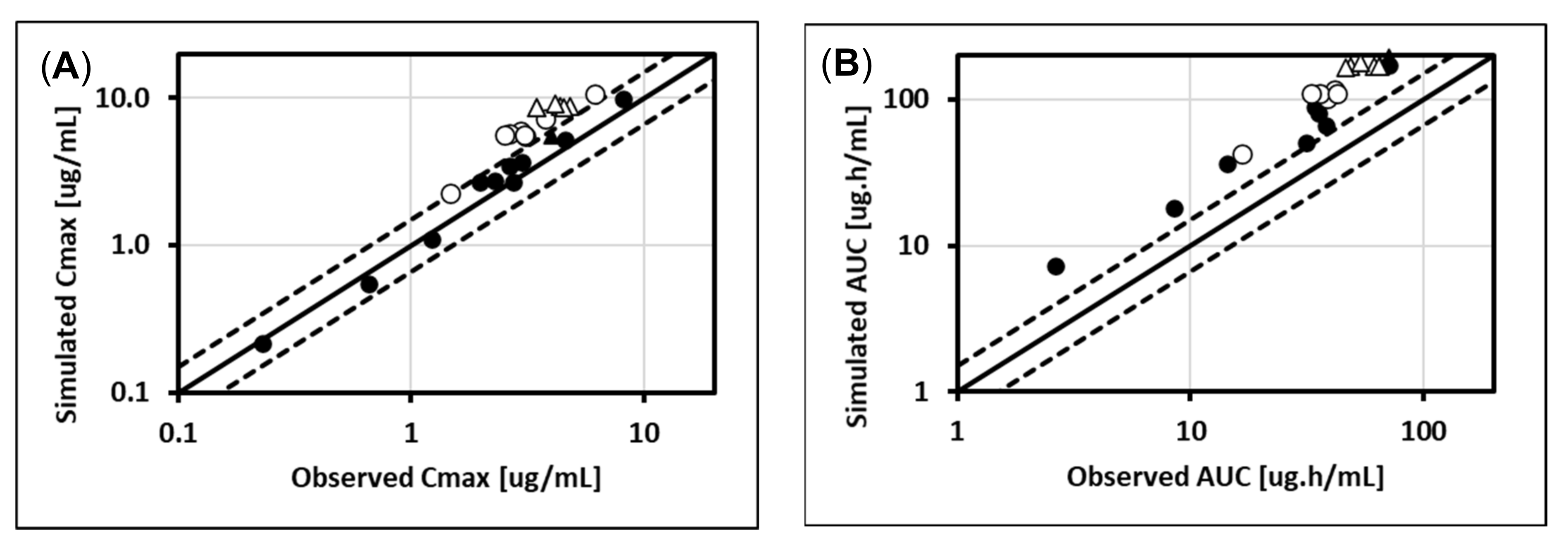
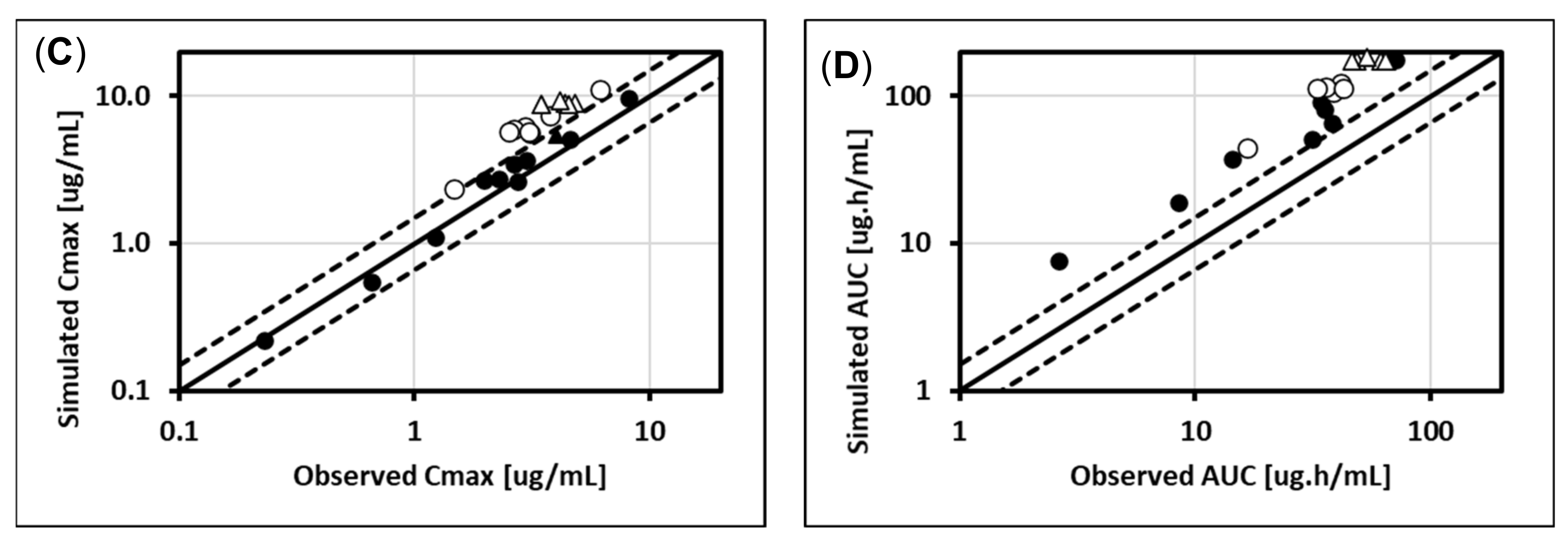
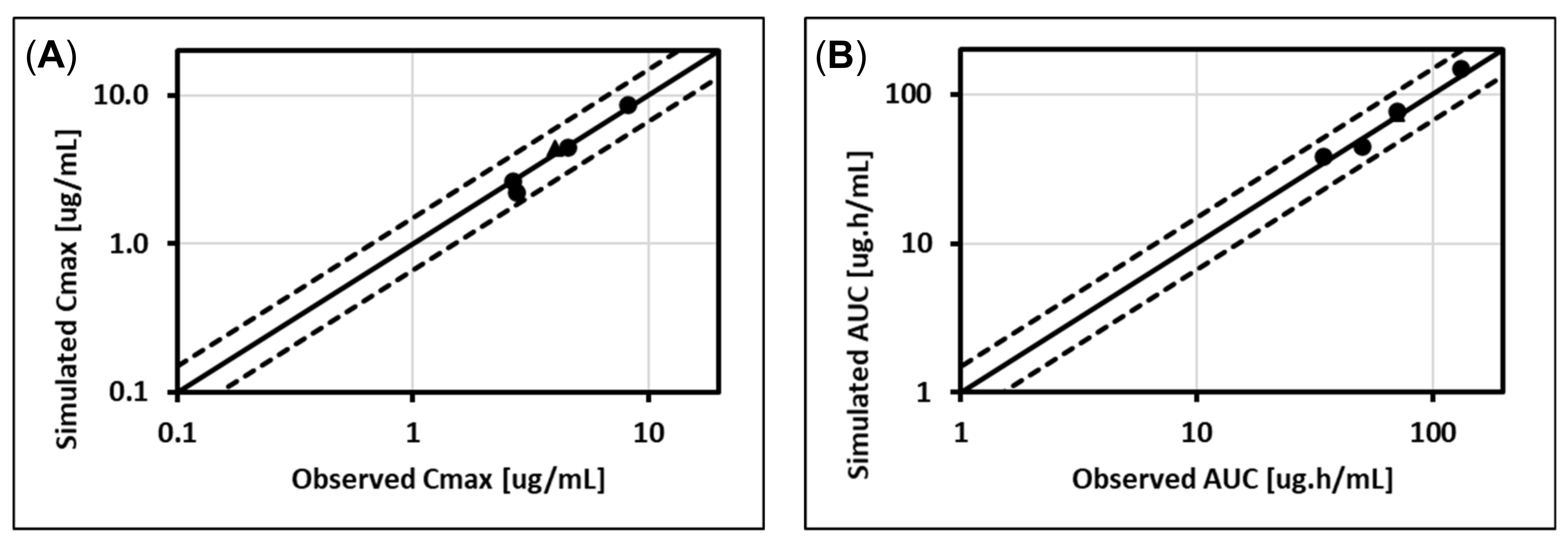
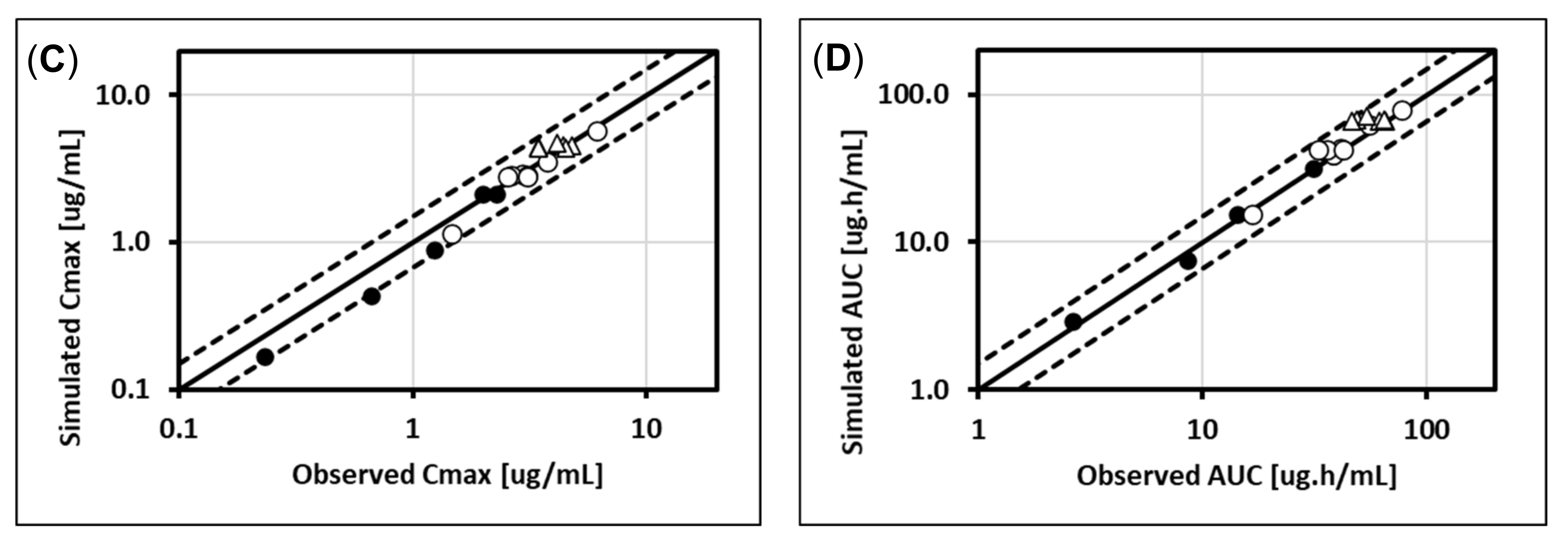
5.1. Lorazepam (UGT2B7, UGT2B15), Oxazepam (UGT1A9, UGT2B15), Naloxone (UGT2B7), and Zidovudine (UGT2B7, CYP3A4)
5.2. Cabotegravir (UGT1A1, UGT1A9)
5.3. Dolutegravir (UGT1A1, CYP3A4)
5.4. Raltegravir (UGT1A1)
6. Discussion
6.1. Prediction of Intestinal Metabolism Mediated by UGTs with PBPK Modeling
6.2. Clinical Relevance of UGT Intestinal Metabolism
6.3. Limitations of the PBPK Approach for Predicting Fg for UGT Substrates
6.4. In Silico Example: Predicted Substrate Specificity for UGT Enzymes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, H.M.; Parrott, N.; Jorga, K.; Lavé, T. A Novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharm. 2006, 45, 511–542. [Google Scholar] [CrossRef]
- Miller, N.A.; Reddy, M.B.; Heikkinen, A.T.; Lukacova, V.; Parrott, N. Physiologically based pharmacokinetic modelling for first-in-human predictions: An updated model building strategy illustrated with challenging industry case studies. Clin. Pharm. 2019, 58, 727–746. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, S.; Bukhari, I.; Albassam, A.; Alenazi, M. An update on the potential role of intestinal first-pass metabolism for the prediction of drug–drug interactions: The role of PBPK modeling. Expert Opin. Drug Met. Toxicol. 2018, 14, 625–634. [Google Scholar] [CrossRef]
- Yang, J.; Jamei, M.; Yeo, K.R.; Tucker, G.T.; Rostami-Hodjegan, A. Prediction of intestinal first-pass drug metabolism. Curr. Drug Metab. 2007, 8, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, A.T.; Fowler, S.; Gray, L.; Li, J.; Peng, Y.; Yadava, P.; Railkar, A.; Parrott, N. In vitro to in vivo extrapolation and physiologically based modeling of cytochrome P450 mediated metabolism in beagle dog gut wall and liver. Mol. Pharm. 2013, 10, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, A.T.; Baneyx, G.; Caruso, A.; Parrott, N. Application of PBPK modeling to predict human intestinal metabolism of CYP3A substrates—An evaluation and case study using GastroPlus™. Eur. J. Pharm. Sci. 2012, 47, 375–386. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Bouchene, S.; Hilgendorf, C.; Dolgos, H.; Peters, S.A. Utility of in vitro systems and preclinical data for the prediction of human intestinal first-pass metabolism during drug discovery and preclinical development. Drug Metab. Dispos. 2013, 41, 2033–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basit, A.; Neradugomma, N.K.; Wolford, C.; Fan, P.W.; Murray, B.; Takahashi, R.H.; Khojasteh, S.C.; Smith, B.J.; Heyward, S.; Totah, R.A.; et al. Characterization of differential tissue abundance of major non-CYP enzymes in human. Mol. Pharm. 2020, 17, 4114–4124. [Google Scholar] [CrossRef]
- Harbourt, D.E.; Fallon, J.K.; Ito, S.; Baba, T.; Ritter, J.K.; Glish, G.L.; Smith, P.C. Quantification of human uridine-diphosphate glucuronosyl transferase 1A isoforms in liver, intestine, and kidney using nanobore liquid chromatography–tandem mass spectrometry. Anal. Chem. 2011, 84, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Nakajin, S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab. Dispos. 2009, 37, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Nagata, M.; Tetsuka, K.; Tamura, K.; Miyashita, A.; Kawamura, A.; Usui, T. Optimized methods for targeted peptide-based quantification of human uridine 5′-diphosphate-glucuronosyltransferases in biological specimens using liquid chromatography–tandem mass spectrometry. Drug Metab. Dispos. 2014, 42, 885–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuma, T. Intestinal glucuronidation metabolism may have a greater impact on oral bioavailability than hepatic glucuronidation metabolism in humans: A study with raloxifene, substrate for UGT1A1, 1A8, 1A9, and 1A10. Int. J. Pharm. 2009, 378, 140–141. [Google Scholar] [CrossRef]
- Ge, S.; Tu, Y.; Hu, M. Challenges and opportunities with predicting in vivo Phase II metabolism via glucuronidation from in vitro data. Curr. Pharm. Rep. 2016, 2, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Argikar, U.A.; Potter, P.M.; Hutzler, J.M.; Marathe, P.H. Challenges and opportunities with non-CYP enzymes aldehyde oxidase, carboxylesterase, and UDP-glucuronosyltransferase: Focus on reaction phenotyping and prediction of human clearance. AAPS J. 2016, 18, 1391–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naritomi, Y.; Nakamori, F.; Furukawa, T.; Tabata, K. Prediction of hepatic and intestinal glucuronidation using in vitro-in vivo extrapolation. Drug Metab. Pharm. 2015, 30, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Docci, L.; Klammers, F.; Ekiciler, A.; Molitor, B.; Umehara, K.; Walter, I.; Krähenbühl, S.; Parrott, N.; Fowler, S. In vitro to in vivo extrapolation of metabolic clearance for UGT substrates using short-term suspension and long-term co-cultured human hepatocytes. AAPS J. 2020, 22, 131. [Google Scholar] [CrossRef]
- McGinnity, D.F.; Soars, M.G.; Urbanowicz, R.A.; Riley, R.J. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab. Dispos. 2004, 32, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Miners, J.O.; Knights, K.M.; Houston, J.B.; Mackenzie, P.I. In vitro–in vivo correlation for drugs and other compounds eliminated by glucuronidation in humans: Pitfalls and promises. Biochem. Pharm. 2006, 71, 1531–1539. [Google Scholar] [CrossRef]
- Zientek, M.A.; Youdim, K. Reaction phenotyping: Advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab. Dispos. 2015, 43, 163–181. [Google Scholar] [CrossRef] [Green Version]
- Nishimuta, H.; Sato, K.; Yabuki, M.; Komuro, S. Prediction of the intestinal first-pass metabolism of CYP3A and UGT substrates in humans from in vitro data. Drug Metab. Pharm. 2011, 26, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Stingl, J.C.; Bartels, H.; Viviani, R.; Lehmann, M.L.; Brockmöller, J. Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: A quantitative systematic review. Pharm. Ther. 2014, 141, 92–116. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Kulkarni, K.; Basu, S.; Zhang, S.; Hu, M. First—pass metabolism via UDP—glucuronosyltransferase: A barrier to oral bioavailability of phenolics. J. Pharm. Sci. 2011, 100, 3655–3681. [Google Scholar] [CrossRef] [Green Version]
- Docci, L.; Umehara, K.; Krähenbühl, S.; Fowler, S.; Parrott, N. Construction and verification of physiologically based pharmacokinetic models for four drugs majorly cleared by glucuronidation: Lorazepam, oxazepam, naloxone, and zidovudine. AAPS J. 2020, 22, 128. [Google Scholar] [CrossRef]
- Reddy, M.B.; Lukacova, V.; Fraczkiewicz, G.; Macwan, J.S.; Del Frari, L. PBPK modeling for binimetinib to understand implications of UGT1A1-mediated metabolism for pharmacokinetics and drug-drug interactions. 2021, in press. [Google Scholar]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: Oral solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar] [CrossRef] [PubMed]
- Agoram, B.; Woltosz, W.S.; Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001, 50, S41–S67. [Google Scholar] [CrossRef]
- Simulations Plus. Better Decisions through Better Science. User Manual for GastroPlus Version 9.8; Simulations Plus: Lancaster, CA, USA, 2020. [Google Scholar]
- Meech, R.; Hu, D.G.; McKinnon, R.A.; Mubarokah, S.N.; Haines, A.Z.; Nair, P.C.; Rowland, A.; Mackenzie, P.I. The UDP-glycosyltransferase (UGT) superfamily: New members, new functions, and novel paradigms. Physiol. Rev. 2019, 99, 1153–1222. [Google Scholar] [CrossRef] [PubMed]
- Margaillan, G.; Rouleau, M.; Fallon, J.K.; Caron, P.; Villeneuve, L.; Turcotte, V.; Smith, P.C.; Joy, M.S.; Guillemette, C. Quantitative profiling of human renal UDP-glucuronosyltransferases and glucuronidation activity: A comparison of normal and tumoral kidney tissues. Drug Metab. Dispos. 2015, 43, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, M.; Ejiri, Y.; Kishimoto, S.; Suzuki, S.; Satoh, T.; Horie, T.; Narimatsu, S.; Naito, S. Expression levels of drug-metabolizing enzyme, transporter, and nuclear receptor mRNAs in a novel three-dimensional culture system for human hepatocytes using micro-space plates. Drug Metab. Pharm. 2011, 26, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuki, S.; Schaefer, O.; Kawakami, H.; Inoue, T.; Liehner, S.; Saito, A.; Ishiguro, N.; Kishimoto, W.; Ludwig-Schwellinger, E.; Ebner, T.; et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: Comparison with mRNA levels and activities. Drug Metab. Dispos. 2012, 40, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Soars, M.G.; Burchell, B.; Riley, R.J. In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J. Pharm. Exp. Ther. 2002, 301, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Drozdzik, M.; Busch, D.; Lapczuk, J.; Müller, J.; Ostrowski, M.; Kurzawski, M.; Oswald, S. Protein abundance of clinically relevant drug—metabolizing enzymes in the human liver and intestine: A comparative analysis in paired tissue specimens. Clin. Pharm. Ther. 2018, 104, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wolford, C.; Basit, A.; Li, A.P.; Fan, P.W.; Murray, B.P.; Takahashi, R.H.; Khojasteh, S.C.; Smith, B.J.; Thummel, K.; et al. Regional proteomic quantification of clinically relevant non-cytochrome P450 enzymes along the human small intestine. Drug Metab. Dispos. 2020, 48. [Google Scholar] [CrossRef]
- Couto, N.; Al-Majdoub, Z.; Gibson, S.; Davies, P.; Achour, B.; Harwood, M.D.; Carlson, G.; Barber, J.; Rostami-Hodjegan, A.; Warhurst, G. Quantitative proteomics of clinically relevant drug-metabolizing enzymes and drug transporters and their inter-correlations in the human small intestine. Drug Metab. Dispos. 2020, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, T.; Uchida, Y.; Miyauchi, E.; Tachikawa, M.; Ohtsuki, S.; Terasaki, T. High expression of UGT1A1/1A6 in monkey small intestine: Comparison of protein expression levels of Cytochromes P450, UDP-glucuronosyltransferases, and transporters in small Intestine of cynomolgus monkey and human. Mol. Pharm. 2017, 15, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, E.; Tachikawa, M.; Declèves, X.; Uchida, Y.; Bouillot, J.-L.; Poitou, C.; Oppert, J.-M.; Mouly, S.; Bergmann, J.-F.; Terasaki, T.; et al. Quantitative atlas of cytochrome P450, UDP-glucuronosyltransferase, and transporter proteins in jejunum of morbidly obese subjects. Mol. Pharm. 2016, 13, 2631–2640. [Google Scholar] [CrossRef]
- Kasteel, E.E.J.; Darney, K.; Kramer, N.I.; Dorne, J.L.C.M.; Lautz, L.S. Human variability in isoform-specific UDP-glucuronosyltransferases: Markers of acute and chronic exposure, polymorphisms and uncertainty factors. Arch. Toxicol. 2020, 94, 2637–2661. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, J.B.; Hou, J.; Dou, T.Y.; Ge, G.B.; Hu, W.Z.; Yang, L. Chemical probes for human UDP-glucuronosyltransferases: A comprehensive review. Biotechnol. J. 2019, 14, e1800002. [Google Scholar] [CrossRef] [Green Version]
- Wegler, C.; Gaugaz, F.Z.; Andersson, T.B.; Wiśniewski, J.R.; Busch, D.; Gröer, C.; Oswald, S.; Norén, A.; Weiss, F.; Hammer, H.S.; et al. Variability in mass spectrometry-based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol. Pharm. 2017, 14, 3142–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, B.; Achour, B.; Artursson, P.; Hop, C.E.C.A.; Lai, Y.; Smith, P.C.; Barber, J.; Wisniewski, J.R.; Spellman, D.; Uchida, Y.; et al. Toward a consensus on applying quantitative liquid chromatography—tandem mass spectrometry proteomics in translational pharmacology research: A white paper. Clin. Pharm. Ther. 2019, 106, 525–543. [Google Scholar] [CrossRef]
- Paine, M.F.; Khalighi, M.; Fisher, J.M.; Shen, D.D.; Kunze, K.L.; Marsh, C.L.; Thummel, J.D.P.; Kenneth, E.; Thummel, K.E. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J. Pharm. Exp. Ther. 1997, 283, 1552–1562. [Google Scholar]
- Di, L.; Obach, R.S. Addressing the challenges of low clearance in drug research. AAPS J. 2015, 17, 352–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratochwil, N.A.; Meille, C.; Fowler, S.; Klammers, F.; Ekiciler, A.; Molitor, B.; Simon, S.; Walter, I.; McGinnis, C.; Walther, J.; et al. Metabolic profiling of human long-term liver models and hepatic clearance predictions from in vitro data using nonlinear mixed-effects modeling. AAPS J. 2017, 19, 534–550. [Google Scholar] [CrossRef]
- Pelkonen, O.; Turpeinen, M. In vitro-in vivo extrapolation of hepatic clearance: Biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 2007, 37, 1066–1089. [Google Scholar] [CrossRef] [PubMed]
- Manevski, N.; Troberg, J.; Svaluto-Moreolo, P.; Dziedzic, K.; Yli-Kauhaluoma, J.; Finel, M. Albumin stimulates the activity of the human UDP-glucuronosyltransferases 1A7, 1A8, 1A10, 2A1 and 2B15, but the effects are enzyme and substrate dependent. PLoS ONE 2013, 8, e54767. [Google Scholar] [CrossRef]
- Rowland, A.; Knights, K.M.; Mackenzie, P.I.; Miners, J.O. The “albumin effect” and drug glucuronidation: Bovine serum albumin and fatty acid-free human serum albumin enhance the glucuronidation of UDP-glucuronosyltransferase (UGT) 1A9 substrates but not UGT1A1 and UGT1A6 activities. Drug Metab. Dispos. 2008, 36, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Walsky, R.L.; Bauman, J.N.; Bourcier, K.; Giddens, G.; Lapham, K.; Negahban, A.; Ryder, T.F.; Obach, R.S.; Hyland, R.; Goosen, T.C. Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: Altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab. Dispos. 2012, 40, 1051–1065. [Google Scholar] [CrossRef]
- Badée, J.; Qiu, N.; Parrott, N.; Collier, A.C.; Schmidt, S.; Fowler, S. Optimization of experimental conditions of automated glucuronidation assays in human liver microsomes using a cocktail approach and ultra-high performance liquid chromatography-tandem mass spectrometry. Drug Metab. Dispos. 2019, 47, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Wood, F.L.; Houston, J.B.; Hallifax, D. Clearance prediction methodology needs fundamental improvement: Trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metab. Dispos. 2017, 45, 1178–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonn, B.; Svanberg, P.; Janefeldt, A.; Hultman, I.; Grime, K. Determination of human hepatocyte intrinsic clearance for slowly metabolized compounds: Comparison of a primary hepatocyte/stromal cell co-culture with plated primary hepatocytes and HepaRG. Drug Metab. Dispos. 2016, 44, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.S.; Yu, H.; Moore, A.; Khetani, S.R.; Tweedie, D. Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome P450 using a novel hepatocyte model, HepatoPac. Drug Metab. Dispos. 2019, 47, 58–66. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Nguyen, K.; Fretland, A.J. Utility of intersystem extrapolation factors in early reaction phenotyping and the quantitative extrapolation of human liver microsomal intrinsic clearance using recombinant cytochromes P450. Drug Metab. Dispos. 2011, 39, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Proctor, N.J.; Tucker, G.T.; Rostami-Hodjegan, A. Predicting drug clearance from recombinantly expressed CYPs: Intersystem extrapolation factors. Xenobiotica 2004, 34, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, K.; von Moltke, L.L.; Court, M.H.; Harmatz, J.S.; Crespi, C.L.; Greenblatt, D.J. Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: Ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab. Dispos. 2000, 28, 1493–1504. [Google Scholar]
- Busse, D.; Leandersson, S.; Amberntsson, S.; Darnell, M.; Hilgendorf, C. Industrial approach to determine the relative contribution of seven major UGT isoforms to hepatic glucuronidation. J. Pharm. Sci. 2020, 109, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.; Jones, C.R.; Ungell, A.L.; Hatley, O.J. Predicting drug extraction in the human gut wall: Assessing contributions from drug metabolizing enzymes and transporter proteins using preclinical models. Clin. Pharm. 2016, 55, 673–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.D.; Ring, N.; Amaral, K.; Doshi, U.; Li, A.P. Human enterocytes as an in vitro model for the evaluation of intestinal drug metabolism: Characterization of drug-metabolizing enzyme activities of cryopreserved human enterocytes from twenty-four donors. Drug Metab. Dispos. 2017, 45, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P.; Alam, N.; Amaral, K.; Ho, M.D.; Loretz, C.; Mitchell, W.; Yang, Q. Cryopreserved human intestinal mucosal epithelium: A novel in vitro experimental system for the evaluation of enteric drug metabolism, cytochrome P450 induction, and enterotoxicity. Drug Metab. Dispos. 2018, 46, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P.; Ho, M.D.; Alam, N.; Mitchell, W.; Wong, S.; Yan, Z.; Kenny, J.R.; Hop, C.E.C.A. Inter-individual and inter-regional variations in enteric drug metabolizing enzyme activities: Results with cryopreserved human intestinal mucosal epithelia (CHIM) from the small intestines of 14 donors. Pharmacol. Res. Perspect. 2020, 8, e00645. [Google Scholar] [CrossRef]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef]
- Reese, M.J.; Savina, P.M.; Generaux, G.T.; Tracey, H.; Humphreys, J.E.; Kanaoka, E.; Webster, L.O.; Harmon, K.A.; Clarke, J.D.; Polli, J.W. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 2013, 41, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, G.D.; Culp, A.; Reese, M.J.; Tabolt, G.; Moss, L.; Piscitelli, S.; Huynh, P.; Wagner, D.; Ford, S.L.; Gould, E.P.; et al. Disposition and metabolism of cabotegravir: A comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica 2015, 46, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.L.; Sutton, K.; Lou, Y.; Zhang, Z.; Tenorio, A.; Trezza, C.; Patel, P.; Spreen, W. Effect of rifampin on the single-dose pharmacokinetics of oral cabotegravir in healthy subjects. Antimicrob. Agents Chemother. 2017, 61, e00487-17. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, M.L.; Hadzic, T.; Kashuba, A.D.M. Clinical pharmacokinetic, pharmacodynamic and drug-interaction profile of the integrase inhibitor dolutegravir. Clin. Pharm. 2013, 52, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Min, S.; Song, I.; Borland, J.; Chen, S.; Lou, Y.; Fujiwara, T.; Piscitelli, S.C. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.; Borland, J.; Chen, S.; Patel, P.; Wajima, T.; Peppercorn, A.; Piscitelli, S.C. Effect of Food on the Pharmacokinetics of the Integrase Inhibitor Dolutegravir. Antimicrob. Agents Chemother. 2012, 56, 1627–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.; Borland, J.; Chen, S.; Lou, Y.; Peppercorn, A.; Wajima, T.; Min, S.; Piscitelli, S.C. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next-generation HIV integrase inhibitor, S/GSK1349572. Brit. J. Clin. Pharm. 2011, 72, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.L.; Patterson, K.B.; Prince, H.M.A.; Sykes, C.; Greener, B.N.; Dumond, J.B.; Kashuba, A.D.M. Single and multiple dose pharmacokinetics of dolutegravir in the genital tract of HIV negative women. Antivir. Ther. 2013, 18, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greener, B.N.; Patterson, K.B.; Prince, H.M.A.; Sykes, C.S.; Adams, J.L.; Dumond, J.B.; Shaheen, N.J.; Madanick, R.D.; Dellon, E.S.; Cohen, M.S.; et al. Dolutegravir pharmacokinetics in the genital tract and colorectum of HIV-negative men after single and multiple dosing. Jaids J. Acquir. Immune Defic. Syndr. 2013, 64, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Borland, J.; Chen, S.; Savina, P.; Wynne, B.; Piscitelli, S. Effects of boceprevir and telaprevir on the pharmacokinetics of dolutegravir. Brit. J. Clin. Pharm. 2014, 78, 1043–1049. [Google Scholar] [CrossRef]
- Ross, L.L.; Song, I.H.; Arya, N.; Choukour, M.; Zong, J.; Huang, S.-P.; Eley, T.; Wynne, B.; Buchanan, A.M. No clinically significant pharmacokinetic interactions between dolutegravir and daclatasvir in healthy adult subjects. BMC Infect. Dis. 2016, 16, 347. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Borland, J.; Arya, N.; Wynne, B.; Piscitelli, S. Pharmacokinetics of dolutegravir when administered with mineral supplements in healthy adult subjects. J. Clin. Pharm. 2015, 55, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Borland, J.; Chen, S.; Guta, P.; Lou, Y.; Wilfret, D.; Wajima, T.; Savina, P.; Peppercorn, A.; Castellino, S.; et al. Effects of enzyme inducers efavirenz and tipranavir/ritonavir on the pharmacokinetics of the HIV integrase inhibitor dolutegravir. Eur. J. Clin. Pharm. 2014, 70, 1173–1179. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Borland, J.; Min, S.; Lou, Y.; Chen, S.; Patel, P.; Wajima, T.; Piscitelli, S.C. Effects of etravirine alone and with ritonavir-boosted protease inhibitors on the pharmacokinetics of dolutegravir. Antimicrob. Agents Chemother. 2011, 55, 3517–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.; Weller, S.; Patel, J.; Borland, J.; Wynne, B.; Choukour, M.; Jerva, F.; Piscitelli, S. Effect of carbamazepine on dolutegravir pharmacokinetics and dosing recommendation. Eur. J. Clin. Pharm. 2016, 72, 665–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.H.; Borland, J.; Chen, S.; Savina, P.; Peppercorn, A.F.; Piscitelli, S. Effect of prednisone on the pharmacokinetics of the integrase inhibitor dolutegravir. Antimicrob. Agents Chemother. 2013, 57, 4394–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Chando, T.J.; Everett, D.W.; Patten, C.J.; Dehal, S.S.; Humphreys, W.G. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab. Dispos. 2005, 33, 1729–1739. [Google Scholar] [CrossRef] [Green Version]
- De Bruyn, T.; Stieger, B.; Augustijns, P.F.; Annaert, P.P. Clearance prediction of HIV protease inhibitors in man: Role of hepatic uptake. J. Pharm. Sci. 2016, 105, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Perloff, E.S.; Duan, S.X.; Skolnik, P.R.; Greenblatt, D.J.; von Moltke, L.L. Atazanavir: Effects on P-glycoprotein transport and CYP3A metabolism in vitro. Drug Metab. Dispos. 2005, 33, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaï, J.; De Bruyn, T.; Thevelin, L.; Augustijns, P.; Annaert, P. Transport-metabolism interplay of atazanavir in rat hepatocytes. Drug Metab. Dispos. 2016, 44, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Isentress [Package Insert]; Merck Co., Inc.: Kenilworth, NJ, USA, 2015.
- Kassahun, K.; McIntosh, I.; Cui, D.; Hreniuk, D.; Merschman, S.; Lasseter, K.; Azrolan, N.; Iwamoto, M.; Wagner, J.A.; Wenning, L.A. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab. Dispos. 2007, 35, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Butterton, J.; Persson, A.; Stonier, M.; Comisar, W.; Panebianco, D.; Breidinger, S.; Zhang, J.; Bertz, R. Pharmacokinetics and safety of twice-daily atazanavir 300 mg and raltegravir 400 mg in healthy individuals. Antivir. Ther. 2010, 15, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, M.; Wenning, L.A.; Mistry, G.C.; Petry, A.S.; Liou, S.Y.; Ghosh, K.; Breidinger, S.; Azrolan, N.; Gutierrez, M.J.; Bridson, W.E.; et al. Atazanavir modestly increases plasma levels of raltegravir in healthy subjects. Clin. Infect. Dis. 2008, 47, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Neely, M.; Decosterd, L.; Fayet, A.; Lee, J.S.; Margol, A.; Kanani, M.; di Iulio, J.; von Schoen-Angerer, T.; Jelliffe, R.; Calmy, A. Pharmacokinetics and pharmacogenomics of once-daily raltegravir and atazanavir in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 4619–4625. [Google Scholar] [CrossRef] [Green Version]
- Paine, M.F.; Shen, D.D.; Kunze, K.L.; Perkins, J.D.; Marsh, C.L.; McVicar, J.P.; Barr, D.M.; Gillies, B.S.; Thummel, K.E. First-pass metabolism of midazolam by the human intestine. Clin. Pharm. Ther. 1996, 60, 14–24. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Walker, A.; Hoffer, C.; Sheffels, P. Intravenous and oral alfentanil as in vivo probes for hepatic and first-pass cytochrome P450 3A activity: Noninvasive assessment by use of pupillary miosis. Clin. Pharm. Ther. 2004, 76, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.S.; Obach, R.S.; Rotter, C.; Miller, H.R.; Chang, G.; Steyn, S.J.; El-Kattan, A.; Troutman, M.D. Physicochemical space for optimum oral bioavailability: Contribution of human intestinal absorption and first-pass elimination. J. Med. Chem. 2010, 53, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Ther. 2005, 106, 97–132. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Tang, W.; Ngui, J.; Tephly, T.; Braun, M. Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol. Sci. 2001, 61, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.R.; Hatley, O.J.D.; Ungell, A.-L.; Hilgendorf, C.; Peters, S.A.; Rostami-Hodjegan, A. Gut wall metabolism. Application of pre-clinical models for the prediction of human drug absorption and first-pass elimination. AAPS J. 2016, 18, 589–604. [Google Scholar] [CrossRef]
- Devineni, D.; Murphy, J.; Wang, S.S.; Stieltjes, H.; Rothenberg, P.; Scheers, E.; Mamidi, R.N.V.S. Absolute oral bioavailability and pharmacokinetics of canagliflozin: A microdose study in healthy participants. Clin. Pharm. Drug Dev. 2015, 4, 295–304. [Google Scholar] [CrossRef]
- Devineni, D.; Vaccaro, N.; Murphy, J.; Curtin, C.; Mamidi, R.N.V.S.; Weiner, S.; Wang, S.-S.; Ariyawansa, J.; Stieltjes, H.; Wajs, E.; et al. Effects of rifampin, cyclosporine A, and probenecid on the pharmacokinetic profile of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in healthy participants. Int. J. Clin. Pharm. Ther. 2015, 53, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Wenning, L.A.; Hanley, W.D.; Brainard, D.M.; Petry, A.S.; Ghosh, K.; Jin, B.; Mangin, E.; Marbury, T.C.; Berg, J.K.; Chodakewitz, J.A.; et al. Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob. Agents Chemother. 2009, 53, 2852–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulton, D.W.; Kasichayanula, S.; Keung, C.F.; Arnold, M.E.; Christopher, L.J.; Xu, X.; LaCreta, F. Simultaneous oral therapeutic and intravenous 14C-microdoses to determine the absolute oral bioavailability of saxagliptin and dapagliflozin. Brit. J. Clin. Pharm. 2013, 75, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Raje, S.; Callegari, E.; Sahasrabudhe, V.; Vaz, A.; Shi, H.; Fluhler, E.; Woolf, E.J.; Schildknegt, K.; Matschke, K.; Alvey, C.; et al. Novel application of the two-period microtracer approach to determine absolute oral bioavailability and fraction absorbed of ertugliflozin. Clin. Transl. Sci. 2018, 11, 405–411. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Cubitt, H.E.; Houston, J.B.; Galetin, A. Prediction of human drug clearance by multiple metabolic pathways: Integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab. Dispos. 2011, 39, 864–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Zhang, L.; Grillo, J.A.; Liu, Q.; Bullock, J.M.; Moon, Y.J.; Song, P.; Brar, S.S.; Madabushi, R.; Wu, T.C.; et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharm. Ther. 2011, 89, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Lewinsky, R.H.; Smith, P.A.; Mackenzie, P.I. Glucuronidation of bioflavonoids by human UGT1A10: Structure-function relationships. Xenobiotica 2005, 35, 117–129. [Google Scholar] [CrossRef]
- Sorich, M.J.; McKinnon, R.A.; Miners, J.O.; Smith, P.A. The importance of local chemical structure for chemical metabolism by human uridine 5′-diphosphate-glucuronosyltransferase. J. Chem. Inf. Modeling 2006, 46, 2692–2697. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Intestine (pmol/mg prot) | Liver (pmol/mg prot) | Small Intestine/Liver Ratio 2 | Small Intestine (pmol/mg prot) | Liver (pmol/mg prot) | Small Intestine/Liver Ratio | Small Intestine/Liver Ratio | Small Intestine (pmol/mg prot) | Liver (pmol/mg prot) | Small Intestine/Liver Ratio | Small Intestine (Copy Number Normalized to GAPDH) | Liver (Copy Number Normalized to GAPDH) | Small Intestine/Liver Ratio | Small Intestine/Liver Ratio Assumed in ACAT Model Built in GP 9.8 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UGT1A1 | 7.2 | 18.3 | 0.0157 | 39.6 | 124 | 0.0128 | 0.0280 | 0.33 | 0.87 | 0.0152 | 582 | 1430 | 0.0326 | 0.0196 |
| UGT1A3 | <0.5 | 9.9 | 1.93 | 20.6 | 0.0037 | 0.00284 | 0.04 | 0.3 | 0.0053 | 24 | 131 | 0.0149 | NA | |
| UGT1A4 | 5.3 | 4.6 | 0.0461 | 1.6 | 84 | 0.0008 | <0.99 | 3.95 | BLQ | 618 | 0.0462 | |||
| UGT1A5 | 72 | 9 | 0.667 | 0.401 | ||||||||||
| UGT1A6 | 2.3 | 5.2 | 0.0177 | <2 | 22.6 | <0.22 | 1.02 | 95 | 468 | 0.0162 | 0.0188 | |||
| UGT1A7 3 | 8.4 | BLQ | <2 | <1 | 26 | 5 | 0.4150 | 4.52 3 | ||||||
| UGT1A8 3 | 6.1 | BLQ | <2 | <1 | 70 | BLQ | NA 3 | |||||||
| UGT1A9 | 6.6 | 26.7 | 0.0099 | <2 | 61.2 | <0.03 | 1.43 | 38 | 1210 | 0.0025 | 0.0156 | |||
| UGT1A10 3 | 4.7 | BLQ | 17.9 | <1 | 4.35 | <1.91 | 968 | BLQ | NA 3 | |||||
| UGT2B7 | 15.7 | 200 | 0.0031 | 0.00589 | 0.77 | 6.01 | 0.0051 | 1930 | 4220 | 0.0366 | 0.0268 | |||
| UGT2B15 | <2 | 99.7 | <0.72 | 3.76 | 738 | 18,500 | 0.00319 | 0.00496 | ||||||
| UGT2B17 | 112 | 54.3 | 0.0825 | 1.91 | 0.21 | 0.364 | 2680 | 197 | 1.09 | 0.655 | ||||
| Source | Harbourt et al. [9] 4 | Sato et al. [11] 4 | Drozdzik et al. [34] 5 | Basit et al. [8] 6 | Ohno and Shizuo [10] 4 | |||||||||
| Sample type | Microsome fraction | Microsome fraction | Tissue homogenate | S9 fraction | Total RNA | |||||||||
| Parameter | Cabotegravir | Dolutegravir | Lorazepam | Oxazepam | Naloxone | Raltegravir | Zidovudine |
|---|---|---|---|---|---|---|---|
| Drug class | Antiretroviral | Antiretroviral | Benzodiazepine | Benzodiazepine | Opioid antagonist | Antiretroviral | Antiretroviral |
| Model reference | Internal Sim+ | [24] | [23] | [23] | [23] | [24] | [23] |
| MW, g/mol | 405.4 | 419.4 | 321 | 287 | 327 | 445.2 | 267 |
| pKa | 4.52 (acid) | 4.58 (Acid), 8.2 (Acid) | 1.3 (base) 11.5 (acid) | 2.57 (base) 11.31 (acid) | 7.9 (base) | 5.5 (acid), 1.53 (base) | 9.1 (acid) |
| LogP | 2.16 2 | 2.16 | 2.4 | 2.4 | 1.92 | 0.58 | 0.06 |
| Solubility, mg/mL (reference pH) | <0.01 free acid | 2.1 × 10−4 at pH = 1.2 | 0.0485 at pH = 7 | 0.057 at pH = 7 | 0.66 at pH = 8.45 | 0.014 (1.2) 0.020 (4.5) 0.1 (6.8) | 25.4 at pH = 7 |
| Permeability (cm/s × 104) | 2.54 | 2.37 | 3.3 | 3.6 | 4 | 1.79 | 1.9 |
| fu,p | 0.01 | 0.009 | 0.11 | 0.6 | 0.56 | 0.17 | 0.74 |
| Rbp | 0.58 | 0.55 | 0.75 | 1.1 | 1.1 | 0.60 | 0.86 |
| Major CL mechanism | Metabolism (~79%), renal (~21%) | Metabolism | Metabolism | Metabolism | Metabolism | Hepatic (88–95.6%), renal (4.4–12%) | Metabolism (~75%), renal (~25%) |
| Hepatic enzymes, fm in % | UGT1A1 (66.8%), UGT1A9 (33.2%) | UGT1A1 (51%), CYP3A4 (21%) 4 | UGT2B7 (42%), UGT2B15 (58%) | UGT1A9 (13%), UGT2B15 (87%) | UGT2B7 (100%) | UGT1A1 (100) | UGT2B7 (85%), CYP3A4 (15%) |
| Simulated Fg | 85.1% | 68–84% (dose dependent) | 95% 3 | 96% 3 | 17% | 51–52% | 93% 3 |
| Simulated Fh | 99.82% | 99% | 96% | 94% | 6% | 79% | 53% |
| Study | Atazanavir Dose, mg | Raltegravir Dose, mg | Observed RCmax | Observed RAUC | Simulated RCmax | Simulated RAUC |
|---|---|---|---|---|---|---|
| Iwamoto et al. [86] | 400 QD | 100 | 1.53 | 1.72 | 1.8–1.9 | 2.0–2.1 |
| Neely et al. [87] 2 | 0/400 QD 2 | 400 BID/400 QD 2 | 1.32 | 1.37 | 1.8 | 1.7–1.8 |
| Zhu et al. [85] | 300 BID | 400 BID | 1.54 | 1.54 | 1.8–1.9 | 1.8–2.0 |
| Name | F | Fa | Fg | Fh | UGTs Involved | Intestinal Metabolism? 2 | Reference |
|---|---|---|---|---|---|---|---|
| Canagliflozin | 0.65 | 1 3 | 0.75 4 | 0.86 | UGT1A9, UGT2B4 | No | Devineni et al. [94] |
| Dapagliflozin | 0.78 | 1 3 | 0.90 | 0.86 | UGT1A9, UGT2B7 | Yes | Boulton et al. [97] |
| Diclofenac | 0.54 | 1 3 | 0.64 | 0.85 | UGT2B7, UGT1A9, UGT1A6, UGT2B15 | Yes | Varma et al. [90] |
| Ertugliflozin | 1.05 | 1.11 | 1.08 | 0.88 | UGT1A9, UGT2B4, UGT2B7 | Yes | Raje et al. [98] |
| Gemfibrozil | 0.98 | 1 3 | 1.09 | 0.90 | UGT2B7 | Yes | Nishimuta et al. [20] |
| Lorazepam | 0.93 | 1 3 | 0.97 | 0.96 | UGT2B4, UGT2B7, UGT2B15 | Yes | Varma et al. [90] |
| Raloxifene | 0.02 | 0.63 | 0.054 | 0.593 | UGT1A1, UGT1A8, UGT1A9, UGT1A10 5 | Yes | Mizuma [12] |
| Troglitazone | 0.45 | 1 3 | 0.56 | 0.80 | UGT1A1, UGT1A10 | Yes | Nishimuta et al. [20] |
| Lorazepam | 0.93 | 1 3 | 0.97 | 0.96 | UGT2B4, UGT2B7, UGT2B15 | Yes | Varma et al. [90] |
| Telmisartan | 0.43 | 0.90 | 0.75 | 0.64 | UGT1A1, UGT1A3, UGT1A9 | Yes | Varma et al. [90] |
| Oxazepam | 0.93 | 0.93 | 1.01 | 0.99 | UGT1A9, UGT2B7, UGT2B15 | Yes | Varma et al. [90] |
| Substrate of 2: | UGT1A1 | UGT1A3 | UGT1A4 | UGT1A6 | UGT1A8 | UGT1A9 | UGT1A10 | UGT2B7 | UGT2B15 |
|---|---|---|---|---|---|---|---|---|---|
| Overall Accuracy | 85% | 85% | 88% | 85% | 87% | 88% | 85% | 84% | 91% |
| Cabotegravir (UGT1A1, UGT1A9) | Yes (96%) | No (78%) | No (99%) | No (97%) | Yes | Yes (88%) | Yes (56%) | Yes (80%) | Yes (52%) |
| Canagliflozin (UGT1A9, UGT2B4) | No (50%) | No (66%) | Yes (89%) | No (83%) | No (98%) | No (96%) | No (75%) | No (70%) | No (92%) |
| Dolutegravir (UGT1A1) | Yes (96%) | No (78%) | No (99%) | No (97%) | Yes | Yes (62%) | Yes (56%) | Yes (80%) | Yes (58%) |
| Lorazepam (UGT2B7, UGT2B15) | No (95%) | No (92%) | No (66%) | No (97%) | Yes (81%) | No (96%) | Yes (71%) | Yes (80%) | No (82%) |
| Naloxone (UGT2B7) | No (99%) | Yes (53%) | No (99%) | No (97%) | Yes (72%) | No (96%) | No (97%) | Yes (93%) | No (98%) |
| Oxazepam (UGT1A9, UGT2B15) | No (97%) | No (92%) | Yes (45%) | No (88%) | No (63%) | No (96%) | Yes (64%) | Yes (93%) | No (98%) |
| Raltegravir (UGT1A1) | Yes (90%) | No (49%) | No (99%) | No (91%) | Yes (75%) | Yes (73%) | No (70%) | Yes (74%) | Yes |
| Tapentadol (UGT1A9, UGT2B7) | No (99%) | Yes (90%) | Yes (59%) | No (97%) | Yes (78%) | Yes (97%) | No (90%) | Yes (85%) | No (67%) |
| Zidovudine (UGT2B7) | No (98%) | No (98%) | No (95%) | No (97%) | No | No (69%) | No (97%) | Yes (66%) | No (84%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, M.B.; Bolger, M.B.; Fraczkiewicz, G.; Del Frari, L.; Luo, L.; Lukacova, V.; Mitra, A.; Macwan, J.S.; Mullin, J.M.; Parrott, N.; et al. PBPK Modeling as a Tool for Predicting and Understanding Intestinal Metabolism of Uridine 5′-Diphospho-glucuronosyltransferase Substrates. Pharmaceutics 2021, 13, 1325. https://doi.org/10.3390/pharmaceutics13091325
Reddy MB, Bolger MB, Fraczkiewicz G, Del Frari L, Luo L, Lukacova V, Mitra A, Macwan JS, Mullin JM, Parrott N, et al. PBPK Modeling as a Tool for Predicting and Understanding Intestinal Metabolism of Uridine 5′-Diphospho-glucuronosyltransferase Substrates. Pharmaceutics. 2021; 13(9):1325. https://doi.org/10.3390/pharmaceutics13091325
Chicago/Turabian StyleReddy, Micaela B., Michael B. Bolger, Grace Fraczkiewicz, Laurence Del Frari, Laibin Luo, Viera Lukacova, Amitava Mitra, Joyce S. Macwan, Jim M. Mullin, Neil Parrott, and et al. 2021. "PBPK Modeling as a Tool for Predicting and Understanding Intestinal Metabolism of Uridine 5′-Diphospho-glucuronosyltransferase Substrates" Pharmaceutics 13, no. 9: 1325. https://doi.org/10.3390/pharmaceutics13091325
APA StyleReddy, M. B., Bolger, M. B., Fraczkiewicz, G., Del Frari, L., Luo, L., Lukacova, V., Mitra, A., Macwan, J. S., Mullin, J. M., Parrott, N., & Heikkinen, A. T. (2021). PBPK Modeling as a Tool for Predicting and Understanding Intestinal Metabolism of Uridine 5′-Diphospho-glucuronosyltransferase Substrates. Pharmaceutics, 13(9), 1325. https://doi.org/10.3390/pharmaceutics13091325