
Stability of Hydrocortisone in Oral Powder Form Compounded for Pediatric Patients in Japan

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Test Solution Preparation
2.2. Hydrocortisone Powder Compounding
2.3. Stability Study
2.4. Dissolution Test
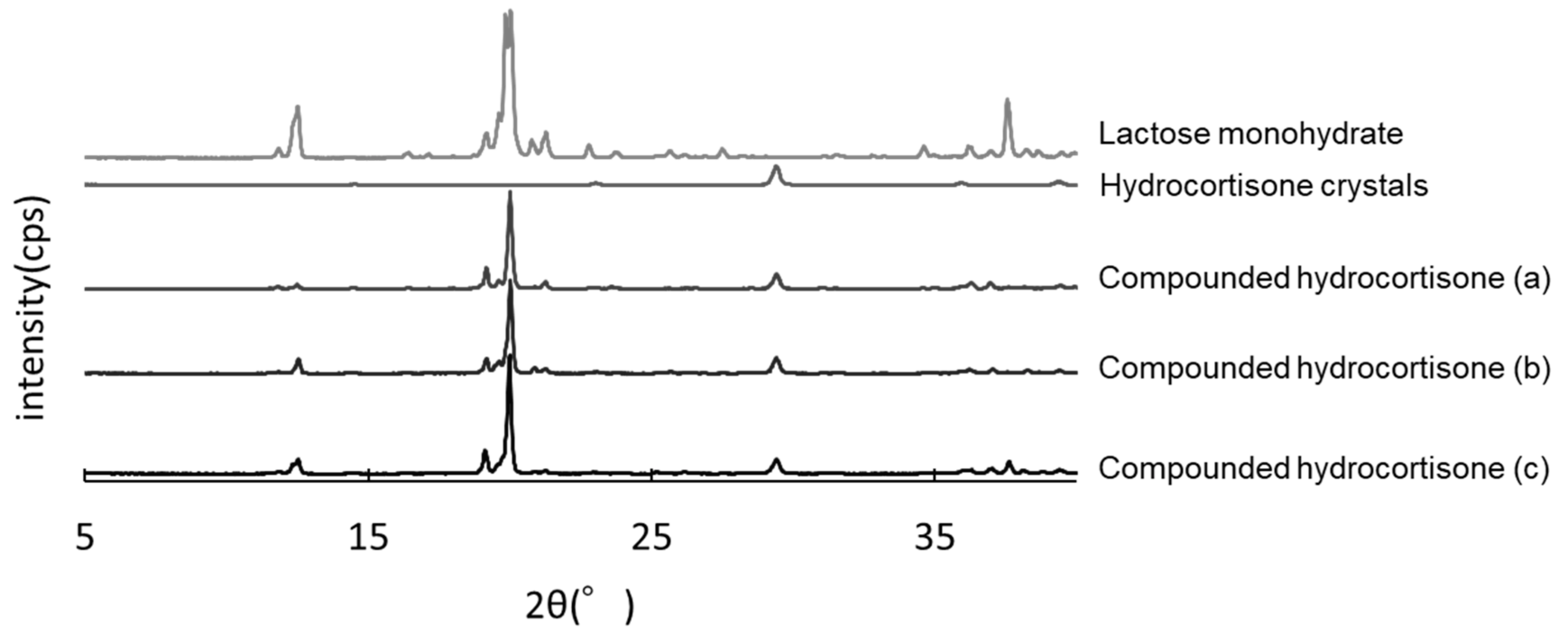
2.5. Powder X-ray Diffraction Analysis
2.6. Assays of Hydrocortisone and Its Impurities
2.6.1. Instrumentation and Chromatographic Conditions
2.6.2. Linearity, Precision, and Accuracy
2.7. Assay for Known and Unknown Hydrocortisone Impurities
3. Results
3.1. Liquid Chromatography Method and Validation
3.2. Stability Study
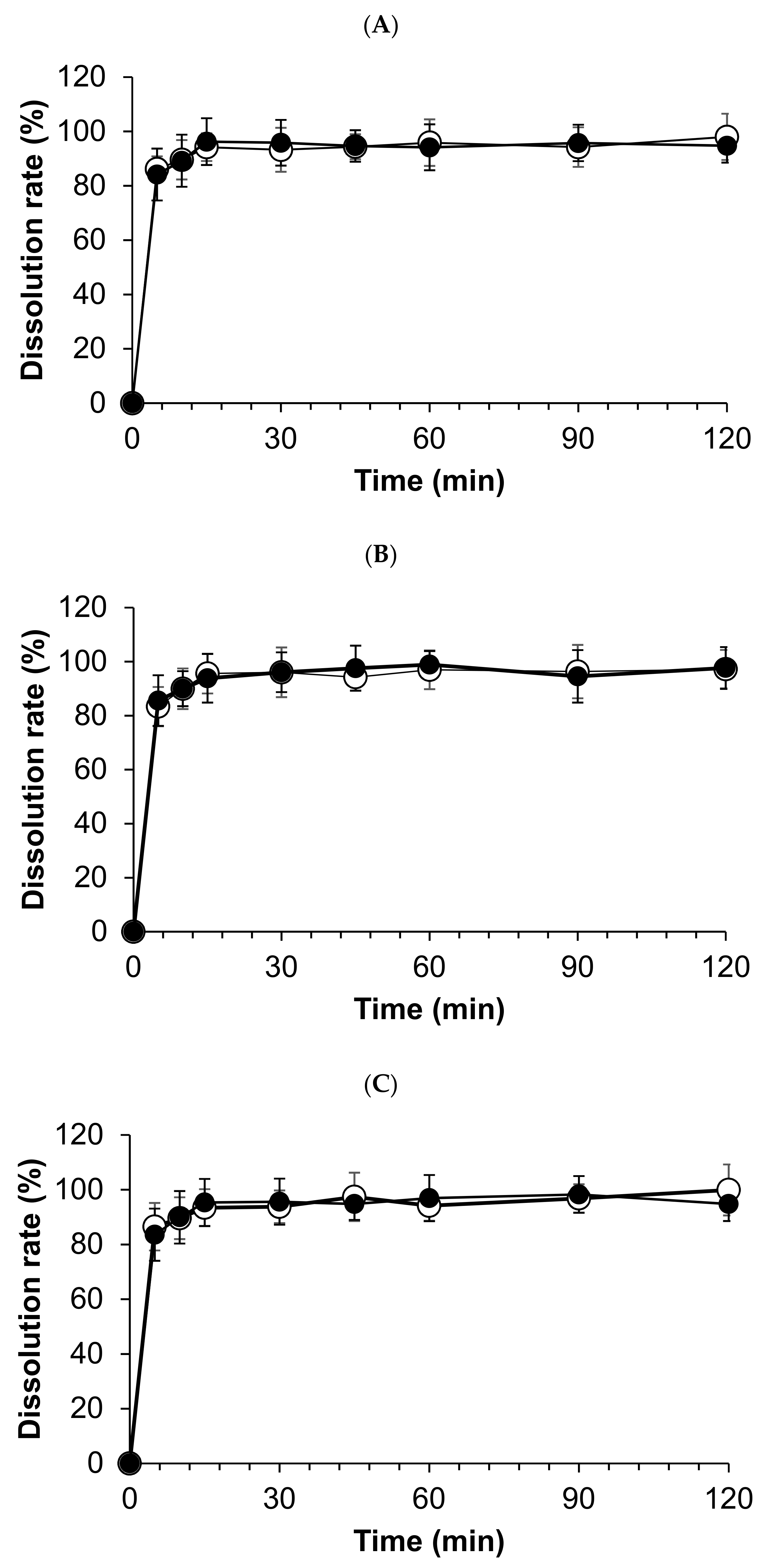
3.3. Dissolution Test
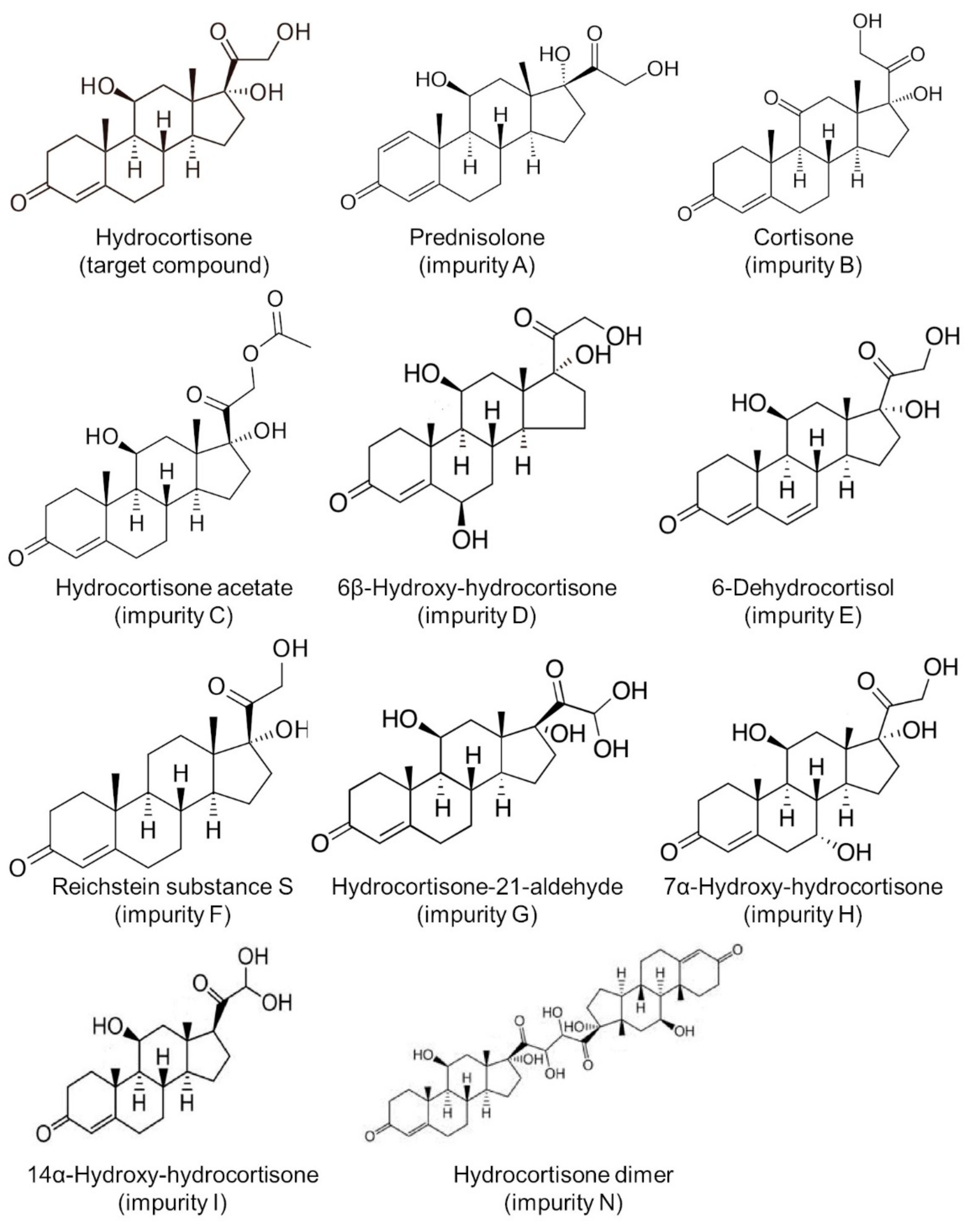
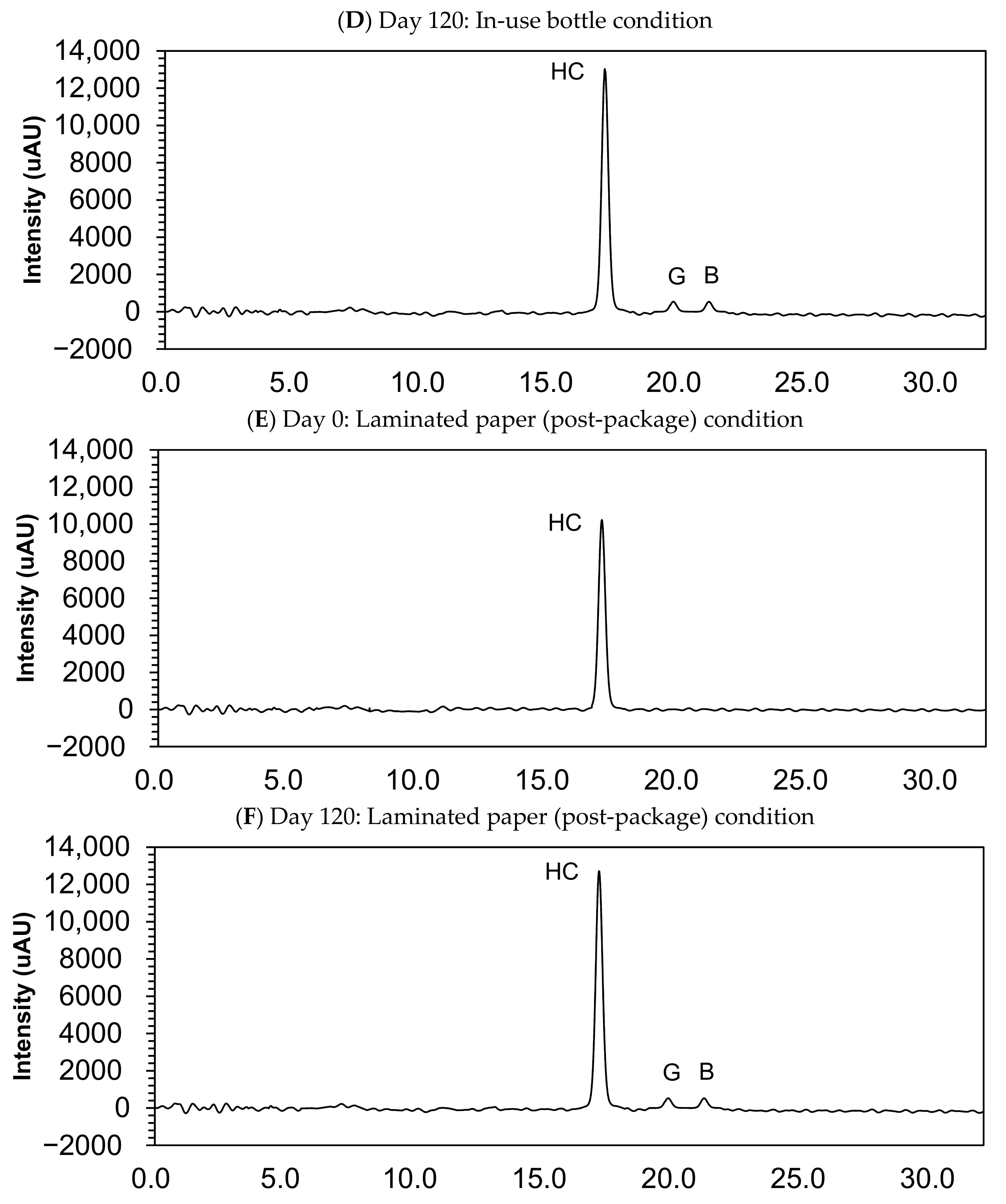
3.4. Impurity Study
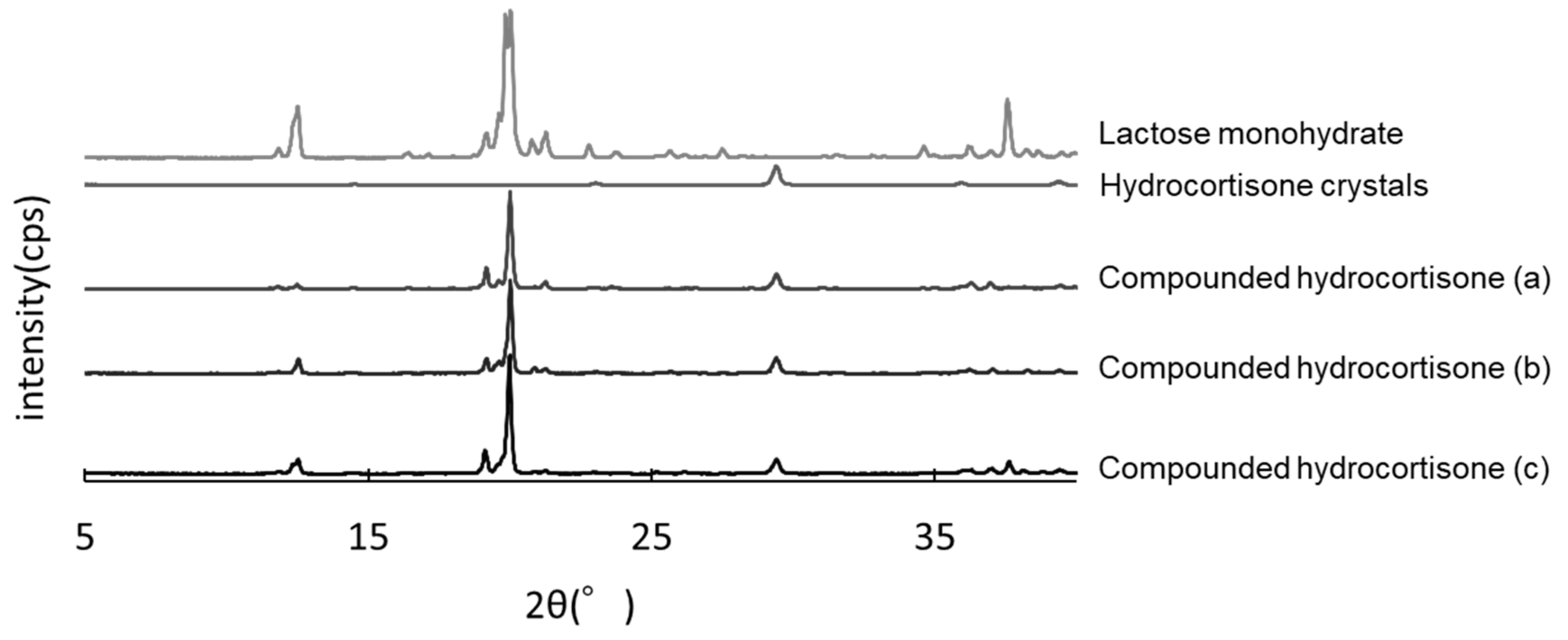
3.5. PXRD Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirkgoz, T.; Guran, T. Primary adrenal insufficiency in children: Diagnosis and management. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 397–424. [Google Scholar] [CrossRef]
- Oprea, A.; Bonnet, N.C.G.; Pollé, O.; Lysy, P.A. Novel insights into glucocorticoid replacement therapy for pediatric and adult adrenal insufficiency. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018818821294. [Google Scholar] [CrossRef]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Porter, J.; With, M.; Ross, R.J. Immediate-release granule formulation of hydrocortisone, Alkindi®, for treatment of paediatric adrenal insufficiency (Infacort development programme). Expert Rev. Endocrinol. Metab. 2018, 13, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Neumann, U.; Whitaker, M.J.; Wiegand, S.; Krude, H.; Porter, J.; Davies, M.; Digweed, D.; Voet, B.; Ross, R.J.; Blankenstein, O. Absorption and tolerability of taste-masked hydrocortisone granules in neonates, infants and children under 6 years of age with adrenal insufficiency. Clin. Endocrinol. 2018, 88, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Neumann, U.; Burau, D.; Spielmann, S.; Whitaker, M.J.; Ross, R.J.; Kloft, C.; Blankenstein, O. Quality of compounded hydrocortisone capsules used in the treatment of children. Eur. J. Endocrinol. 2017, 177, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Webb, E.A.; Watson, C.; Kerr, S.; Davies, J.H.; Stirling, H.; Batchelor, H. Hydrocortisone tablets: Human factors in manipulation and their impact on dosing accuracy. Endocr. Abstr. 2017, 51, OC8.1. [Google Scholar] [CrossRef]
- Barillas, J.E.; Eichner, D.; Van Wagoner, R.; Speiser, P.W. Iatrogenic Cushing syndrome in a child with congenital adrenal hyperplasia: Erroneous compounding of hydrocortisone. J. Clin. Endocrinol. Metab. 2018, 103, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Al-Rayess, H.; Fleissner, K.; Jaber, M.; Brundage, R.C.; Sarafoglou, K. Manipulation of hydrocortisone tablets leads to iatrogenic Cushing syndrome in a 6-year-old girl with CAH. J. Endocr. Soc. 2020, 4, bvaa091. [Google Scholar] [CrossRef]
- Saito, J.; Akabane, M.; Ishikawa, Y.; Iwahashi, K.; Nakamura, H.; Yamatani, A. Retrospective survey of compounded medications for children in Japan. Eur. J. Pharm. Biopharm. 2020, 155, 122–127. [Google Scholar] [CrossRef]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use [ICH]. Evaluation for Stability Data (Q1E). Available online: https://database.ich.org/sites/default/files/Q1E%20Guideline.pdf (accessed on 18 July 2021).
- Madathilethu, J.; Roberts, M.; Peak, M.; Blair, J.; Prescott, R.; Ford, J.L. Content uniformity of quartered hydrocortisone tablets in comparison with mini-tablets for paediatric dosing. BMJ Paediatr. Open 2018, 2, e000198. [Google Scholar] [CrossRef] [PubMed]
- Ledeți, I.; Bengescu, C.; Cîrcioban, D.; Vlase, G.; Vlase, T.; Tomoroga, C.; Buda, V.; Ledeti, A.; Dragomirescu, A.; Murariu, M. Solid-state stability and kinetic study of three glucocorticoid hormones: Prednisolone, prednisone and cortisone. J. Therm. Anal. Calorim. 2020, 141, 1053–1065. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Chen, B.; Chan, T.M.; Rustum, A. Forced degradation of betamethasone sodium phosphate under solid state: Formation, characterization, and mechanistic study of all four 17,20-diastereomers of betamethasone 17-deoxy-20-hydroxy-21-oic acid. J. Pharm. Sci. 2009, 98, 894–904. [Google Scholar] [CrossRef]
- Cortril® Tablets 10 mg [Prescribing Information]; Pfizer Japan Inc.: Tokyo, Japan, 2020.
- Committee for Medicinal Products for Human Use [CHMP]. Assessment Report; Plenadren: London, UK, 2011; Available online: https://www.ema.europa.eu/en/documents/assessment-report/plenadren-epar-public-assessment-report_en.pdf (accessed on 18 July 2021).
- Wollmer, E.; Karkossa, F.; Freerks, L.; Hetberg, A.E.; Neal, G.; Porter, J.; Whitaker, M.J.; Margetson, D.; Klein, S. A Biopredictive In Vitro Approach for Assessing Compatibility of a Novel Pediatric Hydrocortisone Drug Product within Common Pediatric Dosing Vehicles. Pharm. Res. 2020, 37, 203. [Google Scholar] [CrossRef]
- Pilaniya, K.; Chandrawanshi, H.K.; Pilaniya, U.; Manchandani, P.; Jain, P.; Singh, N. Recent trends in the impurity profile of pharmaceuticals. J. Adv. Pharm. Technol. Res. 2010, 1, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakam, L.N.R.; Dongala, T.; Ettaboina, S.K. Novel stability indicating UHPLC method development and validation for simultaneous quantification of hydrocortisone acetate, pramoxine hydrochloride, potassium sorbate and sorbic acid in topical cream formulation. Talanta Open. 2020, 1, 100004. [Google Scholar] [CrossRef]
- Committee for Proprietary Medical Products [CPMP]. Note for Guidance on In-Use Stability Testing of Human. London, UK. 2001. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-use-stability-testing-human-medicinal-products_en.pdf (accessed on 18 July 2021).
- Allen, L.V., Jr.; Bassani, G.S.; Elder, E.J.; Parr, A.F. Strength and stability testing for compounded preparations. U.S. Pharmacop. 2014. Available online: https://www.usp.org/sites/default/files/usp/document/FAQs/strength-stability-testing-compounded-preparations.pdf (accessed on 18 July 2021).
- The Ministry of Health, Labour and Welfare [MHLW]. The Japanese Pharmacopoeia Seventeenth Edition. 2016. Available online: https://www.pmda.go.jp/files/000217650.pdf (accessed on 18 July 2021).
- Fawcett, J.P.; Boulton, D.W.; Jiang, R.; Woods, D.J. Stability of hydrocortisone oral suspensions prepared from tablets and powder. Ann. Pharmacother. 1995, 29, 987–990. [Google Scholar] [CrossRef]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use [ICH]. Guidelines on Impurities in Bulk Drugs with New Active Ingredients (Q3A(R2)). 2011. Available online: https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf (accessed on 18 July 2021).
- Litalien, C.; Autmizguine, J.; Carli, A.; Giroux, D.; Lebel, D.; Leclerc, J.M.; Théorêt, Y.; Gilpin, A.; Bérubé, S. Providing Suitable Pediatric Formulations for Canadian Children: A Call for Action. Can. J. Hosp. Pharm. 2020, 73, 247–256. [Google Scholar]
- Gerrard, S.E.; Walsh, J.; Bowers, N.; Salunke, S.; Hershenson, S. Innovations in Pediatric Drug Formulations and Administration Technologies for Low Resource Settings. Pharmaceutics 2019, 11, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riet-Nales, D.A.; Kozarewicz, P.; Aylward, B.; de Vries, R.; Egberts, T.C.; Rademaker, C.M.; Schobben, A.F. Paediatric Drug Development and Formulation Design—A European Perspective. AAPS PharmSciTech 2017, 18, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Gadge, P.M.; Kenjale, P.P.; Pokharkar, V.B.; Gaikwad, V.L. Global pediatric regulations: An overview. Ther. Innov. Regul. Sci. 2019, 54, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, C.M.; Gilpin, A.; Autmizguine, J.; Denburg, A.; Dupuis, L.L.; Finkelstein, Y.; Gruenwoldt, E.; Ito, S.; Jong, G.; Lacaze-Masmonteil, T.; et al. Improving paediatric medications: A prescription for Canadian children and youth. Paediatr. Child Health 2019, 24, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, V.; Rademaker, C.M.; van Dijk, L.; Mantel-Teeuwisse, A.K. Pediatric drug formulations: A review of challenges and progress. Pediatrics 2014, 134, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchelor, H.; Salunke, S.; Tuleu, C. European Paediatric Formulation Initiative (EuPFI). Formulating better medicines for children-reflections. Int. J. Pharm. 2015, 492, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Conroy, S. Extemporaneous (magistral) preparation of oral medicines for children in European hospitals. Acta Paediatr. 2003, 92, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Chong, G.; Decarie, D.; Ensom, M.H.H. Stability of hydrocortisone in extemporaneously compounded suspension. J. Inform. Pharmacother. 2003, 13, 100–110. [Google Scholar]
- Manchanda, A.; Laracy, M.; Savji, T.; Bogner, R.H. Stability of an alcohol-free, dye-free hydrocortisone (2 mg/mL) compounded oral suspension. Int. J. Pharm. Compd. 2018, 22, 66–75. [Google Scholar]
- SickKids. Hydrocortisone 1 mg/mL Oral Suspension. Toronto, Canada. 2020. Available online: https://www.sickkids.ca/siteassets/care--services/for-health-care-providers/compounding-recipes/hydrocortisone-1mgml-pharmacy-compounding-recipe.pdf (accessed on 18 July 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Retention Time (min) | |
|---|---|---|
| Target compound | Hydrocortisone | 17.1 |
| Impurity A | Prednisolone | 15.3 |
| Impurity B | Cortisone | 21.2 |
| Impurity C | Hydrocortisone acetate | 23.6 |
| Impurity D | 6β-Hydroxy-hydrocortisone | 8.7 |
| Impurity E | 6-Dehydrocortisol | 13.5 |
| Impurity F | Reichstein substance S | 27.5 |
| Impurity G | Hydrocortisone-21-aldehyde | 19.8 |
| Impurity H | 7α-Hydroxy-hydrocortisone | 10.5 |
| Impurity I | 14α-Hydroxy-hydrocortisone | 12.1 |
| Impurity N | Hydrocortisone dimer | 19.1 |
| Study Methods | Storage Conditions | Storage Container | Test Periods (Days) | ||||
|---|---|---|---|---|---|---|---|
| 0 | 30 | 60 | 90 | 120 | |||
| Hydrocortisone Concentrations * | |||||||
| Bottle (closed) | 25 °C ± 2 °C/60% ± 5% relative humidity | Amber/PC bottle | 100.0% | 99.4 ± 2.2% | 99.8 ± 4.1% | 98.9 ± 4.1% | 98.9 ± 3.6% |
| Bottle (in use) | Amber/PC bottle | 100.0% | 98.2 ± 1.3% | 100.8 ± 1.7% | 97.9 ± 3.8% | 98.5 ± 2.2% | |
| Laminated paper | Amber/CP laminated paper | 100.0% | 99.6 ± 1.2% | 99.7 ± 3.4% | 102.1 ± 2.4% | 100.2 ± 3.1% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, J.; Yoshikawa, N.; Hanawa, T.; Ozawa, A.; Matsumoto, T.; Harada, T.; Iwahashi, K.; Nakamura, H.; Yamatani, A. Stability of Hydrocortisone in Oral Powder Form Compounded for Pediatric Patients in Japan. Pharmaceutics 2021, 13, 1267. https://doi.org/10.3390/pharmaceutics13081267
Saito J, Yoshikawa N, Hanawa T, Ozawa A, Matsumoto T, Harada T, Iwahashi K, Nakamura H, Yamatani A. Stability of Hydrocortisone in Oral Powder Form Compounded for Pediatric Patients in Japan. Pharmaceutics. 2021; 13(8):1267. https://doi.org/10.3390/pharmaceutics13081267
Chicago/Turabian StyleSaito, Jumpei, Nozomi Yoshikawa, Takehisa Hanawa, Ayuna Ozawa, Takahiro Matsumoto, Tsutomu Harada, Kana Iwahashi, Hidefumi Nakamura, and Akimasa Yamatani. 2021. "Stability of Hydrocortisone in Oral Powder Form Compounded for Pediatric Patients in Japan" Pharmaceutics 13, no. 8: 1267. https://doi.org/10.3390/pharmaceutics13081267
APA StyleSaito, J., Yoshikawa, N., Hanawa, T., Ozawa, A., Matsumoto, T., Harada, T., Iwahashi, K., Nakamura, H., & Yamatani, A. (2021). Stability of Hydrocortisone in Oral Powder Form Compounded for Pediatric Patients in Japan. Pharmaceutics, 13(8), 1267. https://doi.org/10.3390/pharmaceutics13081267